
Abstract
Background
Current research into the mechanisms of organ atrophy associated with cancer cachexia have centred on the loss of skeletal muscle, as it is one of the most profound physical changes of the disease. However, many patients with cancer cachexia also experience significant atrophy of the heart. The mechanisms causing cardiac tissue wastage in cancer cachexia are largely unknown. However, it is believed to involve a number of molecular interactions between the tumour and host. Increased levels of oxidative stress have been found in cancer cachectic skeletal muscle and has been linked to the activation of the ubiquitin proteosome system (UPS). The aim of the current study was to examine the role of oxidative stress and the UPS in the hearts of mice with cancer cachexia.
Methods
Oxidative damage to DNA (8-OH-2dG), mRNA levels of the ROS-producing enzymes NADPH oxidase (NOX), and xanthine oxidase (XDH), the antioxidant enzyme superoxide dismutase (SOD) and key components of the UPS was measured in the heart of mice with cancer cachexia. Protein expression levels of NOX enzyme subunits and SOD enzyme activity was also measured in the same heart samples.
Results
8-OH-2dG levels were 1.5-fold higher in the heart of mice with cancer cachexia, and this was associated with a 1.7-fold lower level of NOX2 mRNA and twofold higher XDH mRNA in the same hearts. Cancer cachexia was also associated with a 1.5-fold lower level of SOD activity in the heart. Accompanying these pro- and antioxidant differences was a significantly higher level of mRNA for the key UPS elements MURF-1 (4.3=fold) and MAFbx (3.8-fold) in the hearts of mice with cancer cachexia.
Conclusions
The current study demonstrated that cardiac atrophy of cachectic mice is associated with oxidative damage to DNA in the myocardium. The higher levels of XDH mRNA in cachectic hearts suggest that xanthine oxidase may have an important role to play in producing oxidative stress. It appears that the combination of higher XDH expression and lower SOD enzyme activity are key contributors to oxidative stress and cardiac tissue damage in cancer-induced cardiac atrophy. Oxidative stress in the myocardium as with skeletal muscle may also induce increased expression of the E3 ligases MURF-1 and MAFbx as seen in this study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Current research into the mechanisms of organ atrophy associated with cancer cachexia (CC) is centred on the loss of skeletal muscle mass as it is one of the most profound physical changes of the disease. However, studies have also established that CC causes significant atrophy of the heart muscle in animal models [1–3]. Decreases in heart weight by as much as 60 % have been reported in a rat model of CC [3]. Clinical research does not currently exist examining the pathologic consequence of cardiac atrophy in cancer cachexia. However, Tian et al. used echocardiography to analyse the function of cachectic hearts in a mouse model of CC [4]. The study found that, 14 days after the inoculation with tumour cells, the hearts of cancer cachectic mice had reduced heart rate and fractional shortening.
The mechanisms causing cardiac tissue wastage are largely unknown. However, it is believed to involve a number of molecular interactions between the tumour and host. Elevated cytokines in response to tumour presence and tumour-derived factors such as proteolysis inducing factor (PIF) and lipid mobilising factor together produce tissue wasting [5–7]. Host factors such as cytokines including IL-1, IL-6, TNF-α, INF-γ and PIF derived from the tumour have been shown to create a state of oxidative stress and net protein catabolism in skeletal muscle [8]. Similarly increased oxidative stress has been shown to be present in the cachectic heart [9], but how this is linked to the activation of protein breakdown pathways is currently unknown.
A key catabolic pathway known to contribute to both skeletal and cardiac muscle atrophy is the ubiquitin proteosome system (UPS). The UPS is the principle mechanism by which muscle proteins are degraded. The UPS has been found to be upregulated in both experimental models and patients with CC [10, 11]. Enzymes which allow ubiquitin to tag proteins for destruction are termed E3 ligases. Currently, only three of the discovered several hundred E3 ligases have been found to be important in the process of muscle atrophy. These are termed E3 alpha, muscle-specific F-box (MAFbx), and muscle specific ring finger 1 (MURF-1) [12]. Examining a model of congestive heart failure, Adams et al. found a positive correlation between TNF-α and the E3 ligases MAFbx and MURF-1 [13]. Furthermore, the study found that TNF-α was able to bring about troponin I ubiquitinylation leading to impaired contractility of the heart.
