
Abstract
Understanding the genetic risk factors for stroke is an essential step to decipher the underlying mechanisms, facilitate the identification of novel therapeutic targets, and optimize the design of prevention strategies. A very small proportion of strokes are attributable to monogenic conditions, the vast majority being multifactorial, with multiple genetic and environmental risk factors of small effect size. Genome-wide association studies and large international consortia have been instrumental in finding genetic risk factors for stroke. While initial studies identified risk loci for specific stroke subtypes, more recent studies also revealed loci associated with all stroke and all ischemic stroke. Risk loci for ischemic stroke and its subtypes have been implicated in atrial fibrillation (PITX2 and ZFHX3), coronary artery disease (ABO, chr9p21, HDAC9, and ALDH2), blood pressure (ALDH2 and HDAC9), pericyte and smooth muscle cell development (FOXF2), coagulation (HABP2), carotid plaque formation (MMP12), and neuro-inflammation (TSPAN2). For hemorrhagic stroke, two loci (APOE and PMF1) have been identified.
Similar content being viewed by others


Introduction
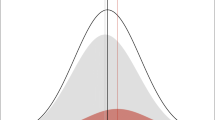
Stroke, characterized by a neurological deficit of sudden onset, typically due to brain infarction (“ischemic stroke”) or, less often, intracerebral hemorrhage, represents the primary neurological cause of acquired disability in adults and a leading cause of death [1]. It is also a major contributor to cognitive decline and dementia [2–4]. Common causes (subtypes) of ischemic stroke include large artery atheroma and cardiac sources of embolism, while small artery disease is a major cause of both ischemic stroke and intracerebral hemorrhage (Fig. 1) [5]. The lifetime risk of stroke has been estimated at one in five for middle-aged women and one in six for middle-aged men in the Framingham Heart Study [6]. A substantial proportion of stroke risk remains unexplained, and a contribution of genetic factors is supported by recent discoveries of common genetic variation associated with stroke risk, identified through large, collaborative, genome-wide association studies (GWAS) [7]. In rare instances, stroke can be directly caused by monogenic disorders, i.e., a rare mutation in one gene is sufficient to cause the disease. In the vast majority of cases, however, genetic risk factors contribute to the risk of stroke as part of a multifactorial predisposition, where each genetic variation is responsible only for modest increases in risk. The advent of high throughput genotyping in the past decade has led to progress in the discovery of genes underlying complex forms of stroke [7]. An important challenge in the identification of genetic determinants of stroke, in contrast with other common vascular or neurological diseases, is the complexity of the phenotype. Indeed, stroke is a highly heterogeneous condition that can be caused by multiple, extremely diverse etiologies (Fig. 1). Of note, genetic risk factors for subarachnoid hemorrhage will not be discussed in this review, as the underlying mechanisms are completely distinct from those of intracerebral hemorrhage and ischemic stroke.

Heterogeneity of the stroke phenotype. Numbers are taken from the latest statement of the American Heart Association/American Stroke Association [116]
The main application expected from these discoveries is to improve our understanding of the biological pathways underlying the disease, and, through this, to accelerate the identification of novel drug targets [8]. While improved risk prediction also remains a long-term goal, its implementation is still complex given the small effect size of genetic risk variants. With the expected increase of identified stroke risk variants through large international consortia, it is however not excluded that in aggregate these variants may eventually contribute to improved risk stratification [8].
Rare Monogenic Causes of Stroke
Monogenic (Mendelian) disorders are responsible for a very small proportion of strokes, probably less than 1 % (this proportion being larger among younger stroke patients) [9]. The mechanisms by which these monogenic disorders result in stroke vary substantially. The main monogenic disorders causing stroke are presented in Table 1. Only monogenic diseases for which stroke is one of the main clinical manifestations are presented, and we have not included inherited cardiopathies, such as familial atrial fibrillation, or vascular malformations, such as familial cavernomas.
As has been shown for other diseases [8], it has been hypothesized that some genes harboring causal mutations for monogenic forms of stroke may also contain common genetic polymorphisms associated with complex stroke risk. There is some preliminary indication of that for the COL4A2 gene, for instance. Indeed, common variants in this gene, which harbors mutations for a Mendelian disease causing intracerebral hemorrhage (Table 1), were found to be associated with an increased risk of multifactorial deep intracerebral hemorrhage [10, 11].
