
Abstract
Aims/hypothesis
Beta cell destruction in type 1 diabetes is apparently mediated by the release of cytokines. We questioned whether cytokine-induced apoptosis preferentially kills replicating beta cells.
Materials and methods
In the first experiment, rat insulinoma (RIN) cells were studied for 36 h by time-lapse video microscopy. Cells were exposed to three doses of a cytokine mixture (maximal concentration: IL-1β 50 U/ml; TNF-α 1,000 U/ml; IFN-γ 1,000 U/ml) or vehicle and analysed for the total cell number (2-h intervals) and timing of each cell death and division. In the second experiment, isolated human islets were incubated with the same cytokine mixture for 24 h and examined for replication and paired (postmitotic) apoptosis.
Results
In the first experiment, after application of cytokines, apoptosis occurred most frequently immediately after the next or subsequent cell mitosis (p<0.05). In the second experiment, cytokines caused increased apoptosis in human islets, with an increase in the proportion of postmitotic apoptotic pairs (p<0.001).
Conclusions/interpretation
Cytokine-induced beta cell death preferentially affects newly forming beta cells, which implies that replicating beta cells might be more vulnerable to cytokine destruction. Efforts to expand beta cell mass in type 1 diabetes by fostering beta cell replication are likely to fail unless cytokine-induced apoptosis is concurrently suppressed.
Similar content being viewed by others


Introduction
Type 1 diabetes is characterised by a nearly complete deficiency of beta cells, resulting in marked insulin deficiency [1–3]. The beta cell deficit is believed to be due to autoimmune induced beta cell apoptosis mediated by the release of inflammatory cytokines, such as IL-1β, TNF-α and IFN-γ from T lymphocytes and macrophages [4]. However, recent studies have shown that most patients with long-standing type 1 diabetes still have some detectable pancreatic beta cells despite continued beta cell apoptosis [3]. This implies ongoing provision of new beta cells even after several years of type 1 diabetes. One obvious question arising from these studies is why beta cell mass fails to regenerate in type 1 diabetes. In the present studies we tested the hypothesis that beta cell regeneration in type 1 diabetes is prevented by increased vulnerability of newly forming beta cells to cytokine-induced cell death.
Subjects, materials and methods
Study design
To address this hypothesis, we evaluated the temporal relationship between cell replication and apoptosis using a time-lapse video microscopy (TLVM) system, which allowed live imaging of a beta cell line (RINm5) in a microculture environment during exposure to different concentrations of a cytokine mixture (IL-1β, IFN-γ, TNF-α) or vehicle. Since beta cell replication has also been described in isolated human islets under culture conditions [5, 6], human islets from pancreases of four heart-beating, brain-dead cadaver donors were used. The pancreases were obtained with informed consent from the donors’ relatives. Human islets were cultured over 24 h in the presence of a cytokine mixture (IL-1β, IFN-γ, TNF-α) or vehicle and stained for apoptosis using the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labelling (TUNEL) method. The percentage of apoptotic cells presenting in pairs was used as a marker of postmitotic apoptosis, as previously described [7, 8].
Time-lapse video microscopy
For TLVM experiments, the microculture dish was mounted on to the motorised stage of an inverted microscope, as described [7]. Microculture conditions were maintained by a dynamic temperature controller and constant inflow of air containing 5% CO2. RIN cells were exposed to three different concentrations of a cytokine mixture (100, 50 and 25% of the following combination: IL-1β, 50 mU/l; TNF-α, 1,000 U/l; IFN-γ, 1,000 U/l) or vehicle over a period of 36 h. Subsequently, images from nine separate locations in the culture chamber were acquired every 10 min, and the rate of cell division, cell death, and the net cell number were analysed by examining the merged image sequences using Image-Pro Plus software (Media Cybernetics, Silver Springs, MD, USA). A successful cell division was judged to have occurred when a single cell divided into two daughter cells. Apoptosis was judged to have occurred when a cell rounded up, the nucleus condensed and subsequently fragmented, and the cell cytoplasm disintegrated via cytoplasmic blebs into apoptotic bodies. In addition, in the experiments with the intermediate cytokine concentration (IL-1β, 25 mU/l; TNF-α, 500 U/l; IFN-γ, 500 U/l), the time interval between replication and subsequent apoptosis in individual replicating cells was analysed.