Oxidative stress in a CC patient population may be an important link between the loss of tissue and initiation of key catabolic pathways [14]. A study by Kaynar et al. found that patients with either non-small cell or small cell lung cancer have elevated levels of xanthine oxidase (XO) activity compared with controls [15]. In the heart, XO is an important enzyme capable of catalyzing the oxidation of hypoxanthine and xanthine to uric acid whilst reducing O2 to O2 − and hydrogen peroxide (H2O2). Traditionally, increases in XO in the failing heart are associated with ventricular remodelling, fibrosis, and hypertrophy [16]. The contribution of XO in cancer-induced cardiac atrophy has not been investigated, despite the enzyme being implicated in myocardial remodelling and dysfunction [17, 18]. In terms of atrophy of the myocardium, like that of skeletal muscle, cardiomyocyte size is dependent on the relative rates of protein synthesis and degradation [19]. Increased levels of oxidative stress have been found in cancer cachectic skeletal muscle and has been linked to the activation of the UPS [20]. However, no such studies have examined the role of oxidative stress in the cancer cachectic heart. The current study aimed to test the hypothesis that increased levels of oxidative stress in the heart muscle of mice with cancer cachexia is associated with and up regulation of the UPS.
2 Materials and methods
2.1 Animals
All experimental procedures were carried out with approval from the Victoria University animal ethics committee (AEETH 07/05). Six-week-old BALB/c nu/nu nude mice purchased from the Animal Resource Centre, Western Australia, and were acclimatised for 1 week prior to injection of the cancer cell lines. Animals were maintained under controlled conditions of a 12-h light/dark cycle at 21 °C. Food and water were provided ad libitum. Food and water were weighed before and after the experimental period to ensure weight loss in the CC group was not due to a decrease in caloric intake. Animals were randomised into three weight-matched groups consisting of a no cancer (control, n = 8), cancer without cachexia control (cancer, n = 12) and cancer with cachexia (cachexia, n = 16).
2.2 Tumour model
Cancer and cancer cachexia were induced as previously described [25]. In brief, the murine adeno-carcinoma cell lines 13 (MAC13) and 16 (MAC16) were cultured in RPMI with 10 % FBS and 0.5 % penicillin/streptomycin (Invitrogen). Mice inoculated with the MAC13 cell line develop a malignant tumour without cachexia and are used as a cancer control (cancer). The MAC16 cell line produces a both solid tumour and cachexia (cachexia). Cells were kept in a humidified incubator at 37 °C in 10 % CO2. Cells were grown to 80 % confluence and removed from cell culture flasks via trypsinisation. These cells were centrifuged at 500×g for 5 min at 4 °C and isolated from the growth media. Cells were then resuspended in sterile PBS pre-incubated to 37 °C and drawn up into a sterile syringe with a 25-gauge needle for subsequent injection into mice. Mouse body weights were recorded daily, and tumour size was measured using calipers. Animals were euthanised before weight loss in the cachexia group reached 25 % of their initial weight or before tumour growth exceeded 1,000 mm3; this occurred at 30 ± 3 days. Tissues were harvested and immediately placed in liquid nitrogen before storing in an −80 °C freezer for later analysis. In the case of the heart, it was removed intact, blotted to remove any blood, and weighed. The atria and any remaining portions of the great vessels were then removed prior to the ventricles being frozen.
