
Abstract
Electrodeposition of tin from acidic gluconate baths (chloride, chloride–sulfate) was investigated. Equilibrium distribution of tin(II) species in both solutions was calculated. Cyclic voltammetric, potentiostatic, and galvanostatic measurements showed that cathodic process can run under limiting current due to the presence of tin complex electroactive species. Average effective diffusion coefficients of metal species were determined. Nucleation of tin occurred through a progressive three-dimensional diffusion growth model. A characteristic of tin deposits was also presented.
Similar content being viewed by others
Introduction
Tin is one of the metals most often used in the commercial electroplating. It is mainly due to its high corrosion resistance, nontoxicity, and good soldering properties. Such features determine relatively wide application area of the electrodeposited tin as tinplates for food industry (containers and packages), protective coatings for copper in electrotechnical industry, layers improving surface solderability of majority of metals, coatings protecting from the corrosion in aggressive atmosphere, or in manufacturing of printed circuit boards and electronic components [1, 2].
Various baths are used for obtaining uniform tin electrodeposits with required quality and morphology. These are acidic tin(II) and alkaline tin(IV) solutions [1, 2]. Nonaggressive alkaline baths work at higher temperatures (60–98 °C) and show good throwing power, although are troublesome in operation, since traces of Sn(II) ions in the electrolyte decrease quality of the cathodic deposits resulting in formation of spongy tin layers. Alkaline electrolytes are usually stannate solutions, but pyrophosphate and polyphosphate baths were also developed. In turn, acidic stannous baths work at ambient temperatures and are easy to control; however, they must contain Sn(II) ions and organic additives in a proper quantitative ratio. The latter can improve adherence and smoothness of the matt tin layers or enable deposition of bright coatings. Generally, acidic tin baths are sulfate, fluoroborate, or alkane-sulfonate electrolytes, while halide solutions are used in a lesser extent.
Composition of the bath affects the mechanism of metal ions reduction. It is commonly accepted that in acidic simple salt systems (sulfate, chloride), Sn2+ ions are the most important species in the charge transfer step, while in strong alkaline baths Sn(OH)6 2− ions participate in the cathodic reaction [2]. Chemistry of the process is more complicated in the electrolytes containing complexing agents [3–6].
Early stages of tin electrodeposition on various substrates using electrolytes of different compositions were also presented in the literature [7–13]. Nucleation models were derived from both chronoamperometric current transients and microscopic (AFM or scanning electron microscopy (SEM)) observations. In the main, tin electrocrystallization in acidic sulfate [7] or tartrate solutions [10] is in agreement with theoretical instantaneous nucleation model followed by three-dimensional (3D) diffusion growth mechanism, but it can change to progressive 3D growth in some conditions [8]. The latter model was also observed in a chloride–citrate system [12] and a hydrophobic room-temperature ionic liquid [13]. Proceeding of the initial stages of tin electrodeposition via instantaneous or progressive nucleation with 2D growth control was reported for sulfate–gluconate baths [11].
Gluconate solutions represent relatively new type of the electrolytes for tin [6, 11] and tin alloys deposition [14–18]. Sodium gluconate is a cheap and environmentally safe additive, which can form soluble complexes with a variety of metal ions in both slightly acidic and alkaline baths. Experiments on tin plating from acidic sulfate–gluconate baths [6, 11] showed that uniform and brilliant deposits can be obtained, while to date no data on the applicability of chloride containing stannous–gluconate baths have been presented. Hence, the aim of this work was to determine the effect of the bath composition on tin electrodeposition from chloride–gluconate and chloride–sulfate–gluconate electrolytes. This paper reports comparative studies of the solutions, since the nature of the electrolyte and presence of various anions (chloride or sulfate) can seriously affect the nucleation stage, morphology, and structure of the deposits, which in turn determine properties of the coatings.
Experimental
