
Abstract
Aims/hypothesis
Local overproduction of nitric oxide is seen in early stages of diabetes, which can react with superoxide (O2 −) to form peroxynitrite (ONOO−). The aim of this study was to examine the effect of scavengers for nitric oxide, O2 −, ONOO− and NOS cofactor tetrahydrobiopterin (BH4) on high glucose-induced cardiac contractile dysfunction.
Methods
Ventricular myocytes were cultured for 24 h with either normal (N, 5.5 mmol/l) or high (25.5 mmol/l) glucose, with or without the nitric oxide scavengers haemoglobin (100 nmol/l), PTIO (100 µmol/l), the NOS inhibitor L-NMMA (100 µmol/l), superoxide dismutase (SOD, 500 U/ml), the ONOO− scavengers urate (100 µmol/l), MnTABP (100 µmol/l), BH4 (10 µmol/l) and its inactive analogue NH4 (10 µmol/l), and the GTP cyclohydrolase I inhibitor DAHP (1 mmol/l). Myocyte mechanics, NOS protein expression and activity were evaluated.
Results
High glucose myocytes showed reduced peak shortening, decreased maximal velocity of shortening/relengthening (± dL/dt), prolonged relengthening (TR90) and normal shortening duration (TPS) associated with reduced cytosolic Ca2+ rise compared to normal myocytes. The high glucose-induced abnormalities were abrogated or attenuated by urate, MnTBAP, L-NMMA, BH4, and SOD, whereas unaffected by haemoglobin, PTIO and NH4. L-NMMA reduced peak shortening while PTIO and DAHP depressed ± dL/dt and prolonged TPS or TR90 in normal myocytes. High glucose increased NOS activity, protein expression of eNOS but not iNOS, which were attenuated by L-NMMA and BH4, respectively.
Conclusion/interpretation
These results suggested that NOS cofactor, NO and ONOO− play a role in glucose-induced cardiomyocyte contractile dysfunction and in the pathogenesis of diabetic cardiomyopathy.
Similar content being viewed by others
Cardiovascular diseases are among the leading causes of high morbidity and mortality in diabetic populations [1]. Diabetic cardiomyopathy often develops in diabetes and is characterized by systolic and diastolic dysfunctions independent of coronary macrovascular disease [2, 3]. Impaired diastolic function is the most prominent mechanical abnormality manifested as prolonged relaxation and decreased compliance. Several mechanisms have been proposed for the pathogenesis of impaired cardiac excitation-contraction (E-C) coupling, including prolonged action potential duration and altered function of Ca2+-regulating proteins such as Na/Ca exchanger and sarco(endo)plasmic reticulum Ca2+-ATPase [4, 5, 6]. However, whether these cellular defects progress as a result of hyperglycaemia or indirectly via glucose toxicity-induced secondary cellular alterations has not been understood completely.
Nitric oxide, an extremely reactive gas with chemical properties of a free radical, is synthesized by a variety of cell types including cardiac, vascular and endothelial cells. It plays an important physiological role in the regulation of cardiac function such as dilation of coronary artery, inhibition of platelet aggregation and biphasic modulation of cardiac contractile function [7, 8]. Nitric oxide also participates in a cascade of pathophysiological processes when formed in excess or in the presence of other pro-oxidants [9]. Enhanced endogenous production of nitric oxide or addition of nitric oxide donors have resulted in compromised cardiac function and enhanced apoptosis [9, 10] illustrating the potential toxic properties of nitric oxide. The toxicity of nitric oxide is believed to be associated with its interaction with iron-sulfur-centered enzymes of the respiratory cycle [11]. This is further complicated by superoxide anion (O2 −), a ubiquitous reaction product of cardiac myocytes during mitochondrial oxidative metabolism. Nitric oxide and O2 − react rapidly (second-order rate constant =6.7×109 M/s) to generate peroxynitrite (ONOO−), which rapidly decomposes to highly oxidant species such as nitronium ion (NO2 +) [12]. ONOO− can directly initiate lipid peroxidation, protein tyrosine nitration and DNA strand breakdown, as well as interact with iron-sulfur-centered enzymes of the respiratory cycle. It has been implicated that ONOO− can mediate the toxic action of nitric oxide. Paradoxically, nitric oxide also protects against apoptosis and O2 −-mediated cell death [13]. Inhibition of nitric oxide synthase (NOS) results in enhanced O2 − and H2O2 production [14], suggesting that a critical balance between nitric oxide and O2 − is required for normal myocyte function.
