
Abstract
Introduction
Luseogliflozin, a sodium glucose cotransporter 2 inhibitor, inhibits reabsorption of glucose in the proximal renal tubule. It was developed for the treatment of type 2 diabetes mellitus.
Methods
For this first human study of luseogliflozin, randomized, single-blind, placebo-controlled, single ascending dose (1–25 mg) and multiple ascending dose (5 or 10 mg/day, 7 days) trials were conducted in healthy male Japanese subjects to investigate safety, pharmacokinetics, and pharmacodynamics.
Results
There were no serious adverse events, adverse events leading to discontinuation, or episodes of hypoglycemia. After administration of a single oral dose of luseogliflozin, its maximum plasma level (C max) and area under the concentration–time curve increased in a dose-dependent manner, and no food effects were observed on pharmacokinetics. The mean time taken to reach C max (T max) ranged from 0.667 to 2.25 h. The mean plasma half-life of luseogliflozin (T 1/2) after multiple dosing for 7 days ranged from 9.14 to 10.7 h, and no detectable accumulation of luseogliflozin was observed. Urinary glucose excretion increased in a dose-dependent manner, ranging from 18.9 to 70.9 g (single-dose study).
Conclusion
Luseogliflozin was well tolerated and showed favorable pharmacokinetic and pharmacodynamic profiles in healthy male Japanese subjects.
Similar content being viewed by others

Introduction
The global prevalence of diabetes is increasing rapidly, with 371 million patients with diabetes recorded in 2012 and 552 million expected in 2030 [1]. The number of patients with diabetes is also increasing in Japan, where the estimated number of patients receiving medical treatment for diabetes was approximately 2.37 million in 2008 [2]. Furthermore, the numbers of Japanese individuals “strongly suspected of having diabetes” and “in whom diabetes cannot be ruled out” were estimated at approximately 8.9 and 13.2 million, respectively, in 2007 [3].
The major symptom of type 2 diabetes mellitus (T2DM), which accounts for approximately 90% of all cases, is chronic hyperglycemia. This leads to microvascular and macrovascular complications, including myocardial infarction and death [4–6]. Previous large clinical trials have emphasized the importance of intensive glycemic control, showing that it could reduce these complications [7–10]. Although various types of oral antidiabetic agents are widely used, including sulfonylurea, biguanide, thiazolidine, dipeptidyl peptidase 4 inhibitors, and α-glycosidase inhibitors, inadequate glycemic control is often found in clinical practice with monotherapy or combination treatments [11–13]. Several other concerns about these antidiabetic agents have also been noted, such as hypoglycemia, weight gain, and gastrointestinal disorders [14]. New classes of antidiabetic agents are therefore needed, with distinct mechanisms of action from those of the currently available agents. Ideally, these new compounds should help achieve effective glycemic control and be well tolerated.
The kidneys contribute to maintenance of normal plasma glucose levels by reabsorbing approximately 180 g of glucose each day [15], ensuring that less than 1% of filtered glucose is excreted in urine. Most of the glucose is reabsorbed in the proximal tubules via the sodium glucose cotransporter (SGLT) [16]. SGLT1 and SGLT2 are subtypes of this transporter, and SGLT2 reabsorbs 90% of filtered glucose, while SGLT1 reabsorbs the remaining 10% [17]. Because SGLT2 acts in an insulin-independent manner, it represents an attractive therapeutic target for T2DM [18]. Some SGLT2 inhibitors, including ipragliflozin, dapagliflozin, and canagliflozin, show potent and selective SGLT2 inhibition in vitro and reduce plasma glucose and HbA1c levels in diabetes animal models as well as in patients with T2DM [19–21]. Dapagliflozin and canagliflozin have already been approved by the European Union and the United States Food and Drug Administration, respectively [22, 23], and are under review by the Ministry of Health, Labour and Welfare, Japan, for the treatment of T2DM.
Luseogliflozin is a novel SGLT2 inhibitor that is currently under development for the treatment of T2DM. It has been shown to increase urinary glucose excretion (UGE) and to decrease plasma glucose levels in various animal models [24]. Luseogliflozin has been shown to act as a potent and selective SGLT2 inhibitor, with a 50% inhibitory concentration (IC50) of 2.26 nM, 1,765-fold lower than its IC50 for SGLT1 [24, 25]. This potency would enable safer usage of lower amounts of luseogliflozin than of other agents in the same class for the treatment of diabetes. The present studies were conducted to investigate safety, pharmacokinetics, and pharmacodynamics by administering single and 7-day multiple doses of luseogliflozin to healthy male Japanese subjects. Here, we report the results of this first-in-human study of luseogliflozin, a novel antidiabetic compound.
Methods
The present studies were conducted in accordance with the standards of the Japanese Pharmaceutical Affairs Law and Good Clinical Practice. Each study protocol was approved by the Institutional Review Board of the Kyushu Clinical Pharmacology Research Clinic. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 and 2008. Informed consent was obtained from all patients prior to their participation in the studies.