Beta cell apoptosis and replication in human islet tissue
Human pancreatic islets were obtained through the National Institute of Health islet distribution programme and all studies were approved by the institutional review board. Human islets were isolated from the whole pancreas retrieved from four heart-beating organ-donors (two male, two female). The donors were 50–61 years old, and BMI was 24–29 kg/m2. None of them had a history of diabetes. After isolation, islets were maintained in RPMI medium (5 mmol/l glucose). Aliquots of human islets were incubated over 24 h with vehicle or a mixture of cytokines containing IL-1β (50 mU/l), TNF-α (1,000 U/l) and IFN-γ (1,000 U/l). The number of apoptotic cells was detected using the TUNEL method (In Situ Cell Death Detection Kit, AP; Roche Diagnostics, Indianapolis, IN, USA). TUNEL-positive cells that were less than a single cell width from each other in cross-sectional area were counted as pairs (n=3 donors), as described [7].
In order to confirm that the apoptosis and replication we observed in the islets was indeed occurring in beta cells, groups of ∼30 islets were plated on extracellular matrix-coated plates derived from bovine corneal endothelial cells, allowing the islet cells to attach to the dishes and spread (Novamed, Jerusalem, Israel), and cultured in 2 ml RPMI (5 mmol/l glucose) medium, as previously described [6]. Five days after plating, when most islets were attached and had flattened, islets were treated with the cytokine mixture or vehicle over 24 h. Subsequently, islets were stained for apoptosis (TUNEL In Situ Cell Death Detection Kit, AP), or for Ki67 using a monoclonal antibody against the human Ki-67 antigen (lot no. 31280059; Zymed, San Francisco, CA, USA), as previously described [6]. The islets were then incubated for 30 min at 37°C with guinea-pig anti-insulin antibody diluted 1:50 (lot no. 50381573; Dako, Carpinteria, CA, USA), followed by a 30-min incubation with a fluorescein-conjugated rabbit anti-guinea pig antibody. The number of Ki67-positive beta cells was quantified after treatment with cytokines and vehicle (n=5 dishes of 30 islets each in both groups).
Cytokines
For human islet experiments, human recombinant IFN-γ (Sigma), human recombinant IL-1γ (Sigma) and human TNF-α (R&D Systems) were used. For TLVM experiments in RIN cells, rat IFN-γ (Sigma), rat IL-1β (Sigma) and rat TNF-α (Sigma) were used. Cytokines were diluted in de-ionised water to prepare stock solutions. The stock solutions were then diluted with the culture medium to obtain the desired final concentration.
Statistics
Results are presented as mean±SEM. Statistics were carried out by repeated-measures ANOVA and Duncan’s post hoc test using Statistica, version 5 (Statsoft, Tulsa, OK, USA). A pvalue< 0.05 was taken to indicate a significant difference.
Results
Time-lapse video microscopy
During control experiments, the RIN cell population increased by 237±13% after 36 h of incubation. In contrast, the number of RIN cells was reduced by 82±5, 86±4 and 21±2% of initial values during cytokine treatment at 100, 50 and 25% of the maximal cytokine concentration, respectively (p<0.001) (Fig. 1). The reduction in cell viability by the cytokine exposure was accompanied by a significant increase in the frequency of apoptosis, which was most apparent after 12–24 h (Fig. 1). In addition, the frequency of replication was dose-dependently reduced by the cytokine exposure (Fig. 1). The mean rates of apoptosis and replication per 12-h experimental period are presented in Table 1. The temporal relationship between cell division and apoptosis was studied in detail during incubation with an intermediate cytokine concentration, because under this condition both replication and apoptosis occur frequently. In these experiments, the mean observation period of RIN cells after replication was 28.5±6.7 h. Apoptosis was documented in 95 out of 180 replicating cells. The mean interval between replication and apoptosis was 13.2±0.9 h. Interestingly, apoptosis was greatest during the first 4 h after replication and after 16–20 h (Fig. 1). The second peak of apoptosis preceded the anticipated next division in this cell line (typical cell cycle length, 26±0.6 h).