Altered nitric oxide signalling and enhanced oxidative stress have been suggested to play a role in the pathogenesis of diabetic cardiac complications [15, 16]. Diabetes could alter the activity and expression of NOS [17, 18, 19]. Local overproduction of nitric oxide has been suggested to play an important role in beta-cell damage and in inducing diabetes. Oxidative stress due to interactions between nitric oxide and oxygen-derived radicals has been speculated to represent a common pathological mechanism in increased cardiovascular risk. Recent evidence has suggested that NOS function could be altered in diabetes due to deficiency of tetrahydrobiopterin (BH4), a critical cofactor for eNOS [20, 21]. Nitric oxide synthase activity is strictly dependent on BH4, the deficiency of which would result in the production of O2− rather than nitric oxide; this phenomenon can be referred to as "eNOS uncoupling" [22]. Peroxymitrite oxidation of BH4 could represent a major pathogenic cause of "eNOS uncoupling" [22]. Both hyperglycaemia and impaired cardiovascular contractile function in diabetes have been shown to be reconciled with the use of NOS inhibitors, superoxide dismutase (SOD) or other antioxidants such as alpha-tocopherol [15], confirming the role of nitric oxide and other reactive oxygen radicals in diabetic heart complications. However, a complete understanding of the mechanisms underscoring the role of nitric oxide in diabetic cardiomyopathy is hampered because few data are available regarding the role of nitric oxide, NOS and nitric oxide-related reactive oxygen species in the onset of diabetic cardiac dysfunctions. We thus took advantage of a cardiac myocyte culture model developed in our laboratory, where the diabetic cardiomyopathy could be phenotypically replicated in normal myocytes by culturing in a high glucose medium [23, 24, 25]. The abnormal cardiac E-C coupling is apparent in high glucose-treated myocytes reminiscent of those from in vivo diabetes [3]. The NOS inhibitor, nitric oxide cofactor and pharmacological scavengers for nitric oxide, O2 − and ONOO− were tested to examine the role of nitric oxide, NOS and ONOO− in the glucose toxicity-induced cardiac mechanical dysfunction simulating diabetic cardiomyopathy.
Methods
Isolation and culture of isolated ventricular myocytes
The experimental procedure used in this study was approved by the Animal Use and Care Committee at University of North Dakota and University of Wyoming. Briefly, adult male Sprague-Dawley rats (200–250 g) were anaesthetized with ketamine/xylazine (5:3, 1.32 mg/kg i.p.). Hearts were rapidly removed and perfused (at 37°C) with Krebs-Henseleit bicarbonate (KHB) buffer (mmol/l: NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, N-[2-hydro-ethyl]-piperazine-N'-[2-ethanesulfonic acid] (HEPES) 10, glucose 11.1, pH 7.4). The heart was then perfused for 20 min with KHB containing 223 U/ml collagenase (Worthington Biochemical, Freehold, N.J., USA) and 0.5 mg/ml hyaluronidase (Sigma Chemical, St. Louis, Mo., USA). After perfusion, the left ventricle was removed and minced. The cells were further digested with 0.02 mg/ml trypsin (Sigma) before being filtered through a nylon mesh (300 µm). Extracellular Ca2+ was added incrementally back to 1.25 mmol/l. Isolated myocytes were then cultured in normal (5.5 mmol/l) or high glucose (25.5 mmol/l) medium supplemented with or without one of the following drugs; the nitric oxide synthase (NOS) inhibitor L-NG-monomethyl-arginine (L-NMMA, 100 µmol/l), the nitric oxide scavenger haemoglobin (100 nmol/l), the nitric oxide trap 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl imidazoline-1-oxyl 3-oxide (carboxy-PTIO, 100 µmol/l) which reacts with nitric oxide to form PTI, superoxide (O2 −) scavenger superoxide dismutase (SOD, 500 U/ml), the NOS cofactor BH4 (10 µmol/l) or peroxynitrite (ONOO−) scavengers urate (100 µmol/l) and manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTABP, 100 µmol/l, which is also a SOD mimetic). To further examine the role of the NOS cofactor BH4, tetrahydroneopterin (NH4, 10 µmol/l), an analogue of BH4, however, without any effect on NOS was co-cultured with the myocytes (obtained from Schircks Laboratories, Jona, Switzerland). In addition, the GTP cyclohydrolase I inhibitor 2,4-diamino-6-hydroxy-pyrimidine (DAHP, 1 mmol/l), which inhibits BH4 synthesis, was also incubated with the N myocytes. Myocytes were maintained for 24 h before used [23].
Cell shortening/relengthening
Mechanical properties of ventricular myocytes were assessed using a video-based edge-detection system (IonOptix, Milton, Mass., USA) [23]. In brief, cells were superfused with a buffer containing (in mmol/l): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, at pH 7.4. The cells were field stimulated at 0.5 Hz. The myocyte was displayed on the computer monitor using an IonOptix MyoCam camera, which rapidly scans the image area at every 8.3 msec such that the amplitude and velocity of shortening or relengthening is recorded with good fidelity.
Intracellular Ca2+ fluorescence measurement
Myocytes were loaded with fura-2/AM (0.5 µmol/l) for 15 min and fluorescence measurements were recorded with an IonOptix dual-excitation fluorescence photomultiplier system [23]. Myocytes were imaged through an Olympus 40× oil objective. Cells were exposed to light emitted by a 75 W lamp and passed through either a 360 or a 380 nm filter (bandwidths were ±15 nm), while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm after first illuminating cells at 360 nm for 0.5 s then at 380 nm for the duration of the recording protocol (333 Hz sampling rate). The 360 nm excitation scan was repeated at the end of the protocol and qualitative changes in intracellular Ca2+ concentration ([Ca2+]i) were inferred from the ratio of the fluorescence intensity at two wavelengths.