Eligibility Criteria
Eligible subjects were healthy Japanese males aged between 20 and 39 years with a body mass index (BMI) between 18.5 and 24.9 kg/m2 and who were considered healthy, based on the results of a screening examination (medical examination, vital signs, and ECG examination) for determination of eligibility performed between 28 and 3 days prior to the day of administration. Subjects were excluded from the studies if they had any clinically significant disease or disorder, were considered to have impaired glucose tolerance, had a serum creatinine level above the upper limit of the reference range in the study institution, tested positive for urinary protein or occult blood (1+ or above), showed significant body weight change (±3 kg) within 4 weeks of the first administration of luseogliflozin or a placebo, used any drugs within 1 week of the first administration of luseogliflozin or a placebo, or if they had drug or food allergies.
Design of the Single Ascending Dose Study
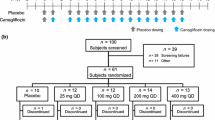
A randomized, single-blind, placebo-controlled, single ascending dose (SAD) study of luseogliflozin was conducted in 57 healthy subjects. The subjects received a single dose of 1, 3, 5, 9, 15, or 25 mg of luseogliflozin or placebo, and were randomly assigned to receive either luseogliflozin or placebo in a ratio of 3:2 in the 1 mg group, 8:2 in the 3, 9, 15, and 25 mg groups, and 8:4 in the 5 mg group. Administration of luseogliflozin was initiated with the lowest dose (Fig. 1a). The safety data and pharmacokinetic data obtained at the lower dose were evaluated before increasing the dose to 3, 9, 15, or 25 mg.

Study design of the SAD study (a) and the MAD study (b). MAD multiple-ascending dose, SAD single-ascending dose
Luseogliflozin (1, 3, 5, 9, 15, or 25 mg) was administered orally with 200 mL of water after fasting for at least 10 h. In addition, analysis of the pharmacodynamic effects of 1, 3, and 9 mg luseogliflozin (see below) indicated that a dose of 5 mg would be appropriate for evaluation of food effects. The subjects who received 5 mg luseogliflozin or placebo under fasting conditions were therefore administered a second dose of 5 mg luseogliflozin or placebo under preprandial conditions 8 days after the first administration (Fig. 1a). On this occasion, administration of 5 mg luseogliflozin was followed by a standardized meal containing approximately 630 kcal, made up of approximately 16% protein, 21% fat, and 63% carbohydrate. The subjects were discharged after safety data were reviewed and evaluated by the investigator on Day 3 (1, 3, 5, and 9 mg groups) or on Day 5 (15 and 25 mg groups), and were required to have a follow-up examination 7 days after administration.
Blood and urine samples were collected for pharmacokinetic and pharmacodynamic analyses. Blood samples for pharmacokinetic studies were collected before administration and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 48 h after administration for all groups, and at the additional time points of 72 and 96 h for the 15 and 25 mg groups. Blood samples for pharmacodynamic studies were collected before administration and at 0.5, 1, 2, 4, 4.5, 5, 6, 8, 12, 12.5, 13, 14, 16, 24, and 48 h after administration for all groups, and also at 96 h for the 15 and 25 mg groups. Urine samples for both pharmacokinetic and pharmacodynamic analyses were collected for 24 h before administration and for 0–2, 2–4, 4–6, 6–8, 8–10, 10–12, 12–14, 14–16, 16–24, and 24–48 h after administration for all groups, and for the additional time periods of 48–72 and 72–96 h for the 15 and 25 mg groups.
Design of the Multiple Ascending Dose Study
A randomized, single-blind, placebo-controlled, 7-day multiple ascending dose (MAD) study of luseogliflozin was conducted in 24 healthy subjects (Fig. 1b). Within each group, the subjects were randomly assigned to receive either luseogliflozin (N = 8) or placebo (N = 4). The results of the present SAD study and other SGLT2 inhibitor studies suggested that 5–10 mg luseogliflozin/day would produce sufficient UGE [26]. The subjects therefore received a daily dose of 5 or 10 mg luseogliflozin or placebo for 7 days orally with 200 mL of water just before food intake (breakfast). Administration of luseogliflozin was initiated with 5 mg, only increasing to 10 mg after safety data had been reviewed by the investigator and 5 mg had been evaluated to be tolerable and safe. On the day of the first and last administration, the subjects were fed the same standardized meal used in the SAD study (see above). The subjects were discharged after their safety data, obtained throughout their hospitalization, were reviewed by the investigator.
For pharmacokinetic assessments, blood samples were collected before the first administration on Day 1, and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 h after the first administration; before administration on Days 2–7; and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, and 96 h after the last administration on Day 7. Urine samples were collected every 24 h from Day -1 to Day 11.