2.3 SOD enzyme activity
Heart tissues were washed in heparinised PBS, and 15 mg of tissue was used for enzyme activity assays. Heart tissue was homogenized in 5 ml of 20 mM HEPES buffer (pH 7.2) containing 1 mM EGTA, 210 mM mannitol and 70 mM sucrose per gram of tissue. Samples were standardised so that 10 ul of sample with 10 mg of protein was added to each well on the 96-well assay plate. The assay kit detecting total SOD activity was undertaken as per the manufacturer’s instructions (Cayman Chemical). The amount of activity was calculated and standardised for protein using the Bradford method (Bio-Rad).
2.4 DNA oxidation assay
An ELISA kit was used to measure the DNA oxidation byproduct 8-hydroxy-2-deoxy guanosine (StressMarq Biosciences). DNA was extracted from 15 mg of tissue using a DNA isolation kit (Promega). Each sample was then diluted so that 50 ug of DNA was used in the 8-hydroxy-2-deoxy guanosine assay. The competitive immunoassay involves the binding of free 8-hydroxy-2-deoxy guanosine to an antibody coated 96-well plate. The assay and sample concentration of 8-hydroxy-2-deoxy guanosine was carried out as per the manufacturer’s instructions.
2.5 RT-PCR
RNA was extracted from frozen heart tissue samples using tri-reagent (Molecular Research Centre). Total RNA concentration yielded per sample was quantified spectrophotometrically at 260 nm. All samples were reverse-transcribed to cDNA using reverse transcription kit (Marligen). cDNA was stored at −20 °C until further analysis. All primer sequences were designed using Primer 3 (primer sequences shown in Table 1). Real-time PCR was performed using a IQ 5 PCR detection system (Bio Rad). A supermix (Bio Rad) was prepared containing 10 ng of sample cDNA, forward and reverse primers diluted to a final concentration of 300 nM, in a total volume of 25 ul. Quantification of mRNA expression was achieved by running samples in duplicate and over a period of 40 cycles. Flourescence data were tabulated and mRNA levels quantified using threshold cycle values. 18S was used as a housekeeping gene to compensate for variations in input RNA amounts by normalizing values to this gene.
2.6 Protein quantification
Heart tissue was homogenized in 20 mM HEPES buffer. The homogenate was centrifuged, and the supernatant was collected and placed on ice. Protein content was assayed using the BCA Protein Assay Kit (Bio Rad). Fifty micrograms of total cellular protein was then loaded on 12 % polyacrylamide gels (NuSep) and separated by standard SDS-PAGE. Gels were then transferred to polyvinylidine fluoride membranes (Bio Rad). Blots were blocked with Tris-buffered saline including 0.1 % Tween 20 and 5 % nonfat dried milk. Primary antibody (Santa-Cruz) against p47phox, NOX2, and GAPDH at a dilution of 1 in 2,000 was added to the membrane overnight at 4 °C. Secondary donkey–antigoat antibodies (Santa-Cruz) conjugated to HRP at a 1 in 10,000 dilution was added to the membrane for 1 h at room temperature. Antigen–antibody complexes were analysed by chemiluminescence using the immune-star HRP detection kit (Bio Rad). Subsequent development and detection of the membrane was achieved with the Chemidoc system (Bio Rad). Protein expression data were then analysed using the Quantity One software (Bio Rad).
2.7 Statistical analysis
The reported values are expressed as mean ± SEM. Data were analyzed using two-way ANOVA, with a Tukey’s test post hoc analysis performed to determine differences between groups where appropriate. Significance was set at a P value equal to or <0.05
3 Results
The animals in this study were randomised to one of three groups being a no-tumour control, cancer or cancer cachexia. Animals in the cachexia group displayed all of the hallmarks of cancer cachexia including the loss of body weight (P = 0.003), skeletal (P = 0.003) and cardiac (P = 0.002) muscle with no significant differences in food and water intake between the groups (Table 2). An analysis was then carried out to ascertain whether key oxidative and proteolytic pathways were upregulated in cardiac tissue.