Western blot analysis for protein expression of eNOS and iNOS
Membrane proteins from normal or high glucose cultured myocytes were extracted as described [26]. Myocytes were collected, sonicated and the supernatants were centrifuged at 7000× g for 30 min at 4°C. Total cell homogenates from the pellets were used for immunoblotting of eNOS and iNOS. We confirmed that these membrane fractions did not contain any detectable collagens. Membrane proteins (50 µg/lane) were separated on 10% SDS-polyacrylamide gels in a minigel apparatus (Mini-PROTEAN II, Bio-Rad) and transferred to polyvinylidene difluoride membranes. The membranes were blocked (4% Block Ace, Dainippon Pharmaceutical; Osaka, Japan) and then incubated for 12 h with anti-eNOS or anti-iNOS mouse IgG monoclonal antibodies (1:1000, BD Transduction Laboratories, Lexington, Ky., USA). Membranes were then washed and incubated with a horseradish peroxidase-conjugated anti-mouse IgG (1:5000) for 1.5 h. After immunoblotting, the film was scanned and the intensity of immuoblot bands was detected with a Bio-Rad Calibrated Densitometer (Model: GS-800).
NOS activity
Nitric oxide synthase activity was evaluated by the 3H-arginine to 3H-citrulline conversion assay as described [10]. Briefly, ventricular myocytes were placed in Hanks' Balanced Salt Solution (HBSS) medium containing 1 µCi/ml 3H-arginine with Trasylol (0.2 KIU/ml). The cells were incubated for 30 min before the reaction was terminated by aspiration of the incubation medium and replacement with iced, HBSS containing 5 mmol/l L-arginine and 4 mmol/l EDTA. Cells were lysed with 20 mmol/l TRIS and centrifuged. An aliquot of the supernatant was diluted with 1:1 (v/v) H2O/Dowex-50 W (20–50, 8% cross-linked), and loaded on a polypropylene BioRad EconoColumn. The effluent was collected and 3H-citrulline was counted by scintillation.
Statistical analyses
Data are presented as means ± SEM. Statistical significance was ascertained by ANOVA. Appropriate follow-up tests for multiple comparisons were chosen depending on whether significance (p<0.05) was identified in main effects and/or interaction terms. Data from each drug were analysed separately. Control groups (i.e., N and high glucose cells) were always recorded on the same day as experimental groups (i.e., with and without drugs) to control for any potential inter-culture variability.
Results
Effect of urate, MnTBAP and SOD on high glucose-induced mechanical abnormalities
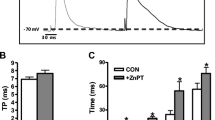
Culturing myocytes for 24 h with either high glucose or any drug tested in this study had no overt effect on cell phenotype. Cell shape, resting cell length, and presence of distinct striations were similar in cells among all groups. Representative traces of cell shortening and relengthening are shown in Fig. 1. Consistent with our previous reports [23, 24], myocytes maintained in high-glucose medium had a reduced peak shortening amplitude associated with decreased maximal velocity of shortening and relengthening (± dL/dt) (p<0.05), normal time-to-peak shortening (TPS), and prolonged time-to-90% relengthening (TR90) (p<0.05) compared to those of normal myocytes. The reduced peak shortening, ± dL/dt and prolonged TR90 in high glucose myocytes were abolished by co-incubation the cells with the ONOO− scavengers urate (100 µmol/l) and MnTBAP (100 µmol/l) as well as the O2 − scavenger SOD (500 U/ml), with the exception that SOD did not prevent the high glucose-induced prolonged TR90. In addition, high glucose myocytes maintained with urate showed a significantly increased peak shortening (p<0.05) compared with urate-treated normal myocytes. Urate, MnTBAP or SOD alone had no effect on cell mechanics in normal myocytes (Figs. 1, 2).

Representative traces of myocyte cell length from normal glucose (NG: 5.5 mmol/l), high glucose (high glucose: 25.5 mmol/l), and high glucose with the ONOO− scavenger MnTBAP (100 µmol/l) groups. B–F Contractile properties of myocytes cultured for 24 h in normal or high glucose with or without MnTBAP (100 µmol/l) or SOD (500 U/ml). Mechanical indices are peak shortening (PS), maximal velocities of shortening/relengthening (± dL/dt), time-to-peak shortening (TPS) and time-to-90% relengthening (TR90). Means ± SEM, n=40–57 cells/group, *p<0.05 vs respective normal group

Contractile properties of myocytes cultured for 18–24 hours in N or high glucose with or without the ONOO− scavenger urate (100 µmol/l) or the nitric oxide scavenger haemoglobin (100 nmol/l). A Peak shortening (PS); B maximal velocities of shortening/relengthening (± dL/dt); C time-to-peak shortening (TPS); D time-to-90% relengthening (TR90); Mean ± SEM, n=42–50 cells/group, *p<0.05 vs respective normal group, #p<0.05 vs non-drug supplemented normal group
Effect of L-NMMA, haemoglobin and PTIO on high glucose-induced abnormal mechanical function
Enhanced NOS activity has been shown in myocytes cultured in a high glucose environment [19]. To demonstrate if altered nitric oxide production plays a role in the high glucose-induced mechanical dysfunction, the membrane permeable NOS inhibitor L-NMMA and the nitric oxide scavenger haemoglobin as well as the nitric oxide trap PTIO were examined. L-NMMA (100 µmol/l) unmasked a high glucose-induced potentiation in PS and ± dL/dt, whereas it prevented high glucose-induced prolongation in TR90 without affecting TPS (Fig. 3). L-NMMA alone depressed peak shortening in normal myocytes. On the other hand, both PTIO (100 µmol/l) and haemoglobin (100 nmol/l) nullified the high glucose-induced decrease in peak shortening but depressed ± dL/dt and prolonged TPS in normal myocytes (p<0.05). In addition, the high glucose-induced prolongation of TR90 was not affected by either PTIO or haemoglobin (Figs. 2, 3).