For pharmacodynamic assessments, blood samples were collected before the first administration on Day 1, and at 0.5, 1, 2, 4, 4.5, 5, 6, 8, 12, 12.5, 13, 14, and 16 h after the first administration; before administration on Days 2–7; and at 0.5, 1, 2, 4, 4.5, 5, 6, 8, 12, 12.5, 13, 14, 16, 24, 96 and 168 h (7 days) after the last administration on Day 7. Urine samples were collected for 24 h before the first administration; for 0–2, 2–4, 4–6, 6–8, 8–10, 10–12, 12–14, 14–16, and 16–24 h after the first administration; every 24 h from Days 2–6; for 0–2, 2–4, 4–6, 6–8, 8–10, 10–12, 12–14, 14–16, and 16–24 h after the last administration on Day 7; and every 24 h on Days 8–14.
Assessment of Safety
All adverse events (AEs) were recorded throughout the study periods; furthermore, a range of assessments including those for body weight and vital signs, 12-lead electrocardiography, and clinical laboratory tests (hematology, biochemistry, urinalysis, and stool analysis) were performed throughout the study periods. Quantitative determination of bacteria in urine was also carried out in the MAD study as an exploratory investigation of the effect of luseogliflozin on urinary bacteria. AEs were defined as any new medical condition developing after administration of luseogliflozin or placebo, or any worsening of a pre-existing condition. AEs were coded using the preferred terms from the Medical Dictionary for Regulatory Activities, version 11.0 (SAD study) or version 11.1 (MAD study).
Bioassay of Luseogliflozin
Blood samples (5 mL) were collected into tubes containing sodium heparin and centrifuged immediately after collection at the clinical facility (4 °C, 3,000 rpm, 15 min) to obtain plasma samples for pharmacokinetics, which were stored at −70 °C until analysis. Urine samples were pooled at 4 °C, and then 4 mL samples for pharmacokinetics were stored at −70 °C until analysis.
The concentrations of luseogliflozin in plasma and urine were determined by validated high-performance liquid chromatography tandem mass spectrometry (HPLC–MS/MS) assays performed by JCL Bioassay Corp. (Nishiwaki, Japan). Luseogliflozin and the internal standard (luseogliflozin-d5; Taisho Pharmaceutical Co. Ltd., Saitama, Japan) were extracted from samples of plasma or urine by solid phase extraction. HPLC was performed on an Inertsil ODS-3 column (2.1 mm × 50 mm, 5-μm particle size) from GL Sciences (Tokyo, Japan) with acetonitrile and 1 mmol/L ammonium acetate solution as the mobile phase, under gradient conditions. MS/MS was performed using an API4000 from AB Sciex (Framingham, MA, USA) in a multiple reaction monitoring mode with a turbo ion spray source under negative ionization conditions, monitoring the transition of the m/z 433 precursor ion to the m/z 104 product ion for luseogliflozin, and the m/z 438 precursor ion to the m/z 104 product ion for the internal standard. A linear calibration curve using peak area was obtained by weighting 1/x 2. The lower limit of quantification (LLOQ) for luseogliflozin in human plasma was 0.05 ng/mL, with linearity to 100 ng/mL using a sample volume of 150 μL. The LLOQ was 0.5 ng/mL in human urine, with linearity to 1,000 ng/mL using a sample volume of 50 μL.
Measurement of Glucose
Blood samples (2 mL) were centrifuged immediately after collection at the clinic (4 °C, 3,000 rpm, 10 min) to obtain plasma, which was stored at 4 °C until glucose analysis. Urine samples (0.5 mL) were obtained from total urine pooled during each collection period and also stored at 4 °C. Plasma and urinary glucose concentrations were measured using an automatic analyzer TBA-120FR (Toshiba Medical Systems Corp., Otawara, Japan) and Glucoroder-NX (A&T Corp., Yokohama, Japan), respectively. These assays were performed by Mitsubishi Chemical Medience Corp. (Tokyo, Japan).
Pharmacokinetic and Pharmacodynamic Assessments
The plasma concentrations of luseogliflozin were used to calculate the maximum concentration (C max), time to maximum concentration (T max), area under the concentration–time curve (AUC), elimination rate constant (λz), elimination half-life (T 1/2), apparent clearance (CL/F), and apparent volume of distribution (Vd/F), by using noncompartmental analysis. In the SAD study, the AUC from 0 to the last quantifiable data point (AUClast) was calculated by the trapezoidal rule, based on plasma concentrations of luseogliflozin. The AUC from 0 to infinity (AUCinf) was calculated by extrapolating the plasma luseogliflozin concentration at the last point of measurement to infinity. In the MAD study, the AUCinf on Day 1 and the AUC during 0–24 h after administration (AUCτ) on Day 7 were calculated.
The urinary concentrations of luseogliflozin and the urine volumes were used to calculate urinary luseogliflozin excretion over each collection period, urinary luseogliflozin excretion rate (as a percentage of total dose), and the daily urinary luseogliflozin excretion rate (as a percentage of daily dose). Total UGE (g) was calculated for each collection period, as well as UGE/h over the period and the 24-h cumulative UGE on Days 1 and 7.