Role of Genetic Risk Factors in Common Multifactorial Stroke
There is evidence from twin studies [12], and from studies on the family history of stroke [13–15], that genetic factors substantially contribute to stroke susceptibility. However, there is important heterogeneity of heritability estimates. Many studies combined ischemic and hemorrhagic stroke, and only a few studies considered ischemic stroke subtypes [16]. Recently, with the advent of genome-wide genotyping (which consists of genotyping several hundreds of thousands or millions of genetic variants distributed across the genome), novel approaches have been developed that enable estimation of the heritability of diseases in the absence of familial information, based solely on genome-wide genotypes [17]. This “pseudo-heritability” corresponds to the proportion of phenotypic variance explained by genome-wide genotypes [18]. Recently, the pseudo-heritability of stroke has been estimated based on data from large genome-wide association studies, confirming a substantial heritability [19, 20], but also important differences according to stroke subtypes. Pseudo-heritability estimates were 40.3 % for large artery ischemic stroke, 32.6 % for cardioembolic ischemic stroke, 16.1 % for small artery occlusion ischemic stroke, 73 % for lobar intracerebral hemorrhage, and 34 % for deep intracerebral hemorrhage. Heritability estimates may increase if phenotyping is more accurate, as was for instance shown for small artery occlusion ischemic stroke, the heritability of which reached 24 % when considering only patients with multiple lacunar infarcts or associated extensive white matter hyperintensities, reflecting underlying cerebral small artery disease [21].
The underlying genetic model is believed to be multifactorial, with numerous genetic polymorphisms that each confers a small increase in risk, and several environmental risk factors also of small effect size, and possible interaction of these risk factors with each other [22, 23]. The most commonly studied type of genetic variation underlying the risk of complex diseases like stroke is single nucleotide polymorphisms (SNPs), of which several million have been identified across the genome.
Given the small effect size of genetic risk variants expected for stroke, large numbers of individuals are required to reach sufficient statistical power, in the range of several thousand at least. Hence, identifying such variants requires large collaborative efforts, made possible through the creation of international consortia, such as the International Stroke Genetics Consortium (ISGC, www.strokegenetics.org), the NINDS Stroke Genetics Network, the Cohorts of Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium, and the METASTROKE consortium [24–26].
Genetic variants predisposing to stroke could act at various levels (Fig. 2), e.g., by increasing the risk of and susceptibility to “conventional” stroke risk factors such as hypertension or diabetes, by influencing specific mechanisms underlying stroke, such as atheroma or atrial fibrillation, by alterating coagulation pathways and predisposing to arterial thrombosis or bleeding, or by modifying tolerance to brain ischemia and more largely brain injury [27].

Genetic risk factors for stroke can act at various levels, example for ischemic stroke. Left side Risk loci identified to be associated with ischemic stroke. Right side Theoretical mechanisms by which genetic factors may modulate ischemic stroke risk
Recently, large collaborative efforts have identified a number of common genetic risk variants associated with an increased risk of stroke, both ischemic and hemorrhagic stroke [28•, 29–34]. Earlier studies had consisted of testing the association of stroke with a few genetic variants in one or few candidate genes, selected based on a priori hypotheses on the mechanisms underlying the disease, leading to disappointing results, as most associations that were identified could not be confirmed in independent samples [35]. Most robust genetic associations with stroke were identified through genome-wide association studies, an agnostic approach that consists of genotyping a very large number of genetic variants (“markers”) across the genome and test their association with a phenotype, without any a priori hypothesis on the underlying biology [22, 36, 37]. This approach has led to the identification of a very large number of genetic associations with various traits and diseases that have been convincingly replicated in independent samples, interestingly mostly near previously unsuspected genes, thus providing new hypotheses on the underlying biology [8].
In the early phase of genome-wide association studies, most genome-wide significant associations for stroke were identified for specific ischemic or hemorrhagic subtypes [28•, 29–31, 38]. However, analysis of shared genetic variation across stroke subtypes has also shown high genetic correlation between some stroke subtypes, such as the large-artery atherosclerosis and small artery disease subtypes of ischemic stroke [24]. Moreover, with increasing sample size through large collaborative efforts, more recently multiple studies have also reported loci associated with all ischemic stroke and all stroke [33•, 34, 39•, 40•, 41•, 42•]. Genetic risk variants associated with all stroke are expected to be either acting at the upstream end of the pathophysiological cascade, by modulating the risk of medical conditions increasing the risk of all types of stroke (such as hypertension) or at the downstream end by modulating the brain’s susceptibility to ischemic injury (e.g., via increased risk of thrombosis) or the brain’s tolerance to brain injury at large (e.g., through affecting neuroprotective pathways, Fig. 2). However, particularly large numbers are likely required to reveal such genetic risk loci for stroke, given their small expected effect size in light of the numerous concomitant risk factors required for this highly heterogeneous condition to occur.
Genetic Risk Factors for Ischemic Stroke
Genome-wide significant associations with ischemic stroke are summarized in Table 2. A p value <5 × 10−8 is required to declare genome-wide significance, to account for approximately one million independent statistical tests performed genome-wide.