Graphs illustrate contractile properties of myocytes cultured for 24 h in normal or high glucose medium with or without the NOS inhibitor L-NMMA (100 µmol/l) or the nitric oxide scavenger PTIO (100 µmol/l). Mechanical indices are peak shortening (PS, panel A), maximal velocities of shortening/relengthening (± dL/dt, Panel B), time-to-peak shortening (TPS, panel C) and time-to-90% relengthening (TR90, panel D). Means ± SEM, n=40–57 cells/group. *p<0.05 vs respective normal group, #p<0.05 vs non-drug supplemented normal group
Effect of BH4, NH4 and DAHP on myocyte mechanics and NOS protein expression
Diabetes has been shown to lead to reduced concentration of the NOS cofactor BH4 [21]. Our study shows that supplementation of BH4 (10 µmol/l) abrogated the high glucose-induced decline in ± dL/dt and prolongation in TR90, whereas it exerted little effect on mechanical properties in normal myocytes (Fig. 4). However, the BH4 analogue pteridine NH4, an antioxidant but not a NOS cofactor, did not affect the high glucose-induced myocyte mechanical dysfunction at equal molar concentration (10 µmol/l). To further examine the role of BH4 in myocyte mechanics, the GTP cyclohydrolase I inhibitor DAHP (1 mmol/l), which inhibits BH4 synthesis [20], was co-incubated with normal myocytes for 24 h. Of interest, DAHP depressed ± dL/dt, prolonged both TPS and TR90 (p<0.05) without affecting peak shortening in normal myocytes (Fig. 5). In addition, high glucose induced significant increase in protein expression of eNOS (p<0.05), which was nullified by BH4. high glucose did not alter protein expression of iNOS although BH4 itself lowered the iNOS protein expression in high glucose myocytes (p<0.05) (Fig. 4).

Effect of the NOS cofactor BH4 (10 µmol/l) on contractile properties and NOS protein expression in myocytes cultured for 24 h in normal or high glucose medium. A Peak shortening (PS); B maximal velocities of shortening/relengthening (± dL/dt); C time-to-peak shortening (TPS); D time-to-90% relengthening (TR90); E Representative gels depicting immunostaining using anti-eNOS and anti-iNOS antibodies; F Protein abundance of eNOS and iNOS in normal and high glucose myocytes incubated with or without BH4 from three isolations. Means ± SEM, n=58–62 cells/group for panel A–D and n=3 for panel F. *p<0.05 vs normal group

Effect of the BH4 analogue NH4 (10 µmol/l) on contractile properties in myocytes cultured for 24 h in normal or high glucose medium. Myocytes were also co-incubated with the GTP cyclohydrolase I inhibitor DAHP (1 mmol/l) for 24 h in nromal medium to inhibit BH4 synthesis. A Peak shortening (PS); B maximal velocities of shortening/relengthening (± dL/dt); C time-to-peak shortening (TPS); D time-to-90% relengthening (TR90); Mean ± SEM, n=48–50 cells/group. *p<0.05 vs normal group, #p<0.05 vs non-drug supplemented normal group
Influence of MnTBAP, PTIO and SOD on high glucose-induced intracellular Ca2+ abnormalities
Our previous studies have shown that the high glucose-induced mechanical dysfunction is caused by abnormal intracellular Ca2+ homeostasis [23, 24, 25]. To evaluate the potential mechanism of action involved in high glucose-induced cardiac defects, the influence of MnTBAP, PTIO and L-NMMA on intracellular Ca2+ properties was tested. Resting [Ca2+]i, electrical stimulus-induced increase in intracellular Ca2+ (Δ[Ca2+]i), and cytosolic free Ca2+ decrease rate (tau) were evaluated in fura-2 loaded myocytes. High glucose myocytes showed similar resting [Ca2+]i, reduced Δ[Ca2+]i associated with slowed cytosolic-free Ca2+ decrease compared to normal myocytes (p<0.05), consistent with reduced peak shortening and prolonged TR90. MnTBAP (100 µmol/l) and L-NMMA (100 µmol/l) abolished the high glucose-induced reduction of Δ[Ca2+]i and prolonged cytosolic free Ca2+ decrease (Fig. 6). PTIO (100 µmol/l) reduced Δ[Ca2+]i in normal myocytes. MnTBAP and PTIO both increased the resting [Ca2+]i in normal myocytes (p<0.05).

