
Abstract
Key message
Genome-wide association analysis in tropical and subtropical maize germplasm revealed that MLND resistance is influenced by multiple genomic regions with small to medium effects.
Abstract
The maize lethal necrosis disease (MLND) caused by synergistic interaction of Maize chlorotic mottle virus and Sugarcane mosaic virus, and has emerged as a serious threat to maize production in eastern Africa since 2011. Our objective was to gain insights into the genetic architecture underlying the resistance to MLND by genome-wide association study (GWAS) and genomic selection. We used two association mapping (AM) panels comprising a total of 615 diverse tropical/subtropical maize inbred lines. All the lines were evaluated against MLND under artificial inoculation. Both the panels were genotyped using genotyping-by-sequencing. Phenotypic variation for MLND resistance was significant and heritability was moderately high in both the panels. Few promising lines with high resistance to MLND were identified to be used as potential donors. GWAS revealed 24 SNPs that were significantly associated (P < 3 × 10−5) with MLND resistance. These SNPs are located within or adjacent to 20 putative candidate genes that are associated with plant disease resistance. Ridge regression best linear unbiased prediction with five-fold cross-validation revealed higher prediction accuracy for IMAS-AM panel (0.56) over DTMA-AM (0.36) panel. The prediction accuracy for both within and across panels is promising; inclusion of MLND resistance associated SNPs into the prediction model further improved the accuracy. Overall, the study revealed that resistance to MLND is controlled by multiple loci with small to medium effects and the SNPs identified by GWAS can be used as potential candidates in MLND resistance breeding program.
Similar content being viewed by others


Introduction
Maize lethal necrosis disease (MLND) has emerged as a devastating disease in eastern Africa since 2011 (Wangai et al. 2012). MLND in eastern Africa was found to result from synergistic interaction between Maize chlorotic mottle virus (MCMV) and Sugarcane mosaic virus (SCMV). Although each of these viruses individually can cause disease, the synergistic interactions are more pronounced. SCMV was reported in Kenya many years ago (Louie 1980). MCMV was first identified in Peru in 1973 (Castillo and Hebert 1974) and has been subsequently reported in the USA, parts of Latin America, and China (Niblett and Clafin 1978; Uyemoto et al. 1980; Xie et al. 2011). Wangai et al. (2012) first reported the MLND and MCMV in Kenya since the MLND has been reported in Uganda, Tanzania, Democratic Republic of the Congo, South Sudan and Ethiopia, seriously threatening maize production and the livelihoods of smallholder farmers in eastern Africa (Adams et al. 2013, 2014).
Maize plants are susceptible to MLND at all growth stages, from seedling to maturity. The diagnostic symptoms of MLND include chlorotic mottling of leaves, necrosis development from the leaf margin to the midrib, and dead heart; later-stage infection could lead to sterile pollen, small cobs with poor seed set, or death of the plants. Possible factors that contributed to the devastating effect of MLND in eastern Africa include new and perhaps highly virulent strains of MCMV and SCMV, conducive environment for survival and spread of insect-vectors of the two viruses (Cabanas et al. 2013), conducive environment for proliferation of the insect vectors of the two viruses, and continuous maize cropping in certain regions leading to build-up of virus inoculum. Studies undertaken jointly by International Maize and Wheat Improvement Center (CIMMYT) and Kenya Agriculture and Livestock Research Organization (KALRO) since 2012 revealed the vulnerability of a large array (nearly 90 percent) of pre-commercial and commercial maize germplasm to the MLND, especially under artificial inoculation. The maize seed industry in eastern Africa is under significant pressure to quickly replace the highly vulnerable commercial hybrids. Therefore, accelerated development and deployment of improved maize varieties with resistance to MLND is now a top priority in eastern Africa. This in turn requires intensive screening of germplasm for identifying sources of resistance, understanding the genetic architecture of MLND resistance, and utilizing molecular markers in breeding programs for fast-tracking development of improved varieties with MLND resistance and other relevant traits for the African smallholder farmers.
Genome-wide association study (GWAS) enables analysis of genetic architecture of complex traits (Yan et al. 2011). Compared to traditional linkage mapping, GWAS offers higher resolution and greater ability for identifying favorable genetic loci responsible for the trait of interest, while saving cost and time (Yu and Buckler 2006). Linkage disequilibrium (LD) decay is rapid in maize due to its high diverse nature. Therefore, large numbers of polymorphic SNPs are required to ensure complete coverage of the genome. Genotyping-by-sequencing (GBS) generates millions of SNPs with affordable cost. To date, GWAS has been successfully applied to identify quantitative trait loci (QTL) or genomic regions conferring resistance to some important diseases of maize, such as Fusarium ear rot (Zila et al. 2013), gray leaf spot (Shi et al. 2014), head smut (Weng et al. 2012), Northern corn leaf blight (Poland et al. 2011), Southern corn leaf blight (Kump et al. 2011), and SCMV (Tao et al. 2013). However, GWAS has not yet been undertaken or reported for identifying genomic regions influencing resistance to MLND.