Statistical Methods
All statistical analyses were performed using SAS software (version 9.1.3; SAS Institute Inc., Cary, NC, USA). All subjects who received luseogliflozin or placebo were included in the safety and the pharmacodynamic analysis sets. In the SAD and MAD studies, the statistical significance of the relationships between luseogliflozin dose and plasma or urine glucose levels were analyzed using analysis of covariance with baseline plasma or urine glucose levels as the covariate. All subjects who received luseogliflozin were included in the pharmacokinetic analysis set. The pharmacokinetic dose proportionality was evaluated by a power model using C max, AUClast, and AUCinf in the SAD study (administration under fasting conditions only). Estimated regression coefficients for log-transformed doses and their 95% confidence intervals were calculated. The effects of food were evaluated by the geometric mean ratio (administration under preprandial/fasting conditions) of C max, AUClast, and AUCinf for 5 mg luseogliflozin, and the 90% confidence interval, calculated using a mixed effect model. The accumulation potential in the MAD study was evaluated by comparing AUCinf on Day 1 with AUCτ on Day 7.
Results
Participants and Baseline Demographics
In the SAD study, 43 and 14 subjects received luseogliflozin and placebo, respectively. In the MAD study, 16 and 8 subjects received luseogliflozin and placebo, respectively. The baseline demographics were comparable between the groups. In the SAD study, the mean age of the subjects ranged from 23.0 to 28.0 years, mean body weight ranged from 57.5 to 65.0 kg, and mean BMI ranged from 20.0 to 22.4 kg/m2. The corresponding ranges in the MAD study were 28.1–29.6 years, 62.3–62.6 kg, and 20.5–21.2 kg/m2. All subjects completed the studies and were included in the populations analyzed.
Safety
In both studies, no serious AEs or AEs leading to discontinuation were observed, and there were no cases of hypoglycemia. In the SAD study, AEs were reported in 8 subjects (7 taking luseogliflozin and 1 placebo). These were “occult blood positive” in 6 subjects who received luseogliflozin doses of 3 (2 subjects), 9, 15, and 25 mg, and placebo; “beta 2 microglobulin urine increased” in 1 subject who received 9 mg luseogliflozin; “diarrhoea” in 1 subject who received 25 mg luseogliflozin; and “urethritis” in 1 subject who received 25 mg luseogliflozin. One subject who received 25 mg luseogliflozin presented with both “occult blood positive” and “diarrhoea”. All AEs were mild, reversible, and did not appear to be dose-related. There were no clinically significant changes in renal function test values (blood and urine creatinine, blood urea nitrogen, blood and urine uric acid, blood cystatin C, urine β-N-acetyl-d-glucosaminidase, urine beta 2 microglobulin, and urinary type IV collagen) or blood and urine electrolytes (sodium, potassium, chlorine, calcium, and phosphorus). No clinically significant changes were observed in body weight, vital signs, or electrocardiography.
In the MAD study, AEs were reported in 3 subjects (2 taking luseogliflozin and 1 placebo). These were “beta 2 microglobulin urine increased” in 1 subject who received placebo; “diarrhoea” in 1 subject who received 10 mg luseogliflozin; and “occult blood positive” in 1 subject who received 10 mg luseogliflozin. All AEs were mild and reversible. Similar to the SAD study, there were no clinically significant changes in renal function test values, blood and urine electrolytes, body weight, vital signs, or electrocardiography. Quantitation of bacteria in urine showed that the bacteria detected after drug administration were indigenous. Escherichia coli and Klebsiella oxytoca, which could cause urinary tract infection, were present at less than 103 colony forming units/mL.
Pharmacokinetics
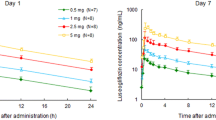
In the SAD study, the mean plasma luseogliflozin concentrations from 0 to 48 h after a single administration of 1–9 mg and from 0 to 96 h after a single administration of 15 and 25 mg under fasting conditions are shown in Fig. 2a and Table 1. Luseogliflozin was absorbed rapidly, with a mean T max between 0.667 and 2.25 h, and the mean T 1/2 ranging from 9.23 to 13.8 h. The mean plasma concentration of luseogliflozin had decreased to 2–3% of C max or lower by 48 h after administration. Luseogliflozin exposure (C max and AUC) increased in a dose-dependent manner. The estimated regression coefficients and 95% confidence intervals for C max, AUClast, and AUCinf determined by the power model were 0.909 (0.845–0.974), 1.02 (0.961–1.07), and 1.00 (0.947–1.06), respectively. Although the 95% confidence interval for C max did not include 1, the 95% confidence intervals for AUClast and AUCinf included 1. Therefore, over the dose range of 1–25 mg, dose proportionality was observed.

Plasma luseogliflozin concentration–time profile in the SAD study (a) and in the MAD study (b). Mean ± SD luseogliflozin concentrations are indicated for each study group and time. h hours, MAD multiple-ascending dose, SAD single-ascending dose
Luseogliflozin 5 mg was administered to the same subjects under fasting conditions and preprandially to examine the effects of food on plasma pharmacokinetics. The ratios of least squares means (preprandial/fasting conditions) for C max, AUClast, and AUCinf were 105–107%. Although the upper limit of the 90% confidence interval for the ratio of C max was just over the criterion for bioequivalence boundary determination of 80–125%, the 90% confidence intervals for the ratio of the AUClast and AUCinf were within the criterion [27]. T max and T 1/2 were generally comparable under preprandial and fasting conditions, indicating that food had little effect on luseogliflozin pharmacokinetics.