Intracellular Ca2+ transient properties in fura-2-loaded ventricular myocytes cultured for 24 h in normal or high glucose medium, with or without MnTBAP (100 µmol/l), PTIO (100 µmol/l) or L-NMMA (100 µmol/l). A Representative traces of fura-2 fluorescence ratio in myocytes from normal, high glucose and high glucose+MnTBAP group; B Baseline intracellular Ca2+ concentrations; C Increase in intracellular Ca2+ transient in response to electrical stimulus; D rate of cytosolic free Ca2+ decrease (tau). Means ± SEM. n=48–55 cells/group. *p<0.05 vs respective normal group, #p<0.05 vs non-drug supplemented normal group
Influence of L-NMMA, PTIO and MnTBAP on NOS activity in N and high glucose myocytes
To evaluate the effect of high glucose on NOS activity, NOS activity was measured with 3H-arginine to 3H-citrulline conversion assay in normal and high glucose myocytes with or without L-NMMA, PTIO, or MnTBAP (100 µmol/l each). None of these drugs elicited any significant effect on the uptake of 3H-arginine into the myocytes as determined by the radioactivity in the 3H-loaded cell pellets after centrifugation. Consistent with the observation from western blot analysis (Fig. 4), high glucose increased NOS activity (p<0.05). L-NMMA (100 µmol/l) inhibited NOS activity in myocytes from both groups (p<0.05), as expected. PTIO did not alter NOS activity in either normal or high glucose group. MnTBAP enhanced NOS activity in normal myocytes (p<0.05) but did not affect NOS activity in high glucose myocytes, therefore nullifying the disparity in NOS activity between normal and high glucose groups (Fig. 7).

NOS activity in ventricular myocytes cultured for 24 h in normal or high glucose medium, with or without L-NMMA (100 µmol/l), PTIO (100 µmol/l) or MnTBAP (100 µmol/l). Means ± SEM. n=13 assays. *p<0.05 vs respective normal group, #p<0.05 vs non-drug supplemented normal group
Discussion
Our findings confirmed that glucose toxicity contributes to the development of cardiac E-C coupling dysfunction reminiscent of in vivo diabetic cardiomyopathy. More importantly, the high glucose-induced cardiac mechanical dysfunctions could be alleviated by the NOS inhibitor L-NMMA, the NOS cofactor BH4, the O2 − scavenger SOD and the ONOO− scavengers urate and MnTBAP (also a SOD mimetic) although not the nitric oxide scavenger haemoglobin nor the nitric oxide trap PTIO. The high glucose-induced myocyte abnormalities were unable to be reconciled by the BH4 analogue pteridine NH4, an antioxidant but not a cofactor for NOS. The involvement of BH4 in the high glucose-induced cardiomyocyte dysfunction was further substantiated by the fact that blockade of BH4 synthesis with the GTP cyclohydrolase I inhibitor DAHP can directly impair cell mechanics in a manner somewhat reminiscent of those induced by high glucose. Increased eNOS but not iNOS protein abundance was observed in association with enhanced NOS activity in high glucose myocytes. These data suggested that NOS, nitric oxide, O2 −, and their reaction product ONOO− could play an important role in the glucose toxicity-induced cardiac contractile dysfunction, and possibly in the pathogenesis of the early-stage of diabetic cardiomyopathy.
Hyperglycaemia is the most important predisposing factor for diabetic cardiomyopathy characterized by reduced contractility and prolonged relaxation [1, 3, 5]. This is supported by the reduced peak shortening, ± dL/dt and prolonged TR90 in high glucose myocytes observed in our study, which could be underscored by reduced intracellular Ca2+ rise and slowed intracellular Ca2+ clearing (Fig. 6) consistent with earlier reports [23, 24, 27, 28]. Although speculations have been made towards the glucose-induced cardiac dysfunction including activation of protein kinase C [29] and altered lipid messenger such as ceramide [30], the role of NOS/nitric oxide signalling in diabetic heart complications did not receive much support over the past years considering that treatment with the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) attenuated hyperglycaemia [31, 32] and improved ventricular function [33] in diabetes. Nevertheless, the direct effect of diabetes on nitric oxide synthesis is still controversial, with either increased [17, 34, 35] or decreased [17, 36, 37] nitric oxide synthesis. Enhanced cardiac NOS protein expression and activity were reported under diabetes in conjunction with elevated nitric oxide production [17, 18, 19, 33], consistent with our current observation that high glucose enhanced the expression and activity of NOS, and the reversal of cardiomyocyte dysfunction with the NOS inhibitor L-NMMA. High glucose has been shown to elicit an overt increase in eNOS mRNA in endothelial cells [17, 34]. No change in cardiac NOS mRNA (eNOS and iNOS) was found at initial stages of diabetes [18, 38], indicating that increase in cardiac NOS mRNA could be a consequence rather than cause of metabolic defects due to hyperglycaemia. Our findings showed enhanced eNOS but not iNOS protein expression in high glucose myocytes, suggesting a potential temporal separation in glucose-induced response between the two enzymes. The expression or activity of NOS and nitric oxide synthesis might not accurately reflect the bioavailability of nitric oxide due to the rapid inactivation of nitric oxide by free radicals [34, 35].