Change in a the relative size of the cell population, b the apoptotic rate (expressed as percentage apoptotic cells per total cell number per 2 h), and c the mitotic rate (expressed as percentage replicating cells per total cell number per 2 h) in RIN cells cultured for 36 h on the stage of a time-lapse video microscope. Cells were cultured during incubation with control solution or different concentrations of a cytokine mixture (maximal concentration: IL-1β 50 U/ml; TNF-α 1,000 U/ml; IFN-γ 1,000 U/ml). Open circle, control; closed circle, cytokines 100%; closed diamond, cytokines 50%; inverted triangle, cytokines 25%. d Frequency of apoptosis per 4-h time interval following cell division in RIN-cells exposed to a mixture of cytokines (IL-1β: 25 mU/l, TNF- α: 500 U/l, IFN-γ: 500 U/l) over 36 h. Results are of four separate experiments with nine different locations each. Data are expressed as means±SEM, statistical analysis was by repeated-measures ANOVA and one-way ANOVA. p<0.0001 (a–c); p=0.046 (d)
Beta cell replication in human islets grown on extracellular matrix
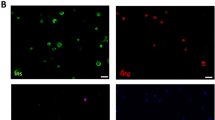
Double staining for insulin and Ki67 identified replicating beta cells both under control conditions and after cytokine exposure (Fig. 2). The frequency of beta cell replication was not significantly different after cytokine treatment compared with vehicle (0.053±0.022 vs 0.091±0.014 Ki67-positive beta cells per islet, respectively; p=0.23).

An Isolated islet from a non-diabetic human subject grown out on an extracellular matrix-coated plate and stained for Ki67 (a) and insulin (b) after 24 h incubation with a mixture of cytokines (IL-1β 50 U/ml; TNF-α 1,000 U/ml; IFN- γ 1,000 U/ml). The arrows point to a Ki67-positive beta cell
Beta cell apoptosis in human islets
TUNEL-positive cells were identified after cytokine treatment as well as under control conditions (Fig. 3). To confirm the hypothesis that replicating beta cells are more vulnerable to cytokine-induced apoptosis in human tissue, a total of 360 isolated human islets from three organ donors were studied. There was some variability in beta cell apoptosis between these three batches of islets under control conditions. The baseline frequency of apoptosis was 6.0±0.8, 3.4±0.4 and 2.2±1.9 TUNEL-positive cells/mean islet area (p<0.01). The number of apoptotic cells per mean islet area was increased ∼three-fold after 24 h of cytokine treatment (p<0.001; Fig. 3). The occurrence of apoptotic cells presenting in pairs was used as an indicator of postmitotic apoptosis, as recently described [7, 8]. This percentage of apoptosis in neighbouring cells increased from 23±3% during control conditions to 32±2% after cytokine exposure (p=0.0038; Fig. 4), consistent with the concept of increased vulnerability of replicating cells to cytokine-induced apoptosis. Double staining for insulin and TUNEL confirmed that apoptotic cells were indeed beta cells (Fig. 4).