Genetic loci found to be associated with cardio-embolic ischemic stroke were already known risk loci for atrial fibrillation (PITX2 and ZFHX3), consistent with the fact that atrial fibrillation is by far the most common source of cardioembolic events [43–45]. Recent data suggest that the PITX2 locus could perhaps also contribute to stroke risk independent of atrial fibrillation [46]. Indeed, Pitx2−/− mutant mice were shown to exhibit reduced and discontinuous smooth muscle actin staining of cerebral vessels and increased cerebral vessel density. Moreover, PITX2 variants were found to be associated with increasing white matter hyperintensity burden on brain MRI in large population-based samples [46]. Evidence for association of the PITX2 locus with small artery occlusion ischemic stroke, however, is lacking to date [29].
In contrast with cardio-embolic IS, genetic variants found to be associated with large artery IS at the genome-wide level (HDAC9, MMP12, CDC5L, and TSPAN2) are all in loci previously unsuspected at the time of discovery [28•, 29, 32]. The HDAC9 locus was subsequently also identified as a risk locus for coronary artery disease [47] and pulse pressure [48]. Interestingly, risk allele carriers of the lead HDAC9 susceptibility variant for large artery ischemic stroke (rs2107595) were found to be associated with increased mRNA levels of HDAC9 [49]. Moreover, compared with Hdac9+/+Apoe−/− mice, Hdac9−/−Apoe−/− mice exhibited reduced atherosclerotic lesion size throughout the aorta [49], making HDAC9 a plausible target for pharmacologic prevention of atherosclerosis. The MMP12 locus was identified by implementing an age-at-onset informed genome-wide association analysis (i.e., a regression analysis conditioning on age-at-onset) and was significantly overexpressed in carotid plaques [28•]. The age-at-onset analysis was driven by the assumption that early onset stroke may have increased genetic liability [50]. Most recently, a large collaborative study conducted by the NINDS-SiGN consortium identified common variants near TSPAN2 to be associated with large artery ischemic stroke [51•]. Other variants near TSPAN2 have been implicated in migraine, but they are not in linkage disequilibrium with (i.e., not correlated with) the stroke risk variants. TSPAN2 has also been implicated in neuro-inflammation [52]. In addition to these genome-wide significant findings, a few candidate gene-based associations (requiring a less stringent threshold for significance) have been robustly replicated in large independent studies, such as the chr9p21 locus (rs2383207) or the ABO locus (rs505922) on chromosome 9 with large artery ischemic stroke [29, 53, 54]. The ABO locus also showed association with cardioembolic ischemic stroke and has recently also been shown to reach genome-wide significance with all ischemic stroke [40•, 54]. Both the chr9p21 and the ABO locus were independently found to be associated with coronary artery disease, and ABO is also a risk locus for venous thromboembolism [47, 55].
Until recently, efforts at identifying genetic risk variants for small artery occlusion ischemic stroke have yielded little, with no genome-wide significant finding in the largest published GWAS, despite a similar sample size compared to the aforementioned ischemic stroke subtypes [29]. As pseudo-heritability estimates were also smaller for this subtype (16 vs. 40 % for large artery and 33 % for cardioembolic ischemic stroke) [19], it has been hypothesized that this could reflect a lesser contribution of genetic factors to this ischemic stroke subtype. Another possible explanation is the heterogeneity and imprecision in the phenotype definition of small artery occlusion ischemic stroke, when following the most commonly used Trial of Org 10172 in Acute Stroke Treatment (TOAST) stroke subtyping algorithm [56]. Genetic liability to small artery occlusion ischemic stroke may also differ according to ethnic background, as this stroke subtype is much more prevalent in Asian populations. A significant association of a variant in the PRKCH gene with small artery occlusion ischemic stroke was described in Japanese and Chinese populations. This variant is monomorphic in European populations and no association was found either with nearby variants in PRKCH and small artery occlusion ischemic stroke in Europeans [29, 57–59]. Interestingly, a recent large genome-wide association study of incident stroke conducted by the CHARGE consortium, followed by replication in studies with prevalent stroke (mostly from the NINDS-SiGN and METASTROKE consortia), identified common variants on chr6p25 near FOXF2 to be associated with all stroke, and this association was particularly strong with small artery occlusion ischemic stroke compared to other stroke subtypes [39•]. The same variants near FOXF2 were also found to be associated with larger white matter hyperintensity burden in older stroke-free community persons. Interestingly, patients with a rare monogenic ophthalmologic condition due to segmental deletions encompassing FOXF2 (Axenfeld-Rieger syndrome) also exhibited extensive white matter hyperintensities. Conditional deletion of Foxf2 in adult mice led to cerebral infarction, reactive gliosis, and microhemorrhage. In zebrafish, foxf2b−/− mutants showed decreased smooth-muscle cell and pericyte coverage, suggestion that FOXF2 may be involved in mural cell differentiation [39•].