The mechanism behind the increased expression or activity of NOS in diabetic hearts is still not known. The high glucose-induced up-regulation of eNOS prevented by the antioxidant lipoic acid and H2O2 could directly induce eNOS mRNA [18], suggesting the potential role of the redox state in the induction of NOS mRNA. ROS has been speculated to play a key role in the pathogenesis of diabetic cardiomyopathy [15]. Parameters of oxidative stress such as lipid hydroperoxides, thiobarbituric acid substances and isoprostanes have been documented in high amounts in diabetic hearts [15, 39]. High glucose has been shown to induce generation of ROS, probably through glucose autoxidation, formation of advanced glycation end-products and activation of NADPH-oxidase [18, 40, 41]. The high glucose-induced ROS especially O2 − is able to quench nitric oxide at a rate that is three times faster than the ability of SOD to degrade O2 −. This could explain the observation that the nitric oxide scavenger haemoglobin and the nitric oxide trap PTIO failed to prevent the high glucose-induced cardiac defect, since nitric oxide is quickly quenched by surrounding O2 − molecules to form ONOO− before getting scavenged or trapped by haemoglobin or PTIO. Our data also showed that L-NMMA depressed contractile amplitude (peak shortening) whereas haemoglobin and PTIO depressed ± dL/dt and prolonged TPS in normal myocytes, indicating that tonic nitric oxide release could be essential for normal cardiac contractile function [7, 8]. These data, in conjunction with the observation that the NOS inhibitor L-NMMA prevented high glucose-induced cardiac dysfunctions, suggested that nitric oxide release, rather than its removal, is more likely to be involved in the glucose toxicity-induced cardiac mechanical dysfunction. Therefore, regulation of NOS activity could contribute, at least in part, to high glucose-induced mechanical dysfunction. Our rationale of enhanced nitric oxide inactivation by O2 − in high glucose myocytes is supported by the ability of urate, MnTBAP and SOD to attenuate or prevent the high glucose-induced cardiac contractile defects and intracellular Ca2+ dysregulation. Both ONOO− and O2 − can exert direct cardiac depressive actions [12] and their productions are enhanced in diabetes [42]. Although the direct effects of ONOO− and O2 − on cardiac myocyte mechanics are beyond the scope of this study, our results provided evidence of the involvement of ONOO− and O2 − in the glucose toxicity-induced cardiac E-C coupling. It is not surprising that PTIO failed to affect NOS activity since it traps nitric oxide instead of acting on NOS enzyme. The enhanced NOS activity in response to MnTBAP is not known but could be related to relieve of the oxidation of ONOO− on NOS. Moreover, our fura-2 fluorescent recording data indicated that regulation of intracellular Ca2+ homeostasis could underscore the mechanical effects of the nitric oxide and ONOO− regulators in the high glucose-induced ventricular dysfunction in cardiac myocytes.
Of interest, the NOS cofactor BH4 restored high glucose-induced mechanical dysfunctions and prevented a high glucose-elicited increase in eNOS expression. This is further supported by the observation that depletion of BH4 by the GTP cyclohydrolase I inhibitor DAHP directly impaired cell mechanics somewhat similar to high glucose (such as prolonged TPS/TR90 and reduced ± dL/dt). The fact that DAHP did not exactly duplicate the high glucose-induced myocyte dysfunction (such as in peak shortening and TPS) indicates that mechanism(s) other than NOS cofactor could also participate in the regulation of cardiomyocyte mechanical function. Deficiency of BH4 has been shown in diabetes [21]. This could be related to the fact that BH4 is a major target of oxidation by ONOO−, the amount of which is significantly increased in diabetes [42]. Treatment with BH4 was reported to restore endothelial dysfunction in diabetes [43]. Without sufficient BH4, the electrons flowing from the reductase domain to the oxygenase domain on eNOS can be diverted to O2 molecule rather than L-arginine, resulting in the production of O2 − rather than nitric oxide, or "eNOS uncoupling" [22]. Our data suggests that "eNOS uncoupling" could play a role in glucose toxicity-associated cardiac dysfunctions. The observation that the BH4 analogue NH4 failed to modify high glucose-induced cardiac dysfunctions suggests that BH4-induced improvement in cardiac mechanical dysfunction under high glucose toxicity could reflect a specific effect on eNOS enzyme rather than being the consequence of a nonspecific antioxidant action.
Experimental limitations
Diabetes mellitus is a complex metabolic disease and its cardiac complications are likely due to multiple factors in addition to hyperglycaemia (such as dyslipidaemia and insulin resistance). It is not possible to replicate these factors all in the same cell culture model. In addition, the enzymatic myocyte isolation procedure could induce iNOS expression and create an artificial effect on iNOS expression in the ventricular myocytes studied. Thus, caution should be taken when extrapolating the current findings to in vivo diabetes or when applying polymixin B to absorb endotoxin during cell isolation to minimize any artificial effects on iNOS. Although in vitro models allow us to change one factor at a time and to evaluate the resultant cellular consequences, obvious disadvantages must be considered such as lack of the in vivo settings of haemodynamics from cardiac as well as non-cardiac vasculatures.
In conclusion, our study suggests that NOS, nitric oxide, O2 − and ONOO− could play roles in the glucose toxicity-induced cardiac dysfunction simulating diabetic cardiomyopathy. Our data indicates that altered NOS signalling related to BH4 deficiency or dysfunction, formation of O2 − or ONOO− and accelerated nitric oxide inactivation could be permissive to high glucose-induced contractile and intracellular Ca2+ dysregulations.