Isolated islets from non-diabetic human subjects stained for apoptosis using the TUNEL method after 24 h incubation with vehicle (a, b) or a mixture (c, d) of cytokines (IL-1β 50 U/ml; TNF-α 1,000 U/ml; IFN- γ 1,000 U/ml). Arrows indicate pairs of apoptotic cells suggesting post-mitotic apoptosis. e Number of TUNEL-positive cells per mean islet area, and f percentage of apoptotic cells occurring in pairs after exposure with cytokines and vehicle. Data are expressed as means±SE. ***p<0.001 (e), # p=0.0038 (f); p-values were calculated using one-way ANOVA

An isolated islet from a non-diabetic human subject grown out on an extracellular matrix-coated plate and stained for TUNEL (a) and insulin (b) after 24 h incubation with a mixture of cytokines (IL-1β 50 U/ml; TNF-α 1,000 U/ml; IFN-γ 1,000 U/ml). The arrows point to a doublet of TUNEL-positive beta cells
Discussion
The number of pancreatic beta cells appears to be dynamic and undergoes changes in the context of different secretory demands [8–10]. In rodents, reducing the number of beta cells by partial pancreatectomy results in a substantial regeneration of beta cell mass in the pancreatic remnant [11, 12]. In humans, the adaptive changes in beta cell mass appear to be smaller in magnitude than in rodents, but recent evidence suggests that some turnover of beta cells is ongoing in adult humans [3, 13]. Despite this apparent capacity of endocrine pancreatic beta cells to regenerate throughout life, patients with type 1 and type 2 diabetes both have a substantial deficit in beta cell mass [2, 13–15]. In type 2 diabetes one major mechanism underlying the deficit in beta cells is beta cell apoptosis induced by oligomers of human islet amyloid polypeptide [8, 13]. Previous work from our group has demonstrated that this process particularly affects newly forming beta cells, thereby preventing sufficient islet regeneration [7]. The present studies extend this concept to the setting of type 1 diabetes, where cytokine-mediated toxicity represents the major mechanism underlying beta cell death.
The presently reported ∼25–30% of apoptotic events occurring in neighbouring cells may have underestimated the true amount of postmitotic apoptosis. Thus, while our analyses allowed only a two-dimensional view of the islets, it is possible that additional apoptotic cells were present above or below the imaged section. Taking this into account, the actual percentage of paired TUNEL-positive cells may be as high as ∼50% [7].
The ability of adult human beta cells to replicate is a matter of ongoing discussion [16]. Indeed, several groups have reported very low frequencies of Ki67 staining in adult human pancreas [3, 13, 17]. This is in contrast to embryonic human pancreas or isolated human islets under culture conditions, where beta cell replication can readily be identified by Ki67 staining [6, 18]. It appears possible that other sources, e.g. new islet formation from ductal precursor cells or transdifferentiation of acinar cells, give rise to new beta cells in adult humans. However, regardless of the sources it is likely that such newly forming beta cells at different stages of differentiation into mature beta cells are highly susceptible to cytokine-induced apoptosis as well. In this regard it is important to emphasise that increased vulnerability of replicating cells to apoptosis is not unique to beta cells, but represents a general phenomenon in biology. This concept forms the rationale of chemotherapy for cancer [19].
The relationship between the frequency of apoptosis and time after replication in the present study allows speculation about the most vulnerable phases of the cell cycle. We observed two distinct peaks in the frequency of apoptosis, the first one shortly after replication and the second one ∼5 h prior to the next anticipated mitosis [7]. A similar biphasic pattern of apoptosis has previously been reported in other cell lines after exposure to radiation or anticancer chemotherapy, and the peaks were attributed to the G1 (postmitotic apoptosis) and G2/M checkpoint (premitotic apoptosis) phases of cell cycle [20–22]. Given the similarity in cell cycle progression among different cell lines, we postulate that the peaks in the frequency of apoptosis observed in RIN cells were associated with the same phases of the cell cycle. The increased vulnerability of beta cells in G1 may reflect the increased protein synthesis required to produce a full-sized mature cell. Beta cells are particularly vulnerable to endoplasmic reticulum (ER) stress and oxidative stress [23–27], both of which are increased under conditions of high protein synthesis. The G2/M checkpoint is well recognised as an arrest point in the cell cycle, when the cell pauses to ensure its health before proceeding to replication [19]. If the cell is judged to be damaged, apoptosis is induced instead of replication.