The mean urinary luseogliflozin excretion rates after a single administration under fasting conditions corresponded to 3.36–4.40% of each dose (data not shown). There were almost no effects of dose on urinary excretion rate.
For the MAD study, mean plasma luseogliflozin concentrations and pharmacokinetic parameters at 5 and 10 mg are shown in Fig. 2b and Table 1, respectively. The plasma luseogliflozin concentration–time profiles were similar on Days 1 and 7 at doses of both 5 and 10 mg. The mean T 1/2 after 7-day repeat administration ranged from 9.14 to 10.7 h, and the data were similar to those obtained on Day 1. The trough concentrations (value just before each administration) of luseogliflozin reached a steady state by Day 7. The AUCinf on Day 1 and AUCτ on Day 7 of plasma luseogliflozin were comparable. No accumulation potential was therefore found after multiple administrations of luseogliflozin for 7 days.
Mean daily urinary luseogliflozin excretion rates, as a percentage of daily dose, on Days 1–7 were 3.74–4.69% for the 5 mg dose and 3.58–4.23% for the 10 mg dose. The mean urinary luseogliflozin excretion rates from Days 1–11 (96 h after the last administration) were 4.58% and 4.12% for 5 and 10 mg, respectively.
Pharmacodynamics
In the SAD study, the mean cumulative UGE for 48 h (1, 3, 5, and 9 mg groups) and 96 h (15 and 25 mg groups) after administration of luseogliflozin are shown in Fig. 3a. The mean UGE for 24 h after administration was 18.9, 36.8, 50.2, 54.3, 60.7, and 70.9 g with 1, 3, 5, 9, 15, and 25 mg luseogliflozin, respectively, and 0.182 g for placebo. These data demonstrated a luseogliflozin-induced dose-dependent increase in UGE, which was most apparent up to 5 mg, with a 2.7-fold increase between 1 and 5 mg, compared to a 1.4-fold increase from 5 to 25 mg. The mean UGE from 24 to 48 h after administration was 2.53, 12.5, 21.3, 37.5, 48.4, and 62.7 g with 1, 3, 5, 9, 15, and 25 mg luseogliflozin, respectively, and 0.110 g for placebo. Increased UGE was therefore observed for at least 48 h after luseogliflozin administration at all doses tested.

Cumulative UGE in the SAD study (a) and daily UGE in the MAD study (b). Mean ± SD UGE are indicated for each study group and time. *Number of subjects is 4 at both 72 h and 96 h after the treatment. † P < 0.05 versus placebo, ANCOVA with baseline as the covariate was applied in the MAD study. ANCOVA analysis of covariance, h hours, MAD multiple-ascending dose, SAD single-ascending dose, UGE urinary glucose excretion
For the MAD study, the mean daily UGE is shown in Fig. 3b. Significant elevations in UGE were observed on all luseogliflozin administration days (1–7), and the mean daily UGE ranged 57.2–65.5 and 62.7–76.9 g with 5 and 10 mg luseogliflozin, respectively, and 0.0327–0.133 g for placebo. The mean daily UGE on Day 7 was comparable with the 5 and 10 mg doses (58.0 and 62.7 g). Although UGE decreased after the last administration, a statistically significant increase (P < 0.05) was sustained up to Day 10 (3 days after the last administration) at both 5 and 10 mg doses, compared with placebo.
UGE rate-time profiles were also analyzed, and Fig. 4a presents these data from the SAD study. The UGE rates increased over 24 h after luseogliflozin administration, compared with placebo. In addition, a greater increase in UGE rate was observed after food intake (at 4 and 12 h after administration). Figure 4b, c show the mean UGE rate-time profiles on Days 1 and 7, respectively, of the MAD study. At both doses (5 and 10 mg), the UGE rates on Day 7 were comparable to those on Day 1.

UGE rate-time profile in the SAD study (a) and on Days 1 (b) and 7 (c) of the MAD study. Mean ± SD UGE rates are indicated for each study group and time. h hours, MAD multiple-ascending dose, SAD single-ascending dose, UGE urinary glucose excretion
The mean plasma glucose concentrations in the SAD and MAD studies are shown in Fig. 5a, b, respectively. Although plasma glucose levels decreased slightly at some time points with luseogliflozin administration compared with placebo, luseogliflozin did not produce clinically significant decreases in fasting plasma glucose levels.

Plasma glucose concentration–time profile in the SAD study (a) and on Day 7 of the MAD study (b). Mean ± SD Plasma glucose concentrations are indicated for each study group and time. h hours, MAD multiple-ascending dose, SAD single-ascending dose
Discussion
The results of the SAD study (1–25 mg) and the 7-day MAD study (5 or 10 mg) demonstrated the tolerability and safety of luseogliflozin in healthy male Japanese subjects. All AEs were mild and no hypoglycemia was observed. Multiple doses of luseogliflozin for 7 days did not increase the frequency or severity of AEs.