In addition to FOXF2, other loci have also recently been reported to be associated with all ischemic stroke or all stroke at a genome-wide significant level. The first to be reported was an association with the chr12p13 locus, near NINJ2, associated with incident stroke and ischemic stroke in particular in prospective population-based cohort studies participating in the CHARGE consortium [34]. However, although the same association was reported in several Asian studies [60], it could not be replicated in a large hospital-based genetic association study of prevalent stroke in European populations [61]. One potential explanation for these discrepant results, beside type I error (false positive finding), is that the chr12p13 locus could be associated with stroke severity and mortality more than with stroke risk. Indeed, in hospital-based cross-sectional studies, given high early mortality rates of stroke, death might occur very early before hospitalization or before samples can be taken. Conversely, in prospective cohort studies, severe strokes leading to early death are included, as blood samples were taken at recruitment in the study, before stroke onset. Allelic heterogeneity at this locus, caused by multiple rare, low frequency, and common variants with disparate effects on risk, may also explain the difficulties in replicating the original GWAS results [62]. The second genetic risk locus for all ischemic stroke (chr12q24.12) was identified in a case–control dataset with over 17,000 ischemic stroke patients and found to be equally associated with all subtypes of ischemic stroke. The single nucleotide polymorphism (SNP) showing the most significant association is in linkage disequilibrium with a non-synonymous variant in SH2B3 and is associated with gene expression of ALDH2, pointing to a potential role of these two genes in the association [33•]. The same locus was previously associated with blood pressure and coronary artery disease [47, 63, 64]. Two other recent studies have also described associations of variants near HABP2 and AQP9 to be associated with all ischemic stroke and all stroke, respectively [40•, 41•]. HABP2, which encodes for an extracellular serine protease involved in coagulation, fibrinolysis, and inflammation, was identified through a young-onset stroke GWAS where only stroke cases with age <60 years were studied, and replicated in an independent dataset [40•]. AQP9 was identified through the first large-scale GWAS on individuals of African ancestry [41•]. Though AQP9 reached genome-wide significance in the discovery stage, it did not show evidence of replication in individuals of European ancestry and still needs to be confirmed in independent large studies in individuals of African ancestry.
Novel genetic risk loci have also recently been discovered for other less common but well characterized ischemic stroke etiologies, such as cervical artery dissections, a major cause of ischemic stroke in young adults [65•]. This large collaborative study from the CADISP consortium found the minor allele of a common variant at PHACTR1 to be associated with a lower risk of cervical artery dissection. Interestingly, the same allele was independently found to be associated with a lower risk of migraine (especially without aura) and with an increased risk of coronary artery disease [47, 52, 65•], suggesting that this locus may play a pivotal role in vascular biology. The cervical artery dissection risk allele is associated with increased expression of PHACTR1 in certain tissues [66]. The function of PHACTR1 is poorly understood. Experimental studies revealed a pivotal role in vascular tube formation and actin polymerization [40•, 41•] Upregulation of PHACTR1 by TGFβ has been described [42•], potentially pointing to a connection with the TGFβ signaling pathway. A role in mechanotransduction has also been suggested [67].
Genetic Risk Factors for Intracerebral Hemorrhage
A highly significant and robust association with intracerebral hemorrhage was demonstrated for the APOE locus in a large candidate gene association study on 2189 cases and 4041 controls [38]. Both APOEε2 and the APOEε4 alleles were associated with lobar intracerebral hemorrhage at a “genome-wide significance level”, with odds ratios of 1.82 (p = 6.6 × 10−10) and 2.20 (p = 2.4 × 10−11), respectively. Associations were even stronger when restricting the analysis to patients with definite or probable underlying cerebral amyloid angiopathy (CAA). APOEε4 was also associated with an increased risk for deep intracerebral hemorrhage, a location not affected by CAA, at a lower significance level (odds ratio at 1.21, p = 2.6 × 10−4), suggesting that mechanisms linking APOEε4 to intracerebral hemorrhage may expand beyond CAA-mediated effects [38, 68]. In a subsequent analysis, the authors also demonstrated a strong association of the APOE locus with hemorrhage size and growth [69].
The International Stroke Genetics Consortium recently published the first genome-wide association analysis of intracerebral hemorrhage, based on 1545 patients with intracerebral hemorrhage (664 lobar and 881 non-lobar) and 1481 controls. This study identified one novel genome-wide significant locus on chromosome 1q22 associated specifically with an increased risk of non-lobar (deep) intracerebral hemorrhage, with replication in an independent sample [30•]. Interestingly, this locus was also recently found to be associated at a genome-wide level with increasing white matter hyperintensity burden, the most plausible pathophysiological link between both associations being an increased liability to cerebral small artery disease [70•, 71]. In a recent population-based GWAS of incident stroke, the same locus also showed highly suggestive, although not genome-wide significant, association with incident ischemic stroke [39•]. The identified genetic risk variants are located in a region that contains PMF1 and SLC25A44, and they are associated with expression levels of a nearby gene, SEMA4A [30•]. The results of this study also emphasize the biological heterogeneity across ICH subtypes, as this association was found exclusively for non-lobar ICH.