Abbreviations
- PTIO:
-
2-(4-carboxyphenyl)-4,4,5,5-tetramethyl imidazoline-1-oxyl 3-oxide
- DAHP:
-
2,4-diamino-6-hydroxy-pyrimidine
- E-C:
-
excitation-contraction
- MnTABP:
-
manganese (III) tetrakis (4-benzoic acid) porphyrin
- ± dL/dt:
-
maximal velocity of shortening and relengthening
- L-NAME:
-
Nω-nitro-L-arginine methyl ester
- L-NMMA:
-
L-NG-monomethyl-arginine
- NOS:
-
nitric oxide synthase
- PS:
-
peak shortening
- ONOO− :
-
peroxynitrite
- O2 − :
-
superoxide anion
- SOD:
-
superoxide dismutase
- BH4 :
-
tetrahydrobiopterin
- NH4 :
-
tetrahydroneopterin
- TPS:
-
time-to-peak shortening
- TR90 :
-
time-to-90% relengthening
References
Sowers JR, Epstein M (1995) Diabetes mellitus and associated hypertension, vascular disease and nephropathy: an update. Hypertension 26:869–879
Galderisi M, Anderson KM, Wilson PWF, Levy D (1991) Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (the Framingham Heart Study). Am J Cardiol 68:85–89
Ren J, Davidoff AJ (1997) Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol Heart Circ Physiol 272:H148–H158
Hofmann PA, Menon V, Gannaway KF (1995) Effects of diabetes on isometric tension as a function of [Ca2+] and pH in rat skinned cardiac myocytes. Am J Physiol Heart Circ Physiol 269:H1656–H1663
Lagadic-Gossmann DL, Buckler KJ, Le Prigent K, Feuvray D (1996) Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am J Physiol Heart Circ Physiol 270:H1529–H1537
Schaffer SW, Ballard-Croft C, Boerth S, Allo SN (1997) Mechanisms underlying depressed Na+/Ca2+ exchanger activity in the diabetic heart. Cardiovasc Res 34:129–136
Kojda G, Harrison D (1999) Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res 43:562–571
Ren J, Esberg LB, Combs CK, Ren BH, Chen AF (2001) Adenovirus gene transfer of recombinant endothelial nitric oxide synthase alters contractile function in isolated ventricular myocyte via a phosphatidylinositol 3-kinase-dependent pathway. Circulation 104:II-436 (Abstract)
Schultz R, Panas DL, Catena R, Moncada S, Olley PM, Lopaschuk GD (1995) The role of nitric oxide in cardiac depression induced by interleukin-1β and tumor necrosis factor-α. Br J Pharmacol 114:27–34
Nickola MW, Wold LE, Colligan PB, Wang GJ, Samson WK, Ren J (2000) Leptin attenuates cardiac contraction in adult rat ventricular myocytes: role of nitric oxide. Hypertension 36:501–505
Hibbs JB Jr, Taintor RR, Vavrin Z, Granger DL, Drapier J-C, Amber J, Lancaster JR Jr (1990) Synthesis of nitric oxide from a terminal guanidine nitrogen atom of L-arginine: a molecular mechanism regulating cellular proliferation that targets intracellular iron. In: Moncada S, Higgs EA (eds) Nitric oxide from L-arginine: a bioregulatory system. Excerpta Medica, Amsterdam, pp 189–224
Schulz R, Dodge KL, Lopaschuk GD, Clanachan AS (1997) Peroxynitrite impairs cardiac contractile function by decreasing cardiac efficiency. Am J Physiol Heart Circ Physiol 272:H1212–H1219
Gutierrez HH, Nieves B, Chumley P, Rivera A, Freeman BA (1996) Nitric oxide regulation of superoxide-dependent lung injury: oxidant-protective actions of endogenously produced and exogenously administered nitric oxide. Free Rad Biol Med 21:43–52
Goda N, Suematsu M, Mukai M, Kiyokawa K, Natori M, Nozawa S, Ishimura Y (1996) Modulation of mitochondrion-mediated oxidative stress by nitric oxide in human placental trophoblastic cells. Am. J. Physiol. Heart Circ Physiol 271:H1893–H1899
Rösen P, Du X, Tschope D (1998) Role of oxygen derived radicals for vascular dysfunction in the diabetic heart: prevention by alpha-tocopherol? Mol Cell Biochem 188:103–111
Rösen P, Ballhausen Th, Stockklauser K (1996) Impairment of endothelium dependent relaxation in the diabetic rat heart: mechanisms and implications. Diabetes Res Clin Pract 31:S143–S155
Pieper GM (1998) A review of alterations in endothelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension 31:1047–1060
Stockklauser-Farber K, Ballhausen T, Laufer A, Rosen P (2000) Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim Biophys Acta 1535:10–20
Hintz KK, Wold LE, Colligan PB, Scott GI, Lee KJ, Sowers JR, Ren J (2001) Influence of ovariectomy on ventricular myocyte contraction in simulated diabetes. J Biomed Sci 8:307–313
Bagi Z, Koller A (2003) Lack of nitric oxide mediation of flow-dependent arteriolar dilation in type I diabetes is restored by sepiapterin. J Vasc Res 40:47–57
Meininger CJ, Marinos RS, Hatakeyama K, Martinez-Zaguilan R, Rojas JD, Kelly KA, Wu G (2000) Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem J 349:353–356
Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG (2001) Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103:1282–1288