It is important to note that for the culture of isolated human islets we used dishes that were coated with an extracellular protein matrix. This was done because this matrix has previously been shown to induce proliferation of adult human beta cells in culture [28]. In contrast, beta cell replication occurs relatively rarely in human islet cells under regular culture conditions or in solid pancreas, when it would be difficult to measure meaningful rates of replication [3, 13].
One potential limitation of studies of apoptosis in isolated human islets is the increased rate of cell death consequent upon isolation and subsequent islet culture, when the vasculature has been lost and when islet cells depend on diffusion for oxygen and nutrients. Presumably islet cells under these conditions may be more vulnerable to cytokine-induced apoptosis. While this may increase the number of islet cells undergoing apoptosis at any particular cytokine concentration, our observation that under these conditions replicating cells are more vulnerable to apoptosis is still presumably valid.
The present findings caution against the concept that restoring beta cell mass in people with type 1 diabetes is best approached by efforts to increase beta cell replication. Indeed, since replicating beta cells have increased vulnerability to cytokine-induced apoptosis, any attempts to stimulate beta cell replication may in turn increase the rate of beta cell apoptosis without affecting beta cell mass. Thus, given the apparent capacity for ongoing beta cell formation in humans, targeting the underlying causes of increased beta cell apoptosis may be an alternative approach to restoring beta cell mass in both type 1 and type 2 diabetes. Recently, endogenous beta cell regeneration in NOD mice with established diabetes has been reported using anti-CD3 immunoblockade [29, 30], and preservation of beta cell function has been achieved in patients with new-onset type 1 diabetes [31]. Furthermore, major progress has been made in the treatment of other chronic autoimmune disorders, such as rheumatoid arthritis and chronic inflammatory bowel disease, using TNF-α antibodies [32, 33]. Given the similarities between these diseases and type 1 diabetes, it is conceivable that this strategy may prevent the autoimmune destruction of new beta cells in type 1 diabetes as well. In reality, given the apparently low rate of new beta cell formation in humans compared with rodents, restoration of beta cell mass in type 1 diabetes will probably require a combination of selective inhibition of chronic autoimmune-induced beta cell apoptosis and enhanced formation of new beta cells.
In conclusion, the present data demonstrate that replicating beta cells have an increased vulnerability to apoptosis, induced by inflammatory cytokines. This may explain the failure of beta cell regeneration in patients with type 1 diabetes despite the attempted formation of new islets. Therefore, therapeutic strategies to increase the number of beta cells in type 1 diabetes may increase the rate of beta cell apoptosis without a significant impact on beta cell mass, unless inappropriately increased rates of beta cell apoptosis through autoimmune-mediated destruction are concurrently suppressed.