Genomic selection or genome-wide selection (GS) is another promising breeding tool to improve the efficiency and speed of the breeding process (Zhao et al. 2012; Beyene et al. 2015). GS involves use of a ‘training population’ of individuals that have been phenotyped and genotyped, for developing the prediction model. In the next step, this model is used to predict genomic estimated breeding values (GEBVs) of the individuals from the ‘estimation set’ which are not phenotyped but genotyped with high-density markers (Meuwissen et al. 2001). Initial GS studies applied to maize agronomic traits like plant height and dry matter yield showed promising results with high prediction accuracies (Riedelsheimer et al. 2012; Zhao et al. 2012). The prediction accuracies on complex diseases like Northern corn leaf blight resistance (Technow et al. 2013) and Fusarium ear rot (Zila 2014) in maize clearly indicated the potential of GS for improving quantitative disease resistance. This motivated us to implement GS on a complex trait like MLND.
In this study, two association mapping (AM) panels, namely IMAS (Improved Maize for African Soils) and DTMA (Drought Tolerant Maize for Africa), were used for understanding the genetic architecture of MLND resistance. The objectives of the study were (1) to evaluate the diverse array of tropical and subtropical maize lines for their responses to MLND under artificial inoculation; (2) to identify genomic regions, SNPs, and putative candidate genes associated with MLND resistance; and (3) to assess the potential of GS for MLND resistance in maize.
Materials and methods
Plant materials and field trials
Two AM panels constituted under two major projects in Sub-Saharan Africa, namely DTMA (Drought Tolerant Maize for Africa) and IMAS (Improved Maize for African Soils), led by the Global Maize Program of the CIMMYT were used in this study. The IMAS-AM and DTMA-AM panels comprised 380 and 235 lines, respectively, representing broadly the tropical/subtropical maize genetic diversity, including germplasm derived from breeding programs targeting tolerance to drought, soil acidity, and low N, resistance to insects and pathogens (Wen et al. 2011).
Collection and maintenance of virus isolates
Stock isolates of MCMV and SCMV were collected from MLN hotspot areas in Kenya. Once confirmed on the presence of SCMV or MCMV by Enzyme-linked immunosorbent assay (ELISA), both viruses were propagated on a susceptible hybrid, H614, in separate greenhouses. Infected leaf samples collected from the field were cut into small pieces and ground in a mortar and pestle in grinding buffer (10 mM potassium-phosphate, pH 7.0). The resulting sap extract was centrifuged for 2 min at 12,000 rpm. Carborundum was added to decanted sap extract at the rate of 0.02 g/ml. The susceptible hybrid H614 at two leaves stage was inoculated by rubbing sap extract onto the leaves. Two separate, sealed greenhouses were maintained for SCMV and MCMV inoculum production. Three weeks before inoculation, ELISA was undertaken on random samples of leaves from the SCMV and MCMV greenhouses to confirm the inoculum purity.
Artificial field inoculation and phenotyping
In order to keep uniform MLND pressure across field trials, the optimized combination of SCMV and MCMV viruses (ratio of 4:1) were mixed and inoculated twice at 5th and 6th week after planting. Plants were inoculated using a motorized, backpack mist blower (Solo 423 MistBlower, 12 L capacity). An open nozzle (2-inche diameter) was used to deliver inoculum spray at a pressure of 10 kg/cm2. The presence of both viruses in the field trials was confirmed by ELISA once disease symptoms were apparent (approximately 2-week post-inoculation).
All inbred lines were evaluated in one-row 3 m plots with two replications in alpha lattice design in three seasons during 2012–2014 at Narok [latitude 01°05′S, longitude 35°52′E, 1827 m above sea level (asl)] and Naivasha (latitude 0°43′S, longitude 36°26′E, 1896 m asl) in Kenya. All standard agronomic management practices were followed. Disease severity was scored for MLN at three-week post-inoculation. Inbreds were rated visually on a 1–5 disease severity scale, where 1 = no visible MLN symptoms, 2 = fine chlorotic streaks mostly on older leaves, 3 = chlorotic mottling throughout the plant, 4 = excessive chlorotic mottling on lower leaves and necrosis of newly emerging leaves (dead hear), and 5 = complete plant necrosis.
Phenotypic data analyses
For data based on ordinal scales, it is important to evaluate whether the data meets the assumptions of the applied statistical model (independent, normally distributed and constant variance; Rawlings et al. 1998). In this study for both the panels, we plotted the residuals against predicted values which revealed that the variance was constant. The histogram plot of the residuals was slightly deviated from normal distribution in DTMA panel compared to IMAS panel (data not shown). Therefore, we used the original data for the analyses without any transformation. Analyses of variance within and across environments was determined by the restricted maximum likelihood method using SAS 9.2 (SAS Institute 2010). Variance components were estimated by following linear mixed model: Y ijko = µ + g i + l j + r kj + b ojk + e ijko , where Y ijko was the phenotypic performance of the ith genotype at the jth environment in the kth replication of the oth incomplete block, µ was an intercept term, g i was the genetic effect of the ith genotype, l j was the effect of the jth environment, r kj was the effect of the kth replication at the jth environment, b ojk was the effect of the oth incomplete block in the kth replication at the jth environment, and e ijko was the residual. Environments and replications were treated as fixed effects and the other effects as random. Heritability on an entry-mean basis was estimated from the variance components as the ratio of genotypic to phenotypic variance. In addition, best linear unbiased estimates (BLUEs) were estimated across environments assuming fixed genotype effects. For association analyses, best linear unbiased prediction (BLUP) of each line was calculated for across environments.