Since SGLT2, especially located at renal proximal tubules, is the target of SGLT2 inhibitors [16, 17], renal tubule function was one of our major concerns. However, in these studies, there were no abnormal changes in markers of renal tubule function. An AE related to renal function, increased urinary beta 2 microglobulin, was observed in one subject who received 9 mg luseogliflozin in the SAD study, while no renal AEs were observed in the MAD study. Since this AE was only observed once, was mild in severity, and reversible without treatment, its clinical effect was considered minor. Furthermore, no clinically significant changes were observed in the other renal function test values (blood and urine creatinine, blood urea nitrogen, blood and urinary uric acid, blood cystatin C, urinary β-N-acetyl-d-glucosaminidase, and urinary type IV collagen) and no clinically significant changes were observed in blood or urinary electrolytes (sodium, potassium, chlorine, calcium, and phosphorus) in these two studies.
Because SGLT2 inhibitors cause glucosuria, they could potentially increase the risk for urinary tract or genital infections [28, 29]. An AE related to such infections, urethritis, was observed in one subject who received 25 mg luseogliflozin in the SAD study, while none were observed in the MAD study. This AE was mild and the subject recovered with medication. In addition, quantitative assessment of bacteria in urine did not produce findings suggestive of an increased risk of urinary tract infection in the MAD study. However, an elevated risk of infection cannot be ruled out as clinical studies of another SGLT2 inhibitor, dapagliflozin, in patients with T2DM showed a higher frequency of urinary tract and genital infections [21, 30–34]. These may not have been observed in the present study owing to a low incidence of these AEs with luseogliflozin use, combined with the small sample size and short administration period employed.
Reducing postprandial glucose excursion is difficult in patients with T2DM. Our results indicated that luseogliflozin may help solve this problem. Standard deviations of C max were small at all tested doses, indicating that luseogliflozin may exhibit consistent efficacy, and T max was short, regardless of dose. These pharmacokinetic profiles indicated that luseogliflozin administration would be expected to provide consistent plasma levels and therefore improve postprandial glucose excursion consistently.
Our pharmacokinetic analyses also indicated that the C max, AUClast, and AUCinf were dose-proportional and not affected by food intake. The mean T 1/2 of luseogliflozin after a single administration (9.23–13.8 h) was not dose-dependent at doses of 9 mg or less, and was slightly longer at doses of 15 mg and 25 mg. These results may also contribute to a stable treatment effect and improved glycemic control in patients with diabetes.
Drug metabolism and elimination were not altered by multiple dosing. In the MAD study, examination of luseogliflozin plasma trough concentrations, AUC data on Days 1 and 7, and mean daily urinary luseogliflozin excretion rates indicated that no abnormal accumulation of this drug occurred.
As expected, a substantial amount of UGE was observed after luseogliflozin administration in healthy subjects. The mean UGE for 24 h in the SAD study increased in a dose-dependent manner, reaching a maximum of approximately 70 g. The mean daily UGE after the last administration of luseogliflozin in the MAD study was about 60 g for both 5 and 10 mg doses. The incremental change in UGE was smaller at higher doses of luseogliflozin than at lower doses. The maximum amount of daily UGE after luseogliflozin administration was comparable to that after administration of other SGLT2 inhibitors (dapagliflozin, canagliflozin, empagliflozin, and ipragliflozin) in healthy subjects, which was approximately 70 g/day after single dose and approximately 50–55 g/day after multiple dose [26, 35–38]. Further, the doses of luseogliflozin used in these studies (up to 25 mg for SAD study and 10 mg for MAD study) were lower than those used in previous tests of SGLT2 inhibitors (50–800 mg at the maximum dose in each study). Luseogliflozin thus possibly had the advantage of producing similar UGE in the presence of lower amounts of compound. However, it is still unclear why luseogliflozin works at lower doses compared to other compounds. A possibility could be that the pharmacokinetic distribution profile of luseogliflozin contributes to its high potency, as observed in an animal study of luseogliflozin that showed higher concentration in the kidney than in the plasma [25].
Luseogliflozin was expected to suppress postprandial plasma glucose levels by increasing UGE. At most postprandial time points, UGE rate was increased and plasma glucose levels were slightly decreased in subjects who received luseogliflozin, compared to placebo; similar results were observed in the canagliflozin study [36]. Elevated postprandial glucose increased the amount of glucose filtered through the kidneys, resulting in an increase in UGE with less reabsorption in the renal tubules in the subjects treated with luseogliflozin. Therefore, luseogliflozin may be optimal in reducing postprandial glucose levels in patients who have normal renal function.