Discussion
In summary, in the past few years, the advent of high-throughput genotyping technologies and the creation of large international consortia have brought important new insight into complex stroke genetics. Twelve new risk loci for ischemic stroke have been discovered, although two of these have either not been confirmed or were subject to controversy, with inconsistent replication. Five of these loci showed association with all ischemic stroke and all stroke; four, with large artery ischemic stroke; and two, with cardioembolic ischemic stroke. One all-stroke risk locus showed predominant association with small artery occlusion ischemic stroke, but did not reach genome-wide significance in this subtype. Two new loci were identified for intracerebral hemorrhage.
These discoveries of common genetic variants associated with stroke and its subtypes have substantially broadened our knowledge of the underlying pathophysiology. Recent findings have also emphasized the need to carefully consider ischemic and hemorrhagic stroke not as single entities, but as composite entities comprised of various underlying diseases, some of which may share common mechanisms. Our understanding of stroke genetics may be enriched by exploring MRI-based endophenotypes for specific stroke subtypes, such as white matter hyperintensity burden, a marker of cerebral small artery disease, which is strongly correlated with small artery occlusion ischemic stroke. Recently, five genome-wide significant risk loci for white matter hyperintensity burden have been identified (chr17q25, chr10q24, chr2p21, chr1q22, and chr2p16) [70•].
So far, most studies of complex stroke genetics have focused on common single-nucleotide polymorphisms, and, as for other complex diseases, these identified common risk variants explain only a small proportion of the disease heritability [72]. Other types of variation, such as low frequency (1–5 %) or rare (<1 %) single nucleotide variants, or structural variation such as copy number variants, have been insufficiently explored. New genome-wide genotyping arrays now also partly cover lower frequency variants, and more importantly, the advent of next generation sequencing technologies has made sequencing of large samples more accessible and thus opened new avenues for studying unexplored rare variants as well as structural variation [73, 74]. So far, no major results have emerged yet for common stroke, but there are encouraging preliminary findings.
One important discovery of genome-wide association studies is the amount of pleiotropy or shared genetic variation between stroke and other complex phenotypes, which also tells us more about disease mechanisms [75]. Substantial overlap was, for instance, observed between the genetic risk of ischemic stroke, and particularly the large artery subtype, with coronary artery disease, thus contributing to a better understanding of common underlying biological pathways [76]. Pleiotropy may be particularly helpful in understanding the biological mechanisms underlying statistical association, when specific risk loci are shared across phenotypes, such as the PHACTR1 gene that is assumed to play a central role in vascular biology.
As part of the International Stroke Genetics Consortium, the METASTROKE collaboration, the NIH-funded SiGN initiative, and the CHARGE consortium, efforts are ongoing to perform genome-wide association studies of stroke in much larger samples of patients, in order to increase power to detect genetic association with stroke and its subtypes. These projects take advantage of the most recent 1000 genomes reference panel (www.1000genomes.org) that enables more reliable imputation (statistical inference) of genotypes for millions of genetic variants that have not been genotyped. Availability of newer reference panels like the Haplotype Reference Consortium panel (http://www.haplotype-reference-consortium.org) that enable imputation of low frequency variants also provide an exciting opportunity. Efforts are now being made to expand genetic studies to non-European ethnic groups, an essential step to enhance the discovery of stroke risk loci, as was shown for other conditions [77]. In parallel data on rare variants obtained through exome chip genotyping, whole exome and whole genome sequencing is being accrued. In addition to uncovering new risk loci, sequencing will likely help fine-map genetic risk loci discovered by genome-wide association studies and facilitate the identification of the underlying causal variant and gene. Our understanding of the genetic underpinnings of stroke will be further enriched by combining genomic information with trancriptomic, epigenomic, proteomic, metabolomic, and other -omic data. These can provide crucial information to identify the causal variant and gene underlying the observed statistical associations, an essential step for the design of experimental studies aiming at deciphering the underlying biology.
References
Papers of particular interests, published recently, have been highlighted as: • Of importance
Johnston SC, Mendis S, Mathers CD. Global variation in stroke burden and mortality: estimates from monitoring, surveillance, and modelling. Lancet Neurol. 2009;8:345–54.
Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American heart association/American stroke association. Stroke. 2011;42:2672–713.
Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. 2009;72:368–74.
Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009;8:1006–18.
Greenberg SM. Small vessels, big problems. N Engl J Med. 2006;354:1451–3.
Seshadri S, Wolf PA. Lifetime risk of stroke and dementia: current concepts, and estimates from the Framingham Study. Lancet Neurol. 2007;6:1106–14.
Falcone GJ, Malik R, Dichgans M, Rosand J. Current concepts and clinical applications of stroke genetics. Lancet Neurol. 2014;13:405–18.
Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14:549–58.
Leys D, Bandu L, Henon H, et al. Clinical outcome in 287 consecutive young adults (15 to 45 years) with ischemic stroke. Neurology. 2002;59:26–33.
Rannikmae K, Davies G, Thomson PA, et al. Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology. 2015;84:918–26.
Weng YC, Sonni A, Labelle-Dumais C, et al. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann Neurol. 2012;71:470–7.
Bak S, Gaist D, Sindrup SH, Skytthe A, Christensen K. Genetic liability in stroke: a long-term follow-up study of Danish twins. Stroke. 2002;33:769–74.
Kiely DK, Wolf PA, Cupples LA, Beiser AS, Myers RH. Familial aggregation of stroke. The Framingham Study. Stroke. 1993;24:1366–71.
Liao D, Myers R, Hunt S, et al. Familial history of stroke and stroke risk. The Family Heart Study. Stroke. 1997;28:1908–12.
Jood K, Ladenvall C, Rosengren A, Blomstrand C, Jern C. Family history in ischemic stroke before 70 years of age: the Sahlgrenska Academy Study on Ischemic Stroke. Stroke. 2005;36:1383–7.
Flossmann E, Schulz UG, Rothwell PM. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke. 2004;35:212–27.
Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9.
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–54. doi:10.1038/ng.548.
Bevan S, Traylor M, Adib-Samii P, et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke. 2012;43:3161–7.
Devan WJ, Falcone GJ, Anderson CD, et al. Heritability estimates identify a substantial genetic contribution to risk and outcome of intracerebral hemorrhage. Stroke. 2013;44:1578–83.
Traylor M, Bevan S, Baron JC, Hassan A, Lewis CM, Markus HS. Genetic Architecture of Lacunar Stroke. Stroke. 2015;46:2407–12.
Zondervan KT, Cardon LR. Designing candidate gene and genome-wide case–control association studies. Nat Protoc. 2007;2:2492–501.
Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45.
Battey TW, Valant V, Kassis SB, et al. Recommendations from the international stroke genetics consortium, part 2: biological sample collection and storage. Stroke. 2015;46:285–90.
Majersik JJ, Cole JW, Golledge J, et al. Recommendations from the international stroke genetics consortium, part 1: standardized phenotypic data collection. Stroke. 2015;46:279–84.
Psaty BM, O’Donnell CJ, Gudnason V, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:273–80.
Hassan A, Markus HS. Genetics and ischaemic stroke. Brain. 2000;123(Pt 9):1784–812.
•Traylor M, Makela KM, Kilarski LL, et al. A novel MMP12 locus is associated with large artery atherosclerotic stroke using a genome-wide age-at-onset informed approach. PLoS Genet. 2014;10:e1004469. This was the first study in stroke genetics that used age-at-onset analysis to identify novel loci for stroke.
Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE Collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11:951–62.
•Woo D, Falcone GJ, Devan WJ, et al. Meta-analysis of Genome-wide Association Studies Identifies 1q22 as a Susceptibility Locus for Intracerebral Hemorrhage. Am J Hum Genet. 2014;94:511–21. This is so far the largest gentic study on intracerebral hemorrhage and identified the locus PMF1 a suceptibility locus for intracerebral hemorrhage.
Bellenguez C, Bevan S, Gschwendtner A, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. 2012;44:328–33.
Holliday EG, Maguire JM, Evans TJ, et al. Common variants at 6p21.1 are associated with large artery atherosclerotic stroke. Nat Genet. 2012;44:1147–51.
•Kilarski LL, Achterberg S, Devan WJ, et al. Meta-analysis in more than 17,900 cases of ischemic stroke reveals a novel association at 12q24.12. Neurology. 2014;83:678–85. This was one of the first large scale study that identified a locus for all ischemic stroke and this locus has been replicated in independent studies.
Ikram MA, Seshadri S, Bis JC, et al. Genomewide association studies of stroke. N Engl J Med. 2009;360:1718–28.
Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61.
Feero WG, Guttmacher AE, Collins FS. Genomic medicine—an updated primer. N Engl J Med. 2010;362:2001–11.
McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69.
Biffi A, Sonni A, Anderson CD, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. 2010;68:934–43.
•Chauhan G, Arnold CR, Chu AY, et al. Identification of additional risk loci for stroke and small vessel disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2016;15:695–707. This is the largest genetic study of incident stroke where genetic evidence has been combined with clinical studies and functional studies on mouse and zebrafish.
•Malik R, Traylor M, Pulit SL, et al. Low-frequency and common genetic variation in ischemic stroke: the METASTROKE collaboration. Neurology. 2016;86:1217–26. This is one of the largest genetic study of prevalent stroke and explored heritability explaoned by low frequency variants.