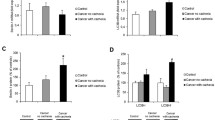
Oxidative damage in the heart muscle was assessed by measuring 8-hydroxy-2-deoxyguanosine (8-OH-2dG), a biomarker of DNA oxidation (Fig. 1). The hearts of cachectic mice displayed a significant 1.5-fold higher 8-OH-2dG (71 ± 26 pg/ml) concentration compared with control (45 ± 18 pg/ml, P = 0.02) and cancer (43 ± 24 pg/ml, P = 0.015) groups. Further studies were then carried out to assess which pathways may be contributing to the oxidative damage in the cancer cachectic hearts.

DNA oxidation by-product 8-hydroxy-2-deoxyguanosine concentration in control, cancer and cachexia. Values are expressed as mean ± SEM. Significantly different from control group (P < 0.02). #Significantly different from cancer group (P < 0.05)
To investigate the potential source of oxidative stress contributing to the oxidative DNA damage found in the cachectic hearts, we investigated the ROS producing enzymes, NADPH oxidase and XO. RT-PCR was performed on the key regulatory subunits (p40 phox and p67 phox) and the catalytic core units (NOX1, NOX2 and NOX4) of NADPH oxidase. There were no changes in the key regulatory or catalytic core units of NADPH oxidase in the heart muscle samples except for NOX2 (Table 3). The current experiment found a significant 1.7-fold lower level of gene expression for NOX2 in cachectic hearts (P = 0.03) compared with the cancer group (Table 3). To assess the possibility of posttranslational modification to the NADPH oxidase protein components, a protein expression analysis was undertaken on p47phox and NOX2. There was no significant difference in either p47phox and NOX2 protein abundance in the cancer cachectic hearts compared with both the control and cancer groups (Table 3).
Subsequently, the XDH enzymatic pathway known to produce ROS was investigated. The RT-PCR results showed a significant twofold higher level of XDH mRNA (P = 0.04) in the heart of cachexia mice in comparison to control and cancer groups (Table 3). Other possible contributors to the state of oxidative stress observed in the cachectic hearts may have been the suppression of the antioxidant enzyme, superoxide dismutase (SOD). Mitochondrial and cytoplasmic SOD displayed no change in mRNA levels (Table 3). However, further investigation of total SOD enzyme activity showed a significant 1.5-fold lower activity in mice with cachexia compared with control (P = 0.02) and cancer (P = 0.001, Fig. 2).

Total SOD activity in control, cancer and cachexia. Values are expressed as mean ± SEM. *Significantly different from control group (P < 0.05). #Significantly different from cancer group (P < 0.05)
Accompanying a state of oxidative stress in cancer cachectic hearts were higher levels of expression of the major muscle proteolytic pathway, the UPS. MURF-1 and MAFbx mRNA expression was significantly higher, 4.3-fold (P = 0.04) and 3.8-fold (P = 0.04), respectively, in the hearts of the cachexia mice compared with the cancer group (Table 3).
4 Discussion
The cardiac atrophy secondary to CC observed in this study is associated with a pro-oxidative shift and consequential oxidative myocardial tissue damage. Furthermore, upregulation of the key ubiquitin ligases in cachectic hearts suggests induction of the UPS may be in response to oxidative stress and subsequent protein damage.
The current study demonstrated the importance of cardiac atrophy as a hallmark of the disease with an average 20 % decrease in heart muscle weight compared with controls.