The present study indicated that luseogliflozin did not reduce plasma glucose levels significantly in healthy subjects. Fasting plasma glucose levels were comparable in subjects treated with luseogliflozin and placebo. This was consistent with the results of other SGLT2 inhibitors [35–38]. Normal homeostatic regulation probably maintained stable plasma glucose levels, compensating for the increased UGE by increasing gluconeogenesis. It has been shown previously that glycerol generated by fat decomposition is used preferentially in gluconeogenesis [39]. This may possibly be an explanation for SGLT2 inhibitor-mediated reduction in body fat levels, as well as an indication that SGLT2 inhibitor use may cause fewer hypoglycemic events.
In addition, a once-daily luseogliflozin dose regimen may provide sufficient UGE. In the SAD study, UGE rates after each food intake were comparable. Furthermore, elevated UGE was also observed the day after administration of luseogliflozin. In the MAD study, the daily UGE during the administration period was stable, and the increased UGE compared to that observed with placebo (P < 0.05) were sustained for up to 3 days after the last administration. Thus, patient compliance may improve if sufficient efficacy can be obtained using a once-daily dose regimen.
The above-mentioned findings suggest that a once-daily luseogliflozin dose regimen may have the potential to reduce plasma glucose levels in patients with T2DM, including mild forms of T2DM and impaired glucose tolerance, without increasing their risk for hypoglycemia. However, since this study was conducted in healthy Japanese males, the pharmacokinetic and pharmacodynamic profile of luseogliflozin in patients with T2DM remains unclear. Further clinical studies in patients with T2DM may provide further insight into the potential of luseogliflozin for the treatment of T2DM.
Conclusion
Luseogliflozin showed good tolerability and safety in a single-dose (1–25 mg) and 7-day multiple-dose (5 and 10 mg) study in healthy male Japanese subjects. Luseogliflozin also showed favorable pharmacokinetic and pharmacodynamic profiles, supporting the feasibility of a once-daily administration regimen. Clinical studies to investigate the safety and efficacy of luseogliflozin in patients with T2DM have recently been completed, and the results may provide further insight into the clinical utility of luseogliflozin.
References
International Diabetes Federation. IDF Diabetes Atlas. 5th edn. 2011/2012 update. Available from: http://www.idf.org/diabetesatlas/. Accessed 19 Nov 2013.
Ministry of Health, Labour and Welfare. Summary of Patient Survey 2008. Available from: http://www.mhlw.go.jp/english/database/db-hss/sps_2008.html. Accessed 19 Nov 2013
National Institute of Health and Nutrition. Outline of the National Health and Nutrition Survey Japan, 2007. Available from: http://www0.nih.go.jp/eiken/english/research/project_nhns.html/. Accessed 19 Nov 2013.
Fukushima S, Nakagami T, Suto C, Hirose A, Uchigata Y. Prevalence of retinopathy and its risk factors in a Japanese population. J Diabetes Investig. 2013;4:349–54.
Hotta N, Nakamura J, Iwamoto Y, et al. Causes of death in Japanese diabetics: a questionnaire survey of 18385 diabetics over a 10-year period. J Diabetes Investig. 2010;1:66–76.
Vinik AI, Erbas T, Casellini CM. Diabetic cardiac autonomic neuropathy, inflammation and cardiovascular disease. J Diabetes Investig. 2013;4:4–18.
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–89.
Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–53.
Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–65.
Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract. 1995;28:103–17.
Nauck MA, Vardarli I. Comparative evaluation of incretin-based antidiabetic medications and alternative therapies to be added to metformin in the case of monotherapy failure. J Diabetes Investig. 2010;1:24–36.
Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999;281:2005–12.
Hoerger TJ, Segel JE, Gregg EW, Saaddine JB. Is glycemic control improving in U.S. adults? Diabetes Care. 2008;31:81–6.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med. 2007;261:32–43.
Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na +/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for d-glucose. J Clin Investig. 1994;93:397–404.
DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab. 2012;14:5–14.
Harada N, Inagaki N. Role of sodium-glucose transporters in glucose uptake of the intestine and kidney. J Diabetes Investig. 2012;3:352–3.
Kurosaki E, Ogasawara H. Ipragliflozin and other sodium-glucose cotransporter-2 (SGLT2) inhibitors in the treatment of type 2 diabetes: preclinical and clinical data. Pharmacol Ther. 2013;139:51–9.
Han S, Hagan DL, Taylor JR, et al. Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes. 2008;57:1723–9.
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo controlled, phase 3 trial. Diabetes Care. 2010;33:2217–24.
Dapagliflozin (Forxiga): EU summary of product characteristics. Available from: http://www.medicines.org.uk/emc/medicine/27188/SPC/Forxiga+5%20mg++%26+10+mg+film+coated+tablets/. Accessed 19 Nov 2013.
Canagliflozin (Invokana): prescribing information. Available from: http://www.invokanahcp.com/prescribing-information.pdf/. Accessed 19 Nov 2013.
Yamamoto K, Uchida S, Kitano K, et al. TS-071 is a novel, potent and selective renal sodium-glucose cotransporter 2 (SGLT2) inhibitor with anti-hyperglycaemic activity. Br J Pharmacol. 2011;164(1):181–91.