Molecular data analyses
DNA of all inbred lines was extracted from greenhouse-grown seedlings at 3–4 leaves stage. DNA was used for genotyping using GBS platform (Elshire et al. 2011) at Cornell University, Ithaca, USA, as per the procedure described in earlier studies (Elshire et al. 2011; Glaubitz et al. 2014). For quality screening in both the AM panels, SNPs which were either monomorphic, had missing value of >5 %, heterozygosity of >5 %, or had a minor allele frequency of <0.02 were discarded from the analysis. After these quality checks, 259,000 and 264,000 high-quality SNPs were retained for GWAS in the IMAS and DTMA-AM panels, respectively.
Genome-wide association study (GWAS)
BLUP of each line was used as phenotypes in AM scans. MLND severity data were corrected for population structure using general linear model (GLM), as well as population structure and kinship (Q + K) using mixed linear model (MLM) algorithm (Flint-Garcia et al. 2005; Yu and Buckler, 2006). GWAS and principal component (PC) analysis was performed using TASSEL ver 4.0 (Bradbury et al. 2007). The first three PCs were used to correct the population structure. The threshold P value (P < 3 × 10−5) was determined by considering the pattern of the Q–Q plot of the model and the point at which the observed F test statistics deviated from the expected F test statistics (Gao et al. 2010; Sukumaran et al. 2012, 2015). The total proportion of phenotypic variance explained by the detected QTL was calculated by fitting all significant SNPs simultaneously in a linear model to obtain \(R_{\rm {adj}}^2\). The proportion of the genotypic variance explained by all QTL was calculated as the ratio of \(p_G = R_{\rm {adj}}^2/h^2\). The 60 bp source sequences of the significantly associated SNPs were used to perform BLAST searches against the ‘B73’ RefGen_v2 (http://blast.maizegdb.org/home.php?a=BLAST_UI). Within the local LD block including associated SNPs, the filtered genes in MaizeGDB (http://www.maizegdb.org) containing directly or adjacent to each associated SNP were considered as possible candidate genes for MLND resistance.
Genomic selection
Ridge regression best linear unbiased prediction (RR-BLUP; Whittaker et al. 2000) was applied on the BLUEs across environments. From the GBS SNP marker data, a sub-set of 2000 SNPs distributed uniformly across genome, with no missing values, and minor allele frequency >0.05 were used for genomic prediction in both the AM panels. Details of the implementation of the RR-BLUP model were described by Zhao et al. (2012). Prediction accuracy of the GS approach was evaluated using the five-fold cross-validation with 1000 times repetitions. The correlation between observed and predicted phenotypes (r MP) was estimated. The accuracy of GS was calculated as r GS = r MP/h (Dekkers 2007), where h refers to the square root of heritability. The genomic prediction was carried out in two scenarios where both the training and estimation populations were derived from (1) within AM panels, (2) across AM panels. Additionally, for both the scenarios, prediction was carried out with and without inclusion of GWAS based MLND resistance associated SNPs. In GS, optimizing the number of markers and the training population size without losing accuracy is crucial. Therefore, we checked the effect of prediction accuracy with different number of SNPs varying from 300 to 14,000, and the number of individuals from 20 to 100 % with the interval of 20 % of the total population size.
Results
In each environment, average MLND severity rate was higher for the DTMA-AM panel, compared to the IMAS-AM panel (Supplementary Figure S1). For both the panels, moderate yet significant correlations were observed among the genotypic values estimated in each environment (Supplementary Table S1). This ruled out the possible bias due to environment-specific disease responses in a combined analysis. Analysis across environments revealed higher average diseases severity in DTMA-AM panel (3.53) than IMAS-AM panel (2.98) in 1–5 disease scale (Table 1). The frequency of the phenotypic values in both the panels followed approximately a normal distribution with larger range of distribution for IMAS panel (Fig. 1). The ANOVA across environments revealed significant genotypic and genotype × environment interaction variances for MLND responses in both the panels (Table 1). The estimate of heritability was high with 0.73 for IMAS and 0.62 for DTMA panel, which reveals predominance of additive control of responses of maize genotypes to MLND resistance.

Phenotypic distribution of MLND scores on a 1–5 scale in the IMAS-AM (A) and DTMA-AM (B) panels (mean values are indicated by arrows)
Among 615 lines evaluated for MLND response, 14 lines were selected as best performing lines (Table 2). Interestingly yellow lines derived from tropical lowland breeding programs from Mexico were the best lines among the selected lines for MLND resistance. African breeding programs where white maize is predominant, and we found five lines which showed relatively better resistance for MLND.