The consequence of such cardiac atrophy was demonstrated by Tian et al. who used echocardiography to analyse the function of cachectic hearts in a mouse model of cancer cachexia [4]. Their study found that, 14 days after the inoculation with tumour cells, the hearts of cancer cachectic mice had reduced heart rate and fractional shortening [4]. To date, the mechanisms influencing cardiac tissue wasting in a state of CC are largely unknown. However, previous studies have shown that patients and animals with CC all show degrees of oxidative stress [21, 22]. To elucidate whether a state of oxidative stress exists in the heart of mice afflicted with CC, the current study measured levels of 8-Oh-2dG and found a significantly higher levels in the cachexia group hearts. The higher 8-Oh-2dG suggests that the activation of pro-oxidant pathways in atrophied hearts of cachectic mice. Importantly, both the release of cytokines such as TNF-alpha and tumour-derived factors such as PIF have been shown to have pro-oxidant effects [20, 21]. Gomes-Marcondes and Tisdale used myotubes in cell culture exposed to FeSO4 and H2O2 which showed that key components of the ubiquitin proteasome system were upregulated by oxidative stress [20]. NOX2 mRNA expression was lower in cachexia hearts compared with the control group, while the Western blot data were unchanged. The results suggest a decreased role for NADPH oxidase in contributing to cellular oxidative stress in cachectic hearts. The results are also in keeping with a previous study by Sullivan-Gunn showing a decrease in NOX2 levels in skeletal muscles of cancer cachectic mice [23]. The animals in this study were allowed to lose up to 25 % of their initial body weight, thus constituting late-stage cachexia. Considering that NADPH oxidase enzyme activity has been shown to degrade cultured myotubes in vitro [23], a time course study would be beneficial in understanding whether the current results are specific to cardiac atrophy in late cachexia. Another key pathway of ROS formation in the myocardium is XDH that was measured using RT-PCR. The results showed a twofold higher level of XDH in the cachexia group hearts. The activation of XDH has been implicated in a number of disease states involving the myocardium such as heart failure and myocardial dysfunction [16, 17]. A study by Ekelund et al. highlighted the importance of XO in cardiac disease by showing that inhibition of the enzyme by allopurinol is capable of increasing left ventricular function and decreasing myocardial oxygen consumption in a dog model of heart failure [24].
In evaluating the potential contribution of antioxidant systems in the cachectic heart, SOD was analysed both at a gene and enzyme activity level. The SOD enzyme is a key endogenous antioxidant in the myocardium for the elimination of superoxide and has been implicated in the pathogenesis of CC [25]. Interestingly, while the cytosolic and mitochondrial SOD mRNA levels were unchanged in cachectic hearts, total SOD enzyme activity was significantly lower compared with controls. This suggests that the SOD enzyme is not compensating for increased levels of ROS. Thus, the concurrent higher levels of XDH and lower SOD activity may significantly contribute to a disruption in the balance between pro-oxidant and antioxidant mechanisms, leading to a state of oxidative stress as evidenced by greater oxidative damage to DNA (8-Oh-2dG) in the cachectic heart. With respect to how oxidative stress may contribute to cardiac atrophy in CC, it is currently unknown; however, evidence suggests induction of the UPS may be an important link between oxidative stress and atrophy in cachectic muscle [20]. Otsuka et al. found that in patients’ hearts undergoing dilated cardiomyopathy, oxidative stress and the UPS activation are colocalised [26]. Increases in levels of XO and altered SOD enzyme function in the myocardium are traditionally linked to cardiac fibrosis and cardiomypathies [16, 27]. However, the current study shows an association between oxidative stress and cardiac atrophy, suggesting an alternative pathologic role for oxidants in the myocardium. In elucidating the mechanisms by which ROS contributes to the loss of lean body mass, Gomes-Marcondes and Tisdale attempted to replicate the oxidative stress seen in cancer cachexia by using murine myotubes in cell culture [20]. The myotube cells were exposed to H2O2 which induced oxidative stress. Their results showed that the induction of oxidative stress caused an increase in the expression of important UPS elements including the 20S proteasome and the E214k conjugating enzyme [20].