Kakinuma H, Oi T, Hashimoto-Tsuchiya Y, et al. (1S)-1,5-anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-D-glucitol (TS-071) is a potent, selective sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for type 2 diabetes treatment. J Med Chem. 2010;53:3247–61.
Komoroski B, Vachharajani N, Boulton D, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85:520–6.
English translation of Attachment 1 of Division-Notification 0229 No. 10 of the Pharmaceutical and Food Safety Bureau, dated February 29, 2012. Guideline for Bioequivalence Studies of Generic Products. Available from: http://www.nihs.go.jp/drug/be-guide(e)/Generic/GL-E_120229_BE.pdf. Accessed 28 Jan 2014.
Nicolle LE, Capuano G, Ways K, Usiskin K. Effect of canagliflozin, a sodium glucose co-transporter 2 (SGLT2) inhibitor, on bacteriuria and urinary tract infection in subjects with type 2 diabetes enrolled in a 12-week, phase 2 study. Curr Med Res Opin. 2012;28:1167–71.
Nyirjesy P, Zhao Y, Ways K, Usiskin K. Evaluation of vulvovaginal symptoms and Candida colonization in women with type 2 diabetes mellitus treated with canagliflozin, a sodium glucose co-transporter 2 inhibitor. Curr Med Res Opin. 2012;28:1173–8.
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–7.
Wilding JP, Norwood P, T’joen C, Bastien A, List JF, Fiedorek FT. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–62.
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–33.
Johnsson KM, Ptaszynska A, Schmitz B, Sugg J, Parikh SJ, List JF. Urinary tract infections in patients with diabetes treated with dapagliflozin. J Diabetes Complicat. 2013;27:473–8.
Johnsson KM, Ptaszynska A, Schmitz B, Sugg J, Parikh SJ, List JF. Vulvovaginitis and balanitis in patients with diabetes treated with dapagliflozin. J Diabetes Complicat. 2013;27:479–84.
Kasichayanula S, Chang M, Hasegawa M, et al. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13:357–65.
Sha S, Devineni D, Ghosh A, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13:669–72.
Sarashina A, Koiwai K, Seman LJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter-2 (SGLT-2) inhibitor, in healthy Japanese subjects. Drug Metab Pharmacokinet. 2013;28:213–9.
Kadokura T, Saito M, Utsuno A, et al. Ipragliflozin (ASP1941), a selective sodium-dependent glucose cotransporter 2 inhibitor, safely stimulates urinary glucose excretion without inducing hypoglycemia in healthy Japanese subjects. Diabetol Int. 2011;2:172–82.
Carlson MG, Snead WL, Campbell PJ. Fuel and energy metabolism in fasting humans. Am J Clin Nutr. 1994;60:29–36.
Acknowledgments
The clinical trials described in this study and the article processing charges were sponsored by Taisho Pharmaceutical Co., Ltd., Tokyo, Japan. The authors express their appreciation and gratitude to the healthy male Japanese subjects who participated in the studies and are grateful to the investigators and the clinic staff for their conduct of the studies. Editorial assistance in the preparation of this manuscript was provided by Taisho Pharmaceutical Co., Ltd. All named authors meet the ICMJE criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. This study was sponsored by Taisho Pharmaceutical Co., Ltd., Tokyo, Japan.
Conflict of interest
Takashi Sasaki has received joint research fund from Canon Inc. and consultancy fees from Taisho Pharmaceutical Co., Ltd. Yutaka Seino has received consultancy fees or lecture fees from Sanofi, Novo Nordisk, Eli Lilly and Company, GlaxoSmithKline, Astellas Pharma, Takeda Pharmaceuticals, Boehringer Ingelheim, Johnson & Johnson, Becton–Dickinson and Company, Bristol-Myers Squibb, AstraZeneca, and Taisho Pharmaceutical Co., Ltd. Atsushi Fukatsu has received consultancy fees from Taisho Pharmaceutical Co., Ltd. Soichi Sakai is an employee of Taisho Pharmaceutical Co., Ltd. Yoshishige Samukawa is an employee of Taisho Pharmaceutical Co., Ltd.
Compliance with ethics guidelines
The present studies were conducted in accordance with the standards of the Japanese Pharmaceutical Affairs Law and Good Clinical Practice. Each study protocol was approved by the Institutional Review Board of the Kyushu Clinical Pharmacology Research Clinic. All procedures followed were in accordance with the ethical standards of the responsible committee on human dexperimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 and 2008. Informed consent was obtained from all patients prior to their participation in the studies.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial registration: JapicCTI-132353 and JapicCTI-132354.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article
Cite this article
Sasaki, T., Seino, Y., Fukatsu, A. et al. Safety, Pharmacokinetics, and Pharmacodynamics of Single and Multiple Luseogliflozin Dosing in Healthy Japanese Males: A Randomized, Single-Blind, Placebo-Controlled Trial. Adv Ther 31, 345–361 (2014). https://doi.org/10.1007/s12325-014-0102-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-014-0102-3