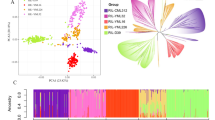
Principal component analysis revealed the presence of a clear population structure in both the panels with respect to first three PCs (Fig. 2a, c), as well as by several of the first ten PCs as revealed by their density distribution (Fig. 2b, d). In IMAS panel, lines derived from the CIMMYT physiology program and from the South African Agriculture Research Council’s (ARC’s) breeding program formed distinct clusters (Fig. 2a). In the DTMA panel too, the lines developed by CIMMYT physiology program formed a distinct group (Fig. 2c).

Population structure based on principal component (PC) analysis of IMAS-AM (a) and DTMA-AM (c) panels. Violin plot showing the density distribution of the first ten principal components for the genotypes from IMAS-AM (b) and DTMA-AM (d) panel. (In the IMAS-AM panel, the seven groups represent lines from the breeding program of G1 CIMMYT gene bank, G2 Physiology, G3 Zimbabwe, G4 Kenya, G5 Lowland tropical, G6 MAS-DT, and G7 ARC South Africa; and in the DTMA-AM panel, the seven groups represent lines from the breeding program of G1 Tropical, G2 Physiology, G3 Zimbabwe, G4 Kenya, G5 Subtropical, G6 Entomology, and G7 Columbia)
From the GBS data, we selected a set of ~260 K high-quality polymorphic SNPs for GWAS. Manhattan plots of the GWAS results for both IMAS and DTMA panels are shown in Fig. 3. In the IMAS-AM panel, we detected 18 significant marker–trait associations for MLND resistance (Table 3, P < 3 × 10−5). These significantly associated SNPs individually explained 8–10 % of the total genotypic variance, whereas together explained 30 % of the total proportion of genotypic variance for resistance to MLND. In the DTMA-AM panel, we detected six significant marker–trait associations which individually explained 14–18 % of the total genotypic variance and together explained 37 % of the total proportion of genotypic variance for MLND resistance (Table 4). Comparison of the significant SNPs in the two AM panels revealed that there were no common marker–trait associations across panels; however, there was some similarity on number of SNPs falling into same chromosome bins. We used B73 maize genome reference sequence to identify putative candidate genes based on the SNPs significantly associated with MLND resistance (Tables 3, 4). From both the AM panels, a set of putative candidate genes were identified; based on their functions, these can be grouped as either R genes or plant defense responsive genes.

Quantile–quantile plots (a, c), and Manhattan plots of a mixed linear model for MLND resistance in the IMAS-AM and DTMA-AM panels. Plots above red horizontal line showed the genome-wide significance with stringent threshold of P = 3 × 10−5. The different colors indicate the 10 different chromosomes of maize (color figure online)
The accuracy of genomic predictions within the panel was higher for the IMAS-AM over the DTMA-AM panel (Fig. 4). The prediction accuracy was improved in both the panels by inclusion of the MLND resistance associated SNPs. The prediction accuracy across AM panels was 0.41 which increased to 0.56 with the inclusion of MLND resistance associated SNPs into the prediction model. The prediction accuracy was severely affected by population size, whereas the effect was relatively low with decrease in the number of markers (Fig. 5).

Distribution of the accuracy of genomic predictions for scenario 1 (prediction based on random markers) and scenario 2 (prediction based on random and significant MLND-associated markers) within and across IMAS-AM and DTMA-AM panels, as revealed by five-fold cross-validation for MLND resistance

Effect of the number of markers, and the number of individuals on the accuracy of genomic prediction for MLND resistance in the IMAS association mapping panel
Discussion
Maize lethal necrosis disease is not only due to individual effect of either SCMV or MCMV, but it also includes their interaction effects which together lead to substantial yield loss and threatening the food security currently in eastern Africa (Ali and Yan 2012). The genetics of SCMV and other potyviruses has been extensively studied in maize with diverse germplasm (as reviewed by Redinbaugh and Pratt 2009). The genetics and inheritance of MLND resistance is not known and is expected to be very complex due to combination of two viruses. GWAS and GS are the best tools used to study such complex traits (Riedelsheimer et al. 2012).
In GWAS, the power of QTL detection largely depends not only on the sample size but also on the trait architecture and heritability (Yu et al. 2008); therefore, precise phenotypic evaluation for the trait of interest is critical. To obtain reliable phenotypic data, we have used a broad array of tropical and subtropical maize germplasm and evaluated the same for MLND severity under optimized artificial inoculation procedure for three environments in Kenya. Heritability was moderately high in both AM panels. The significant genotypic variation observed in both the panels also reflected the high quality of the phenotypic data, thereby enabling identification of genomic regions with substantive power.
Population structure and linkage disequilibrium
The lines used in this study represents various breeding programs from Kenya, Zimbabwe, South Africa, Nigeria, Malawi and Columbia, as well as from CIMMYT gene bank and some specific programs such as CIMMYT Physiology program, Latin American tropical lowland breeding program, and the mid-elevation Africa adapted breeding program (Fig. 2). As a result, confounding structure exists in these panels and false-positive associations would be expected if the data is not corrected for population structure (Yan et al. 2009). The use of first three PCs along with relative kinship matrix in the Q + K model enabled us to correct for spurious associations which is also evident in quantile plots (Fig. 3a, c).