The UPS E3 ligases MURF-1 and MAFbx are expressed in cardiomyocytes and are an important component of cardiac protein turnover [28]. Increased mRNA expression of these ligases is linked to the degradation of cardiac troponin I, α-actin-2 and MyoD leading to impaired contractility [13, 29]. This study shows that both MURF-1 and MAFbx are upregulated at the mRNA level (Table 3) which is in keeping with results seen in studies analysing cachectic heart muscle tissue [30]. Interestingly, Cosper and Leinwand using CD2F1 hybrid mouse model of CC did not see any change in the UPS but found an upregulation in autophagy-related protein degradation [2]. However, the study by Tian et al. using a mouse model of CC similarly to this study found that mRNA of UPS E3 enzymes to be increased in cachectic hearts [30]. Furthermore, Tian and colleagues found that there was increased protein ubiquitinylation in the hearts of cancer cachexia mice compared with controls suggesting that indeed the UPS has an important role to play in cardiac atrophy secondary to CC [30]. A limitation of the current study is the changes in MURF-1 and MAFbx that are at the mRNA level and so correlative in nature. Future investigations would include the evaluation of UPS activation, the state of activation of other proteolytic systems and the phosphorylation levels of Akt and Foxo proteins upstream of ubiquitin ligase activation.
5 Conclusion
The current study used a mouse model of CC producing significant body weight loss and a reduction in heart mass by 20 %. The experimental data demonstrated that cardiac atrophy of cachectic mice is related to a state of oxidative DNA damage in the myocardium. Levels of XDH mRNA were higher in CC hearts, suggesting that XO may have an important role to play in producing oxidative stress. Most evidently, it appears that the combination of higher XDH expression and lower SOD enzyme activity are key contributors to oxidative stress and cardiac tissue damage in cancer-induced cardiac atrophy. Oxidative stress in the myocardium as with skeletal muscle may also induce increased expression of the E3 ligases MURF-1 and MAFbx as seen in higher mRNA levels of both in this study. Further research is needed in a time-dependent manner to ascertain whether these changes are seen in early cachexia and are indeed initiating mechanisms.
References
Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest. 2004;114:370–8.
Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011;71:1710–20.
Costelli P, Tullio RD, Baccino FM, Melloni E. Activation of Ca(2+)-dependent proteolysis in skeletal muscle and heart in cancer cachexia. Br J Cancer. 2001;84:946–50.
Tian M, Nishijima Y, Asp ML, Stout MB, Reiser PJ, Belury MA. Cardiac alterations in cancer-induced cachexia in mice. Int J Oncol. 2010;37:347–53.
Cariuk P, Lorite MJ, Todorov PT, Field WN, Wigmore SJ, Tisdale MJ. Induction of cachexia in mice by a product isolated from the urine of cachectic cancer patients. Br J Cancer. 1997;76:606–13.
Hirai K, Hussey HJ, Barber MD, Price SA, Tisdale MJ. Biological evaluation of a lipid-mobilizing factor isolated from the urine of cancer patients. Cancer Res. 1998;58:2359–65.
Todorov P, Cariuk P, McDevitt T, Coles B, Fearon K, Tisdale M. Characterization of a cancer cachectic factor. Nature. 1996;379:739–42.
Barreiro E, de la Puente B, Busquets S, Lopez-Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2005;579:1646–52.
Marin-Corral J, Fontes CC, Pascual-Guardia S, Sanchez F, Olivan M, Argiles JM, et al. Redox balance and carbonylated proteins in limb and heart muscles of cachectic rats. Antioxid Redox Signal. 2010;12:365–80.
Bossola M, Muscaritoli M, Costelli P, Grieco G, Bonelli G, Pacelli F, et al. Increased muscle proteasome activity correlates with disease severity in gastric cancer patients. Ann Surg. 2003;237:384–9.
Khal J, Wyke SM, Russell ST, Hine AV, Tisdale MJ. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br J Cancer. 2005;93:774–80.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8.
Adams V, Linke A, Wisloff U, Doring C, Erbs S, Krankel N, et al. Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: Effect on myocardial contractility. Cardiovasc Res. 2007;73:120–9.