•Cheng YC, Stanne TM, Giese AK, et al. Genome-wide association analysis of young-onset stroke identifies a locus on chromosome 10q25 Near HABP2. Stroke. 2016;47:307–16. This is the first large scale genetic study of young-onset stroke in individuals with stroke onset at less than 60 years.
•Carty CL, Keene KL, Cheng YC, et al. Meta-analysis of genome-wide association studies identifies genetic risk factors for stroke in African Americans. Stroke. 2015;46:2063–8. This is the first large scale genetic study of stroke on individuals of African ancestry.
Gretarsdottir S, Thorleifsson G, Manolescu A, et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann Neurol. 2008;64:402–9.
Lemmens R, Buysschaert I, Geelen V, et al. The association of the 4q25 susceptibility variant for atrial fibrillation with stroke is limited to stroke of cardioembolic etiology. Stroke. 2010;41:1850–7.
Gudbjartsson DF, Holm H, Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876–8.
French CR, Seshadri S, Destefano AL, et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J Clin Invest. 2014;124:4877–81.
Deloukas P, Kanoni S, Willenborg C, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
Kato N, Loh M, Takeuchi F, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47:1282–93.
Azghandi S, Prell C, van der Laan SW, et al. Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke. 2015;46:197–202.
Cheng YC, Cole JW, Kittner SJ, Mitchell BD. Genetics of ischemic stroke in young adults. Circ Cardiovasc Genet. 2014;7:383–92.
•Pulit SL et al. Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study. Lancet Neurol. 2015;15:174–84. This is the largest genetic study of stroke, where detailed subtying of stroke was available using the causative classification of stoke (CCS) system.
Anttila V, Winsvold BS, Gormley P, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet. 2013;45:912–7.
Gschwendtner A, Bevan S, Cole JW, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol. 2009;65:531–9.
Williams FM, Carter AM, Hysi PG, et al. Ischemic stroke is associated with the ABO locus: the EuroCLOT study. Ann Neurol. 2013;73:16–31.
Germain M, Chasman DI, de Haan H, et al. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet. 2015;96:532–42.
McArdle PF, Kittner SJ, Ay H, et al. Agreement between TOAST and CCS ischemic stroke classification: the NINDS SiGN study. Neurology. 2014;83:1653–60.
Wu L, Shen Y, Liu X, et al. The 1425G/A SNP in PRKCH is associated with ischemic stroke and cerebral hemorrhage in a Chinese population. Stroke. 2009;40:2973–6.
Serizawa M, Nabika T, Ochiai Y, et al. Association between PRKCH gene polymorphisms and subcortical silent brain infarction. Atherosclerosis. 2008;199:340–5.
Kubo M, Hata J, Ninomiya T, et al. A nonsynonymous SNP in PRKCH (protein kinase C eta) increases the risk of cerebral infarction. Nat Genet. 2007;39:212–7.
Zhang Z, Xu G, Wei Y, Zhu W, Fan X, Liu X. Impact of chromosome 12p13 variants on ischemic stroke risk. Int J Neurosci. 2016;126:856–62.
International_Stroke_Genetics_Consortium, Wellcome_Trust_Case-Control_Consortium_2. Failure to validate association between 12p13 variants and ischemic stroke. N Engl J Med. 2010;362:1547–50.
Bis JC, DeStefano A, Liu X, et al. Associations of NINJ2 sequence variants with incident ischemic stroke in the Cohorts for Heart and Aging in Genomic Epidemiology (CHARGE) consortium. PLoS One. 2014;9:e99798.
Ehret GB, Munroe PB, Rice KM, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9.
Wain LV, Verwoert GC, O’Reilly PF, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet. 2011;43:1005–11.
•Debette S, Kamatani Y, Metso TM, et al. Common variation in PHACTR1 is associated with susceptibility to cervical artery dissection. Nat Genet. 2015;47:78–83. This is the first large-scale genetic study of stroke related etiologies and identified a novel locus for cervical artery dissection.
Beaudoin M, Gupta RM, Won HH, et al. Myocardial infarction-associated SNP at 6p24 Interferes With MEF2 binding and associates With PHACTR1 expression levels in human coronary arteries. Arterioscler Thromb Vasc Biol. 2015;35:1472–9.
Kelloniemi A, Szabo Z, Serpi R, et al. The early-onset myocardial infarction associated PHACTR1 gene regulates skeletal and cardiac alpha-actin gene expression. PLoS One. 2015;10:e0130502.
Rannikmae K, Kalaria RN, Greenberg SM, et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry. 2014;85:300–5.
Biffi A, Anderson CD, Jagiella JM, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol. 2011;10:702–9.
Verhaaren BF, Debette S, Bis JC, et al. Multi-ethnic genome-wide association study of cerebral white matter hyperintensities on MRI. Circ Cardiovasc Genet. 2015;8:398–409. This is the largest genetic study of white matter hyperintensity burden, which is an MRI based marker of cerebral small vessel disease.