The mapping resolution and the required marker density for GWAS is largely depends on the extent of LD in the population (Yu and Buckler 2006; Myles et al. 2009). The extent of LD for the panels used in this study was examined in detail in earlier study (Vinayan et al. 2013). The average r 2 values between neighboring markers were 0.29 and 0.24 for IMAS-AM and DTMA-AM panel, respectively. This moderate LD estimate in both the panels suggests the diverse nature of the tropical/subtropical maize germplasm used in this study, which on the other hand leads to high mapping resolution. The observed r 2 between adjacent markers was comparable to (r 2 = 0.28) the earlier studies (Van Inghelandt et al. 2011; Massman et al. 2013). Although it is estimated that at least one million SNPs are required to efficiently detect all minor QTL (Gore et al. 2009), the observed average LD estimates in our study indicates that at least medium to large effect QTLs should be detected.
Genome-wide association study for main effect QTL
Twenty-four SNPs significantly associated with MLND resistance are localized to eight out of ten different chromosomes (Tables 3, 4). In IMAS-AM panel, the total genotypic variance explained by each significantly associated SNPs was <10 %, consequently each of the QTL defined by these SNPs can be regarded as relatively minor QTL. On the contrary in DTMA-AM panel, we observed all six detected QTL explained >10 % of the total genotypic variance.
In IMAS-AM panel, eight SNPs detected on chromosome 3 are localized to the linkage map bins 3.04 and 3.05 which reportedly had resistance genes to multiple potyviruses, including SCMV, MDMV (Maize dwarf mosaic virus), MCDV (Maize chlorotic dwarf virus), MSV (Maize mosaic virus), and WSMV (Wheat streak mosaic virus; Lübberstedt et al. 2006; Jones et al. 2011; Zambrano et al. 2014). In addition, these two genomic regions are also found to confer resistance to other fungal diseases like Southern corn leaf blight, Northern corn leaf blight and gray leaf spot (Belcher 2009). Comparison of the GWAS detected SNPs position with previous QTL studies revealed that the SNP S2_211771737 was overlapped with the MMV resistance QTL(Zambrano et al. 2014). Overall coincidence of MLND resistance associated SNPs with several other virus resistance loci supports the clustering nature of QTL for multiple virus resistance. In conclusion, the identified SNPs can be used as diagnostic markers, and targeted selection of these SNPs alleles are useful in improvement of MLND resistance levels in elite breeding lines.
Putative candidate genes
Putative candidate genes identified on chromosomes 2 and 3 were primarily involved in cell-to-cell transport of micro and macromolecules (Tables 3, 4). Plant viruses need to be able to move between mesophyll cells and also in and out of phloem tissue for systematic infection. It is assumed that plants resist the virus infection by controlling the virus movement inside the host and this mechanism is clearly demonstrated by RTM system in Arabidopsis (Chrisholm et al. 2000). Similarly, there is high probability that the putative candidate genes identified in this study might be involved in MLND resistance/vulnerability by controlling the movement of one or both the viruses in the plants; however, it needs to be confirmed by independent validation studies.
The plant defence mechanism against viruses is mediated by resistance (R) genes and is well characterized in several crop plants (Spassova et al. 2001; Stange et al. 2004; Vidal et al. 2002). In maize, two NBS-LRR genes are mapped into bin 3.05 of chromosome 3 (Xiao et al. 2007). On the other hand, the R genes often express complete resistance in the form of hypersensitive response by which the infected cells are killed by programmed cell death. In line with this observation, we identified one candidate gene GRMZM2G109805 on chromosome 5 which directly involved in hypersensitive reaction. Clear hypersensitive reaction and leaf death symptoms were also observed in MLND infected plants which suggest the possible role of these genes in plants resistance against viruses.
For viruses, host factors are important to complete their life cycle. Mutations in these host factors forms a recessive inherited virus resistance genes. We found one candidate gene GRMZM2G018943 functions as a translation initiation factor eIF-2B is also due to similar type of mutations. Previously, few recessive inherited virus resistance genes were also reported for potyvirus and other viruses (Ingvardsen et al. 2010). These recessive genes contribute for certain level of resistance to MLND by associating with other minor QTL of SCMV or MCMV. However, it should be point out that these candidate genes should be further validated before integrating them in a breeding program.
Two candidate genes with putative protein serine/threonine kinase activity have a role in signaling interactions during the perception of pathogens and consequent activation of defence responses (Romeis et al. 2000; Zhou et al. 1995). Three identified putative candidate genes with ice-binding functions are type of antifreeze proteins which belongs to group of pathogenesis-related proteins (Griffith and Yaish 2004; Hon et al. 1995) indicating their possible role in plant defence against MLND.