Abbreviations
- TLVM:
-
time-lapse video microscopy
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labelling
References
Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Gepts W (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14:619–633
Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC (2005) Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 48:2221–2228
Donath MY, Storling J, Maedler K, Mandrup-Poulsen T (2003) Inflammatory mediators and islet beta cell failure: a link between type 1 and type 2 diabetes. J Mol Med 81:455–470
Beattie GM, Cirulli V, Lopez AD, Hayek A (1997) Ex vivo expansion of human pancreatic endocrine cells. J Clin Endocrinol Metab 82:1852–1856
Maedler K, Fontana A, Ris F et al (2002) FLIP switches Fas-mediated glucose signaling in human pancreatic beta cells from apoptosis to cell replication. Proc Natl Acad Sci U S A 99:8236–8241
Ritzel RA, Butler PC (2003) Replication increases beta cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes 52:1701–1708
Butler AE, Janson J, Soeller WC, Butler PC (2003) Increased beta cell apoptosis prevents adaptive increase in beta cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52:2304–2314
Bonner-Weir S, Deery D, Leahy JL, Weir GC (1989) Compensatory growth of pancreatic beta cells in adult rats after short-term glucose infusion. Diabetes 38:49–53
Finegood DT, Scaglia L, Bonner-Weir S (1995) Dynamics of beta cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 44:249–256
Lee HC, Bonner-Weir S, Weir GC, Leahy JL (1989) Compensatory adaption to partial pancreatectomy in the rat. Endocrinology 124:1571–1575
Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE (1993) A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 42:1715–1720
Butler AE, Janson J, Bonner-Weir S et al (2003) Beta cell deficit and increased beta cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Kloppel G, Drenck CR, Oberholzer M, Heitz PU (1984) Morphometric evidence for a striking B-cell reduction at the clinical onset of type 1 diabetes. Virchows Arch A Pathol Anat Histopathol 403:441–452
Junker K, Egeberg J, Kromann H, Nerup J (1977) An autopsy study of the islets of Langerhans in acute-onset juvenile diabetes mellitus. Acta Pathol Microbiol Scand A 85:699–706
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Bouwens L, Pipeleers DG (1998) Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia 41:629–633
Bouwens L, Lu WG, De Krijger R (1997) Proliferation and differentiation in the human fetal endocrine pancreas. Diabetologia 40:398–404
Kohn KW, Jackman J, O’Connor PM (1994) Cell cycle control and cancer chemotherapy. J Cell Biochem 54:440–452
Shinomiya N, Kuno Y, Yamamoto F et al (2000) Different mechanisms between premitotic apoptosis and postmitotic apoptosis in X-irradiated U937 cells. Int J Radiat Oncol Biol Phys 47:767–777
Johnson VJ, He Q, Kim SH, Kanti A, Sharma RP (2003) Increased susceptibility of renal epithelial cells to TNFalpha-induced apoptosis following treatment with fumonisin B1. Chem Biol Interact 145:297–309
Eastman A (2004) Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem 91:223–231
Eizirik DL, Mandrup-Poulsen T (2001) A choice of death—the signal-transduction of immune-mediated beta cell apoptosis. Diabetologia 44:2115–2133
Oyadomari S, Araki E, Mori M (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta cells. Apoptosis 7:335–345
Gurgul E, Lortz S, Tiedge M, Jorns A, Lenzen S (2004) Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 53:2271–2280
Lortz S, Gurgul-Convey E, Lenzen S, Tiedge M (2005) Importance of mitochondrial superoxide dismutase expression in insulin-producing cells for the toxicity of reactive oxygen species and proinflammatory cytokines. Diabetologia 48:1541–1548
Azevedo-Martins AK, Lortz S, Lenzen S et al (2003) Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-kappaB activation in insulin-producing cells. Diabetes 52:93–101
Beattie GM, Rubin JS, Mally MI, Otonkoski T, Hayek A (1996) Regulation of proliferation and differentiation of human fetal pancreatic islet cells by extracellular matrix, hepatocyte growth factor, and cell–cell contact. Diabetes 45:1223–1228
Chatenoud L, Primo J, Bach JF (1997) CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 158:2947–2954
Belghith M, Bluestone JA, Barriot S et al (2003) TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 9:1202–1208
Keymeulen B, Vandemeulebroucke E, Ziegler AG et al (2005) Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 352:2598–2608
Olsen NJ, Stein CM (2004) New drugs for rheumatoid arthritis. N Engl J Med 350:2167–2179
Sands BE, Anderson FH, Bernstein CN et al (2004) Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 350:876–885
Acknowledgement
We are grateful Dr Bernhard J. Hering (Diabetes Institute for Immunology and Transplantation, University of Minnesota) for providing human islets. These studies were supported by grants from the US Public Health Service NIH DK 59567 (PCB), the Larry Hillblom Foundation and the Deutsche Forschungsgemeinschaft (Me 2096/2-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meier, J.J., Ritzel, R.A., Maedler, K. et al. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia 49, 83–89 (2006). https://doi.org/10.1007/s00125-005-0069-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0069-3