Fornage M, Debette S, Bis JC, et al. Genome-wide association studies of cerebral white matter lesion burden: the CHARGE consortium. Ann Neurol. 2011;69:928–39.
Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–50.
Panoutsopoulou K, Tachmazidou I, Zeggini E. In search of low-frequency and rare variants affecting complex traits. Hum Mol Genet. 2013;22:R16–21.
Kiezun A, Garimella K, Do R, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet. 2012;44:623–30.
Sivakumaran S, Agakov F, Theodoratou E, et al. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet. 2011;89:607–18.
Dichgans M, Malik R, Konig IR, et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. Stroke. 2014;45:24–36.
Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–81.
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707–10.
Hara K, Shiga A, Fukutake T, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360:1729–39.
Nozaki H, Nishizawa M, Onodera O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2014;45:3447–53.
Terwindt GM, Haan J, Ophoff RA, et al. Clinical and genetic analysis of a large Dutch family with autosomal dominant vascular retinopathy, migraine and Raynaud’s phenomenon. Brain. 1998;121(Pt 2):303–16.
Grand MG, Kaine J, Fulling K, et al. Cerebroretinal vasculopathy. A new hereditary syndrome. Ophthalmology. 1988;95:649–59.
Jen J, Cohen AH, Yue Q, et al. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology. 1997;49:1322–30.
Richards A, van den Maagdenberg AM, Jen JC, et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–70.
Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–60.
Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–94.
Gaustadnes M, Ingerslev J, Rutiger N. Prevalence of congenital homocystinuria in Denmark. N Engl J Med. 1999;340:1513.
Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med. 1998;338:1042–50.
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
Rolfs A, Fazekas F, Grittner U, et al. Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients study. Stroke. 2013;44:340–9.
Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009;40:788–94.
Vanakker OM, Leroy BP, Coucke P, et al. Novel clinico-molecular insights in pseudoxanthoma elasticum provide an efficient molecular screening method and a comprehensive diagnostic flowchart. Hum Mutat. 2008;29:205.
Debette S, Germain DP. Neurologic manifestations of inherited disorders of connective tissue. Handb Clin Neurol. 2014;119:565–76.
van den Berg JS, Hennekam RC, Cruysberg JR, et al. Prevalence of symptomatic intracranial aneurysm and ischaemic stroke in pseudoxanthoma elasticum. Cerebrovasc Dis. 2000;10:315–9.
Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32.
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31–7.
Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–80.
North KN, Whiteman DA, Pepin MG, Byers PH. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Ann Neurol. 1995;38:960–4.
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–85.
Gray JR, Bridges AB, West RR, et al. Life expectancy in British Marfan syndrome populations. Clin Genet. 1998;54:124–8.
Schievink WI, Michels VV, Piepgras DG. Neurovascular manifestations of heritable connective tissue disorders. A review. Stroke. 1994;25:889–903.
Wityk RJ, Zanferrari C, Oppenheimer S. Neurovascular complications of marfan syndrome: a retrospective, hospital-based study. Stroke. 2002;33:680–4.
Majamaa K, Moilanen JS, Uimonen S, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet. 1998;63:447–54.
Testai FD, Gorelick PB. Inherited metabolic disorders and stroke part 1: Fabry disease and mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Arch Neurol. 2010;67:19–24.
Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008;1142:133–58.
Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid beta -protein. J Biol Chem. 2001;276:32860–6.
Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol. 2011;7:1–9.
De Jonghe C, Zehr C, Yager D, et al. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis. 1998;5:281–6.
Bornebroek M, De Jonghe C, Haan J, et al. Hereditary cerebral hemorrhage with amyloidosis Dutch type (AbetaPP 693): decreased plasma amyloid-beta 42 concentration. Neurobiol Dis. 2003;14:619–23.
Palsdottir A, Snorradottir AO, Thorsteinsson L. Hereditary cystatin C amyloid angiopathy: genetic, clinical, and pathological aspects. Brain Pathol. 2006;16:55–9.
Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–71.
Vahedi K, Massin P, Guichard JP, et al. Hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy. Neurology. 2003;60:57–63.
Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol. 2007;62:177–84.
Plaisier E, Alamowitch S, Gribouval O, et al. Autosomal-dominant familial hematuria with retinal arteriolar tortuosity and contractures: a novel syndrome. Kidney Int. 2005;67:2354–60.
Lanfranconi S, Markus HS. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke. 2010;41:e513–518.
Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ganesh Chauhan and Stéphanie Debette declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Stroke
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chauhan, G., Debette, S. Genetic Risk Factors for Ischemic and Hemorrhagic Stroke. Curr Cardiol Rep 18, 124 (2016). https://doi.org/10.1007/s11886-016-0804-z
Published:
DOI: https://doi.org/10.1007/s11886-016-0804-z