Genomic selection
The results from this study give first insights into the potential of genome-based prediction of MLND resistance in maize. The potential of GS has been assessed for simple and complex traits in maize (Crossa et al. 2010, 2013; Zhao et al. 2012). GS allows capture contribution of even small effect QTL and lead to high prediction accuracy. Using a cross-validation approach, genomic predictions explained ~56 and ~36 % of the variation in IMAS-AM and DTMA-AM panel, respectively. This is in accordance with the previous study for complex disease like Northern corn leaf blight (Technow et al. 2013). The differences in the prediction accuracy between two AM panels can be attributed to their sample size, genetic variance, and trait heritability (Table 1). On the other hand, the differences may also reflect the changes in population structure and LD estimates. Surprisingly, prediction accuracy across panel was lower than IMAS panel which might be attributed to higher magnitude of genotypic variance observed for within panel than across panel (data not shown). Inclusion of MLND-associated SNPs into training population led only slight increase in the prediction accuracy in both the panels, indicating that prediction accuracy is mainly attributable to many small effects QTL distributed across genome.
Routine implementation of GS in breeding program is affected by resource allocation especially on cost of genotyping and phenotyping. RR-BLUP is known to perform well under low marker density (Habier et al. 2007), and accordingly, we observed marginal decrease in prediction accuracy when number of markers were reduced from 14,000 to 1000 (Fig. 5). Our finding also corroborates the earlier studies in maize (Zhao et al. 2012). However, accuracy was severely affected with the decrease in the size of training population. This clearly suggests the need of optimum size of training population which approximates n ~ 230 for MLND in IMAS-AM panel in the current study; however, this could vary depending on the germplasm used and the trait under study.
Possible routine use of GS in breeding for resistance to MLND depends on its relative advantage over phenotypic selection. Phenotypic selection accuracy, estimated as h, was 0.85 and 0.79 for IMAS and DTMA-AM panels, respectively. However, in maize, up to three cycles of GS per year are possible (Lorenzana and Bernardo 2009). Therefore, compared to phenotypic selection, GS would be more efficient in terms of genetic gain per year than per cycle.
Conclusion
In this study, we used two AM panels together comprised 615 lines to understand the genetic architecture of MLND resistance in tropical and subtropical maize germplasm. GWAS scan identified 24 SNPs associated with resistance to MLND. GS results revealed higher selection gain per year for marker-based selection compared to phenotypic based selection for MLND resistance. Further research is warranted on validating the effects of the identified candidate genes and their functional variants to confirm that these genes engender resistance to MLND in maize. We identified few lines which can serve as a potential donor in improving susceptible commercial lines into MLND resistant lines either through marker-assisted recurrent selection or GS.
Author contribution statement
BD, DM, GM, BMP, and MG—conceived the experiment; BD, GM, MG, and DM—conducted the field evaluations and phenotyping; MG, KS, and RB—coordinated the GBS experiments; MG—carried out the GWAS analyses; MG, BD, DM, GM, MO, BMP, JMB, KS, and RB—interpreted the results and drafted the manuscript.
Abbreviations
- MLND:
-
Maize lethal necrosis disease
- GBS:
-
Genotyping-by-sequencing
- LD:
-
Linkage disequilibrium
- MLM:
-
Mixed linear model
- MAF:
-
Minor allele frequency
- MCMV:
-
Maize chlorotic mottle virus
- SCMV:
-
Sugarcane mosaic virus
- ELISA:
-
Enzyme-linked immunosorbent assay
References
Adams IP, Miano DW, Kinyua ZM, Wangai A, Kimani E, Phiri N, Reeder R, Harju V, Glover R, Hany U, Souza-Richards R, Deb Nath P, Nixon T, Fox A, Barnes A, Smith J, Skelton A, Thwaites R, Mumford R, Boonham N (2013) Use of next-generation sequencing for the identification and characterization of Maize chlorotic mottle virus and Sugarcane mosaic virus causing maize lethal necrosis in Kenya. Plant Pathol 62:741–749
Adams IP, Harju VA, Hodges T, Hany U, Skelton A, Rai S, Deka MK, Smith J, Fox A, Uzayisenga B, Ngaboyisonga C, Uwumukiza B, Rutikanga A, Rutherford M, Ricthis B, Phiri N, Boonham N (2014) First report of maize lethal necrosis disease in Rwanda. New Dis Rep 29:22
Ali F, Yan J (2012) Disease resistance in maize and the role of molecular breeding in defending against global threat. J Integr Plant Biol 54:134–151
Belcher AR (2009) The physiology and host genetics of quantitative resistance in maize to the fungal pathogen Cochliobolus heterostrophus. Dissertation, North Carolina State University, Raleigh