Vaughan VC, Martin P, Lewandowski PA. Cancer cachexia: impact, mechanisms and emerging treatments. J Cachexia Sarcopenia Muscle. 2013;4:95–109
Kaynar H, Meral M, Turhan H, Keles M, Celik G, Akcay F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu-Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett. 2005;227:133–9.
Bergamini C, Cicoira M, Rossi A, Vassanelli C. Oxidative stress and hyperuricaemia: pathophysiology, clinical relevance, and therapeutic implications in chronic heart failure. Eur J Heart Fail. 2009;11:444–52.
Hayashi K, Kimata H, Obata K, Matsushita A, Fukata A, Hashimoto K, et al. Xanthine oxidase inhibition improves left ventricular dysfunction in dilated cardiomyopathic hamsters. J Card Fail. 2008;14:238–44.
Landmesser U, Spiekermann S, Dikalov S, Tatge H, Wilke R, Kohler C, et al. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation. 2002;106:3073–8.
Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, et al. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–4.
Gomes-Marcondes MC, Tisdale MJ. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002;180:69–74.
Barreiro E, de la Puente B, Minguella J, Corominas JM, Serrano S, Hussain SN, et al. Oxidative stress and respiratory muscle dysfunction in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171:1116–24.
Murr C, Fuith LC, Widner B, Wirleitner B, Baier-Bitterlich G, Fuchs D. Increased neopterin concentrations in patients with cancer: indicator of oxidative stress? Anticancer Res. 1999;19:1721–8.
Sullivan-Gunn MJ, Campbell-O’Sullivan SP, Tisdale MJ, Lewandowski PA. Decreased NADPH oxidase expression and antioxidant activity in cachectic skeletal muscle. J Cachexia Sarcopenia Muscle. 2011;2:181–8.
Ekelund UE, Harrison RW, Shokek O, Thakkar RN, Tunin RS, Senzaki H, et al. Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ Res. 1999;85:437–45.
Russell ST, Eley H, Tisdale MJ. Role of reactive oxygen species in protein degradation in murine myotubes induced by proteolysis-inducing factor and angiotensin II. Cell Signal. 2007;19:1797–806.
Otsuka K, Terasaki F, Shimomura H, Tsukada B, Horii T, Isomura T, et al. Enhanced expression of the ubiquitin-proteasome system in the myocardium from patients with dilated cardiomyopathy referred for left ventriculoplasty: an immunohistochemical study with special reference to oxidative stress. Heart Vessel. 2010;25:474–84.
Ito H, Torii M, Suzuki T. Decreased superoxide dismutase activity and increased superoxide anion production in cardiac hypertrophy of spontaneously hypertensive rats. Clin Exp Hypertens (New York, NY : 1993). 1995;17:803–16.
Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. 2006;41:567–79.
Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci U S A. 2004;101:18135–40.
Tian M, Asp ML, Nishijima Y, Belury MA. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int J Oncol. 2011;39:1321–6.
Acknowledgments
E. Hinch was the recipient of a Deakin University postgraduate research scholarship. V. Vaughan is the recipient of the Victorian Cancer Agency Palliative and Supportive Care Scholarship through the Victorian Cancer Agency funded by the State Government of Victoria, Australia, and the Bellberry Support Scholarship through Bellberry Ltd. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle, 2010;1:7–8 (von Haehling S, Morley JE, Coats AJ, and Anker SD).
Conflict of interest
E. Hinch, M. Sullivan-Gunn, V. Vaughan, M. McGlynn and P. Lewandowski declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 139 kb)
About this article
Cite this article
Hinch, E.C.A., Sullivan-Gunn, M.J., Vaughan, V.C. et al. Disruption of pro-oxidant and antioxidant systems with elevated expression of the ubiquitin proteosome system in the cachectic heart muscle of nude mice. J Cachexia Sarcopenia Muscle 4, 287–293 (2013). https://doi.org/10.1007/s13539-013-0116-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-013-0116-8