Ren J, Dominguez LJ, Sowers JR, Davidoff AJ (1996) Troglitazone attenuates high glucose-induced abnormalities in relaxation and intracellular calcium in rat ventricular myocytes. Diabetes 45:1822–1825
Ren J, Gintant GA, Miller RE, Davidoff AJ (1997) High extracellular glucose impairs cardiac E-C coupling in a glycosylation-dependent manner. Am J Physiol Heart Circ Physiol 273:H2876–H2883
Davidoff AJ, Ren J (1997) Low insulin and high glucose induce abnormal relaxation in cultured adult rat ventricular myocytes. Am J Physiol Heart Circ Physiol 272:H159–H167
Norby FL, Wold LE, Duan J, Hintz KK, Ren J (2002) IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol Endocrinol Metab 283:E658–E666
Barbagallo M, Shan J, Pang PK, Resnick LM (1995) Glucose-induced alterations of cytosolic free calcium in cultured rat tail artery vascular smooth muscle cells. J Clin Invest 95:763–767
Smogorzewski M, Galfayan V, Massry SG (1998) High glucose concentration causes a rise in [Ca2+]i of cardiac myocytes. Kidney Int 53:1237–1243
Yuan SY, Ustinova EE, Wu MH, Tinsley JH, Xu W, Korompai FL, Taulman AC (2000) Protein kinase C activation contributes to microvascular barrier dysfunction in the heart at early stages of diabetes. Circ Res 87:412–417
Colligan PB, Relling DP, Ren J (2202) Ceramide reduces high glucose-induced diabetic cardiomyopathy in adult rat ventricular myocytes. Cell Mol Biol 48:OL251–OL257
Kolb H, Kiesel U, Kroncke KD, Kolb-Bachofen V (1991) Suppression of low dose streptozotocin induced diabetes in mice by administration of a nitric oxide synthase inhibitor. Life Sci 49:PL213–PL217
Papaccio G, Esposito V, Latronico MV, Pisanti FA (1995) Administration of a nitric oxide synthase inhibitor does not suppress low-dose streptozotocin-induced diabetes in mice. Int J Pancreatol 17:63–68
Smith JM, Paulson DJ, Romano FD (1997) Inhibition of nitric oxide synthase by L-NAME improves ventricular performance in streptozotocin-diabetic rats. J Mol Cell Cardiol 29:2393–2402
Cosentino F, Hishikawa K, Katusic ZS, Lüscher TF (1997) High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 96:25–28
Graier WF, Simecek S, Kukovetz WR, Kostner GM (1996) High D-glucose-induced changes in endothelial Ca2+/EDRF signaling are due to generation of superoxide anions. Diabetes 45:1386–1395
Balon, TW, Nadler JL (1997) Evidence that nitric oxide increases glucose transport in skeletal muscle. J Appl Physiol 82:359–363
Trachtman, H, Futterweit S, Crimmins DL (1997) High glucose inhibits nitric oxide production in cultured rat mesangial cells. J Am Soc Nephrol 8:1276–1282
Felaco M, Grilli A, De Lutiis MA, Patruno A, Libertini N, Taccardi AA, Di Napoli P, Di Giulio C, Barbacane R, Conti P (2001) Endothelial nitric oxide synthase (eNOS) expression and localization in healthy and diabetic rat hearts. Ann Clin Lab Sci. 31:179–186
Pekiner B, Ulusu NN, Das-Evcimen N, Sahilli M, Aktan F, Stefek M, Stolc S, Karasu C; Antioxidants in Diabetes-Induced Complications Study Group (2002) In vivo treatment with stobadine prevents lipid peroxidation, protein glycation and calcium overload but does not ameliorate Ca2+-ATPase activity in heart and liver of streptozotocin-diabetic rats: comparison with vitamin E. Biochim Biophys Acta 1588:71–78
Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zhou YS, Pinsky D, Stern D (1994) Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 269:9889–9897
Giardino I, Edelstein D, Brownlee M (1996) BCL-2 expression or antioxidants prevent hyperglycaemia-induced formation of intracellular advanced glycation end-products in bovine endothelial cells. J Clin Invest 94:110–117
Tannous M, Rabini RA, Vignini A, Moretti N, Fumelli P, Zielinski B, Mazzanti L, Mutus B (1999) Evidence for iNOS-dependent peroxynitrite production in diabetic platelets. Diabetologia 42:539–544
Pieper GM (1997) Acute amelioration of diabetic endothelial dysfunction with a derivative of the nitric oxide synthase cofactor, tetrahydrobiopterin. J Cardiovasc Pharmacol 29:8–15
Acknowledgements
The skillful technical assistance from J. Duan, K.K. Hintz and G.E. McFadden is greatly appreciated. This work was supported by grants from American Diabetes Association (7-0-RA-21) and NIH/NCRR (RR-16474). L.B. Esberg is a Ronald McNair Scholar at the University of North Dakota.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Esberg, L.B., Ren, J. Role of nitric oxide, tetrahydrobiopterin and peroxynitrite in glucose toxicity-associated contractile dysfunction in ventricular myocytes. Diabetologia 46, 1419–1427 (2003). https://doi.org/10.1007/s00125-003-1183-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1183-8