Beyene Y, Semagn K, Mugo S, Tarekegne A, Babu R, Meisel B, Sehabiague P, Makumbi D, Magorokosho C, Oikeh S, Gakunga J, Vargas M, Olsen M, Prasanna BM, Banziger M, Crossa J (2015) Genetic gains in grain yield through genomic selection in eight bi-parental maize populations under drought stress. Crop Sci 55:154–163
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cabanas D, Watanabe S, Higashi CHV, Bressan A (2013) Dissecting the Mode of Maize Chlorotic Mottle Virus Transmission (Tombusviridae: Machlomovirus) by Frankliniella williamsi (Thysanoptera: Thripidae). J Econ Entomol 106:16–24
Castillo J, Hebert T (1974) Nueva enfermedad virosa afectando al maiz en el Peru. Fitopatologia 9(2):79–84 (in Spanish)
Chrisholm ST, Mahajan SK, Whitham SA, Yamamoto ML, Carrington JC (2000) Cloning of the Arabidopsis RTM1 gene, which controls restriction of long-distance movement of the tobacco etch virus. Proc Natl Acad Sci USA 97:489–494
Crossa J, Campos G, Pérez P, Gianola D, Burgueño J, Araus JL, Makumbi D, Singh RP, Dreisigacker S, Yan J, Arief V, Banziger M, Braun H-J (2010) Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 186:713–724
Crossa J, Beyene Y, Kassa S, Pérez P, Hickey JM, Chen C, de los Campos G, Burgueño J, Windhausen VS, Buckler E, Jannink J-L, Lopez Cruz MA, Babu R (2013) Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3 3:1903–1926
Dekkers JCM (2007) Prediction of response to marker-assisted and genomic selection using selection index theory. J Anim Breed Genet 124:331–341
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Flint-Garcia Sherry A, Thuillet A, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich J, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064
Gao X, Becker LC, Becker DM, Starmer JD, Province MA (2010) Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol 34:100–105
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9(2):e90346
Gore MA, Chia J-M, Elsaire LJ, Sun Q, Ersoz ES et al (2009) The first-generation haplotype map of maize. Science 326:1115–1117
Griffith M, Yaish MW (2004) Antifreeze proteins in overwintering plants: a tale of two activities. Trends Plant Sci 9:399–405
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics 177:2389–2397
Hon WC, Criffith M, Mlynarz A, Kwok YC, Yang DSC (1995) Antifreeze proteins in winter rye are similar to pathogenesis-related proteins. Plant Physiol 109:879–889
Ingvardsen CR, Xing Y, Frei UK, Lübberstedt T (2010) Genetic and physical fine mapping of Scmv2, a potyvirus resistance gene in maize. Theor Appl Genet 120:1621–1634
Jones MW, Boyd EC, Redinbaugh MG (2011) Responses of maize (Zea mays L.) near isogenic lines carrying Wsm1, Wsm2, and Wsm3 to three viruses in the Potyviridae. Theor Appl Genet 123:729–740
Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza-Rosas MA, Zwonitzer JC, Kresovich S, McMullen MD, Ware D (2011) Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet 43:163–168
Lorenzana R, Bernardo R (2009) Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor Appl Genet 120:151–161
Louie R (1980) Sugarcane mosaic virus in Kenya. Plant Dis 64:944–947
Lübberstedt T, Ingvardsen C, Melchinger AE, Xing Y, Salomon R, Redinbaugh MG (2006) Two chromosome segments confer multiple potyvirus resistance in maize. Plant Breed 125:352–356
Massman JM, Gordillo A, Lorenzana RE, Bernardo R (2013) Genomewide predictions from maize single-cross data. Theor Appl Genet 126:13–22
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE, Buckler ES (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21:2194–2202
Niblett CL, Clafin LE (1978) Maize lethal necrosis-a new virus disease of maize in Kansas. Plant Dis Rep 62:15–19
Poland JA, Bradbury PJ, Buckler ES, Nelson RJ (2011) Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc Natl Acad Sci USA 108:6893–6898
Rawlings JO, Pantula SG, Dickey DA (1998) Applied regression analysis: A research tool, 2nd edn. Springer, New York
Redinbaugh MG, Pratt RC (2009) Virus resistance. In Handbook of Maize: Its Biology. Springer, New York, pp 251–270
Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, Altmann T, Stitt M, Willmitzer L, Melchinger AE (2012) Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat Genet. doi:10.1038/ng.1033
Romeis T, Piedras P, Jones JDG (2000) Resistance gene-dependent activation of a calcium-dependent protein kinase in the plant defense response. Plant Cell 12:803–815
SAS Institute Inc (2010) SAS 9.2 Intelligence Platform: System Administration Guide. SAS Institute Inc, Cary
Shi L, Xiangling L, Weng J, Zhu H, Liu C, Haoa Z, Zhoua Y, Zhanga D, Lia M, Cia X, Lia X, Zhang S (2014) Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize (Zea mays L.). Crop J 2:132–143
Spassova MI, Prins TW, Folkertsma RT, Klein-Lankhorst RM, Hile J, Goldbach RW, Prins M (2001) The tomato gene Sw5 is a member of the coiled coil, nucleotide binding, leucine-rich repeat class of plant resistance genes and confers resistance to TSWV in tobacco. Mol Breed 7:151–161
Stange C, Matus JT, Elorza A, Arce-Johnson P (2004) Identification and characterization if a novel tobacco mosaic virus resistance N gene homologue in Nicotiana tabacum plants. Funct Plant Biol 31:149–158