Investigations of tin electrodeposition were carried out from the baths containing: 0.1 M tin(II) chloride SnCl2, 0.5 M ammonium salt (chloride or sulfate), 0.5 M boric acid H3BO3, and 0.2 M sodium gluconate C6H11O7Na. Blank solutions were prepared using ammonium chloride instead of stannous salt of the same concentration (0.1 M). pH of the baths was 3.50 ± 0.05. Reagents of analytical purity were used.
Electrochemical measurements were carried out in a three-electrode cell using a glassy carbon working electrode (0.2 cm2), a platinum plate (2 cm2) as a counter electrode, and an Ag/AgCl electrode as a reference electrode (all potentials in the further text are referred to this electrode). Potentiostatic tin deposition (30 min) was performed in the same system, but α-brass (10 wt.% Zn) plates (0.8 cm2) as cathode substrates were used. Before each experiment, the glassy carbon electrode with a mirror finish was chemically cleaned, while brass sheets were chemically polished in a mixture of concentrated acids (HNO3/H3PO4/CH3COOH with 1:3:1 volume ratio). After deposition, cathodes were dried at 60 °C to the constant mass. Solution volume of 25 cm3 was used in each electrochemical test.
Various electrochemical techniques were used. Cyclic, linear, and stripping voltammograms were registered at potential scan rate of 20 mV s−1. First cyclic voltammetry (CV) sweep was always performed from the initial potential of 0.5 V (Ag/AgCl) towards more negative values. Stripping analysis was performed immediately after potentiostatic tin deposition (for 40 s) at fixed potential without removing the working electrode from the solution. Anodic stripping curves were registered from deposition potentials up to 0.5 V. Nucleation of tin was investigated using chronoamperometry realized at various constant potentials. Galvanostatic method was also used for electrodeposition studies. Autolab potentiostat/galvanostat (PGSTAT30) controlled by a microcomputer was applied in all measurements.
All experiments were performed at room temperature. No bath agitation was used.
Morphology of the layers was examined using both optical (Nikon) and scanning electron (Hitachi) microscopes. Chemical composition of the deposits was determined by scanning electron microscopy/energy dispersive spectroscopy (SEM–EDS) analyzer. Structure of the deposits was analyzed by X-ray diffractometry (Rikagu diffractometer, CuKα radiation).
Results and discussion
Distribution of tin(II) species
Metal ions are present in solutions in various forms, i.e., free ions, neutral, and/or charged complexes. Specific distribution of the individual species depends mainly on the total concentration of ions, type and concentration of other compounds added to the bath, pH, and temperature. In aqueous solutions, tin(II) can appear as free Sn2+ ions and soluble complexes with formulas dependent on the salt type and solution pH [19]. Precipitation of sparingly soluble tin(II) hydroxide is also observed.
In this research work, gluconate solutions containing SnCl2 and NH4Cl or (NH4)2SO4 were used; hence, species listed in Table 1 were taken into account over the pH range from 2.0 to 5.0. Despite that ammonium salts were present in the electrolytes, tin(II)–ammonia complexes were not considered, since they do not form even in the excess of aqueous ammonia [23].
From the mass balance of stannous, chloride, sulfate, and gluconate (Glu−) ions, a series of general polynomial equations was derived:
Using equilibrium quotients assigned to the reactions presented in Table 1, the equations were solved at given total concentrations of the components and pH. Calculations were performed using HySS2009 software (Hyperquad Simulation and Speciation 2009 Protonic Software).
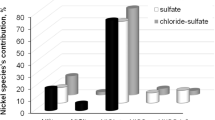
Figure 1 shows that specific composition of the baths is quite complicated. At pH 3.5, SnGlu2 complex represents 57.8 and 66.6 % of total amount of tin(II) ions in the chloride and chloride–sulfate solutions, respectively. It is consistent with Survila et al. data [6], who found that such species is a major form of Sn(II) in tin–sulfate–gluconate system in the pH range of 3–5. Two chloride complexes, SnCl2 (13.7 %) and SnCl3 − (12.0 %) as well as SnGlu+ ions (8.6 %), show relatively considerable percentages in the chloride bath, while in the chloride–sulfate electrolyte solely SnGlu+ (14.4 %) and two tin–sulfate complexes (both in approximately 5.5 % fractions) may be found in noteworthy amounts. In all cases, various hydroxy-complexes and free metal ions show negligible percentages of total Sn(II) concentration (below 0.1 %). Despite the low solubility product of tin(II) hydroxide, no precipitation of Sn(OH)2 in gluconate solutions was predicted by the equilibrium speciation.