Sukumaran S, Xiang W, Bean SR et al (2012) Association mapping for grain quality in a diverse sorghum collection. Plant Genome J 5:126
Sukumaran S, Dreisigacker S, Lopes M, Chavez P, Reynolds MP (2015) Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor Appl Genet 128:353–363
Tao YF, Jiang L, Liu QQ, Zhang Y, Zhang R, Ingvardsen CR, Frei UK, Wang BB, Lai JS, Lubberstedt T, Xu ML (2013) Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol 13:162
Technow F, Bürger A, Melchinger AE (2013) Genomic prediction of northern corn leaf blight resistance in maize with combined or separated training sets for heterotic groups. G3(3):197–203
Uyemoto JK, Bockelman DL, Clafin LE (1980) Severe outbreak of maize lethal necrosis disease in Kansas. Plant Dis 64:99–100
Van Inghelandt D, Reif JC, Dhillon BS, Flament P, Melchinger AE (2011) Extent and genome-wide distribution of linkage disequilibrium in commercial maize germplasm. Theor Appl Genet 123:11–20
Vidal S, Cabrera H, Andersson RA, Fredriksson A, Valkonen JPT (2002) Potato gene Y-1 is an N gene homolog that confers cell death upon infection with potato virus Y. Mol Plant Microbe Interact 15:717–727
Vinayan MT, Babu R, Jyothsna T, Zaidi PH, Blümmel M (2013) A note on potential candidate genomic regions with implications for maize stover fodder quality. Field Crops Res 153:102–106
Wangai AW, Redinbaugh MG, Kinyua ZM, Miano DW, Leley PK, Kasina M, Mahuku G, Scheets K, Jeffers D (2012) First report of Maize chlorotic mottle virus and Maize lethal necrosis in Kenya. Plant Dis 96:1582
Wen W, Araus JL, Trushar S, Cairns J, Mahuku G, Banziger M, Torres JL, Sanchez C, Yan J (2011) Molecular characterization of a diverse maize inbred line collection and its potential utilization for stress tolerance improvement. Crop Sci 51:2569–2581
Weng J, Liu X, Wang Z, Wang J, Zhang L, Hao Z, Xie C, Li M, Zhang D, Bai L, Liu C, Zhang S, Li X (2012) Molecular mapping of the major resistance quantitative trait locus qHS2.09 with simple sequence repeat and single nucleotide polymorphism markers in maize. Phytopathology 102:692–699
Whittaker JC, Thompson R, Denham MC (2000) Marker-assisted selection using ridge regression. Genet Res 75:249–252
Xiao W, Zhai J, Fan S, Li L, Dai J, Xu M (2007) Mapping of genome-wide resistance gene analogs (RGAs) in maize (Zea mays L.). Theor Appl Genet 115:501–508
Xie L, Zhang JZ, Wang Q, Meng CM, Hong J, Zhou XP (2011) Characterization of Maize Chlorotic Mottle Virus associated with maize lethal necrosis disease in China. J Phytopathol 159:191–193
Yan JB, Shah T, Warburton M, Buckler ES, McMullen MD, Crouch J (2009) Genetic characterization of a global maize collection using SNP markers. PLoS One 4:e8451
Yan J, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Yu JM, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:1–6
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551
Zambrano JL, Jones MW, Brenner E, Francis DM, Tomas A, Redinbaugh MG (2014) Genetic analysis of resistance to six virus diseases in a multiple virus-resistant maize inbred line. Theor Appl Genet 127:867–880
Zhao Y, Gowda M, Liu W, Würschum T, Maurer HP, Longin FH, Ranc N, Reif JC (2012) Accuracy of genomic selection in European maize elite breeding populations. Theor Appl Genet 124:769–776
Zhou J, Loh Y-T, Bressan RA, Martin GB (1995) The tomato gene Pti1 encodes a serine/threonine kinase that is phosphorylated by Pto and is involved in the hypersensitive response. Cell 83:925–935
Zila CT (2014) Traditional and genomic methods for improving Fusarium ear rot resistance in maize. Dissertation, North Carolina State University. http://www.lib.ncsu.edu/resolver/1840.16/9195
Zila CT, Samayoa LF, Santiago R, Butrón A, Holland JB (2013) A genome-wide association study reveals genes associated with Fusarium ear rot resistance in a maize Core Diversity Panel. G3 3:2095–2104
Acknowledgments
The present study was supported by various projects, especially the DTMA, IMAS, WEMA, MLND-Africa projects funded by the Bill & Melinda Gates Foundation, USAID, and Syngenta Foundation for Sustainable Agriculture, besides the CGIAR Research Program on MAIZE. The authors would wish to thank CIMMYT field technicians at the different experiment stations in Kenya for managing trials; the management of Kenya Agricultural and Livestock Research Organization (KALRO) for giving us access to the experiment station; and CIMMYT laboratory technicians in Kenya for preparing samples for genotyping; and we also thank Dr. Edward S Buckler and the Institute of Genomic Diversity, Cornell University for the high-density genotyping (GBS) and imputation service.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by A. A. F. Garcia.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gowda, M., Das, B., Makumbi, D. et al. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor Appl Genet 128, 1957–1968 (2015). https://doi.org/10.1007/s00122-015-2559-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2559-0