Distribution of tin species in 0.1 M SnCl2–0.2 M gluconate solutions with addition of 0.5 M ammonium salt: a chloride bath, b chloride–sulfate bath
Potentiodynamic experiments
At the beginning of the studies, the glassy carbon (GC) working electrode was tested in blank solutions, i.e., solutions containing ammonium salts instead of corresponding tin chloride (0.1 M). Results in Fig. 2 show only cathodic currents corresponding to the hydrogen evolution. They appeared below −1.40 and −1.48 V in the chloride and chloride–sulfate blank solutions, respectively. Theoretical calculations indicate that, at pH 3.5, hydrogen can evolve at potentials much below the equilibrium value of −0.43 V (vs. Ag/AgCl) due to high cathodic overpotential expected on the GC substrate (up to 2.0 V) that is dependent on the type of the glassy carbon material [24].

Linear sweep voltammograms of blank solutions on GC surface (20 mV/s)
All plots show that reduction of hydrogen ions on GC was affected by the presence of different ions in the electrolytes, and sulfate anions shifted the curve towards more negative potentials. It was assumed that ions inhibit cathodic reaction due to their competitive and specific adsorption on the electrode surface. It is also possible that reduction of hydrogen ions disturbs dissociation equilibria in the solutions at the cathode surface. It influences on the continuous course of the reaction; hence, its rate is determined by the release of cations from undissociated HSO4 − ions or HGlu molecules. In turn, chloride ions do not form protonated species and similar effect was not observed.
Cyclic voltammograms registered in stannous solutions were started from 0.5 V, and then, potential was scanned in the negative direction and reversed at various potentials (from −0.6 to −1.8 V). Figure 3 shows exemplary curves determined for the vertex potentials of −1.0 and −1.4 V. Electrodeposition of metal on the glassy carbon electrode started at potentials of about −0.70 ± 0.04 V, which were much more positive than the values for hydrogen ion reduction in the blank solutions. Course of the curves typical for electrodeposition process was visible during the reverse scan (Fig.3a). Characteristic crossover between the branches for the negative and positive sweeps is associated with the nucleation overpotential, since the potential of tin deposition on tin itself is more positive than for the metal deposition on a foreign substrate [25]. The crossover potentials E n were about 0.05 V lower than the potentials of the metal deposition starting and were practically independent on the switch potentials. Second crossover potentials E c (at −0.55 to −0.58 V) on the cyclic voltammograms can correspond to the equilibrium potential, but this was not the case since such values were more negative than the equilibrium potential of the Sn/Sn(II) electrode immersed in the 0.1 M tin–gluconate solution (i.e., −0.46 V vs. Ag/AgCl).

Cyclic voltammograms for tin(II) baths (GC electrode; 20 mV/s) at various vertex potentials: a −1.0 V, b −1.4 V
Tin deposition potentials (peak C1) were not dependent on the bath composition, while the rate of the process was affected by the type of anions in the bath. In the chloride system, enhanced tin deposition was observed, while the presence of sulfate anions inhibited the cathodic process despite of the same total concentration of tin salt. It was confirmed by the area of the single anodic peak (registered in the backward scan, at the potential of −0.4 V) corresponding to the formation of Sn(II) ions from the previously deposited metal.
Cathodic currents observed at potentials below −1.20 V (C2 in Fig.3b) were ascribed to the hydrogen coevolution.
Figure 4 shows anodic peaks representing a part of cyclic voltammograms registered for various vertex potentials. In both solutions, more negative vertex potential was accompanied by the increase in the anodic peak area and shifting of the maxima of the anodic peak towards more positive potentials. In the chloride bath, one broad (−0.55 to −0.18 V) and asymmetrical anodic peak was developed. Sulfate addition changed a little position of single anodic peak towards lower potentials, and less shift of the peak maximum with decreased vertex potential was observed.

Anodic part of cyclic voltammograms registered at various vertex potentials for 0.1 M Sn(II)–0.2 M gluconate baths: a chloride, b chloride–sulfate (GC substrate; 20 mV/s)
Results of anodic stripping obtained for tin deposition at various constant potentials (Fig. 5) were different. Comparison of the anodic stripping curves shows that in the sulfate presence, decrease in the deposition potential was accompanied by the inhibition of the metal deposition (revealed by less amount of dissolved metal), while in the chloride solution analogous behavior changed rapidly for deposition potentials more negative than −1.2 V and sudden increase in the amount of deposited metal was observed. To clarify such behavior, deposition of tin at constant potentials was carried out.

Anodic stripping voltammograms (20 mV/s) registered for tin deposition at various potentials (GC substrate, 40 s) from 0.1 M Sn(II)–0.2 M gluconate baths: a chloride, b chloride–sulfate
Potentiostatic deposition
Tin was deposited on the brass substrate at some constant potentials. Figure 6a shows the dependence of the cathodic current efficiency on the deposition potentials and bath composition. Cathodic current efficiencies were calculated according to the mass gain of the cathode Δm and total charge flowed through the circuit Q during electrolysis:

Dependencies of a cathodic current efficiency on the tin deposition potential (brass substrate; deposition time: 30 min), b Q A/Q C ratio on the vertex potential determined from CV (GC substrate; 20 mV/s)
where k Sn is electrochemical equivalent of tin.
Obtained results are consistent with the anodic stripping data but are different from the results of the cyclic voltammetry represented by the anodic to cathodic charge ratio (Fig. 6b). It was also observed that at constant deposition potential, electrodeposition of tin was inhibited by the sulfate ions, while comparable values of Q A/Q C were obtained for both baths. This shows that the current efficiency estimated upon voltammetric data [4] cannot be used as reliable information since scanning of the potential changes rapidly conditions of the plating.
For both baths, shift of the plating potential towards more negative values initially decreased efficiency of the process (Fig. 6a) due to the increased hydrogen component of the cathodic current. At the most negative potentials, mass of the deposit produced from the chloride bath increased again. In turn, the decreasing tendency of Q A/Q C with the change of the vertex potential was not altered (Fig. 6b). It may apparently show that tin mass gains for the plating potentials below −1.20 V resulted from the secondary processes occurring in the system caused, for example, by the gas evolution at the cathode surface.
Figure 7 shows the amount of the deposited metal as a function of the plating conditions. The results are consistent with the data presented in Fig. 6a. However, they show in fact that more negative potentials do not correspond with higher amounts of deposited metal as it could be concluded from CV curves (Fig. 4), and practically, the same mass of tin in the individual baths was obtained for the relatively wide range of the potentials (from −0.7 V up to −1.2 V for chloride–sulfate bath and up to −1.1 V for chloride bath). It may suggest that tin deposition run under limiting current determined by the release of the electroactive tin species from the complexes (Fig. 1).

The influence of plating potential on the mass of tin deposited from various baths (deposition time, 30 min)
Morphology of the deposits was observed under both optical and scanning electron microscopes. Some photographs of tin surface are presented in Figs. 8 and 9. Generally, dense tin layers visible at low magnifications consisted of separate grains with the shape dependent on the applied potential and bath composition. It was found that decrease in the plating potential was accompanied by the change of tin grains from polyhedral to flaky. Simultaneously, sulfate ions favored formation of more fine-grained deposits.

Morphology of tin surface deposited on brass electrode from chloride–gluconate bath at constant potentials: a −0.8 V, b −1.1 V

Morphology of tin surface deposited (for 30 min) on brass electrode from chloride–sulfate–gluconate bath at constant potentials: a −0.8 V, b −1.1 V
Increased mass of the deposit observed in the chloride–gluconate system at the most negative potentials seemed to be related to formation of secondary products resulting from the hydrogen evolution and, thus, alkalization of the solution at the cathode surface [26]. However, no oxygen in such deposits was detected during SEM–EDS analysis. Inorganic tin compounds were also not found during XRD investigations.
Higher amounts of the obtained deposits were accompanied by the worsening of the quality of tin layers. Porous or spongy deposits were produced at potentials below −1.2 V, whereas at higher potentials matt and dense coatings were obtained. It was not observed in the sulfate containing bath due to inhibiting effect of SO4 2− anions and/or better buffering action of such solution (equilibria presented in Table 1 and in Fig. 1). It is worth to note that in the chloride–gluconate bath and at low potentials (below −1.2 V), tin showed tendency to formation long dendritic grains inside the spongy deposits (results not shown). Intensive growth of the grains towards bulk solution can enhance deposition of the metal due to serious development of the cathode surface available for the reduction of Sn2+ cations. It corresponds to the increased mass of the deposits. In turn, sulfate ions adsorbing on the electrode inhibit dendritic growth of tin and the plating occurs under stable conditions.
Figure 10 shows diffraction patterns of the coatings obtained in various conditions. Single-phase tin deposits with tetragonal structure were obtained. It is consistent with the thermodynamical data [23], since β-Sn is a stable phase at temperatures above 13 °C. Decrease in the deposition potential was accompanied by the development of some characteristic planes of tin crystal structure, which were not detected in the metal layers produced at −0.7 V, despite the comparable masses of tin coatings. It is consistent with the SEM observations of the deposits. No effect of the bath composition on the type of the crystallographic structure of tin was found.

X-ray diffraction patterns of tin deposits
Nucleation stage
The initial stage of tin deposition on the glassy carbon electrode was studied by means of potentiostatic step technique realized from the initial potential of 0 V at which no reduction of tin species or any other electrode reaction can occur (Fig. 3). Figure 11 (left side) shows parts of the current transients recorded at various deposition potentials (−0.7 to −1.2 V) for 40 s. The curves registered in the potential range up to −1.0 V show a gradual increase in the cathodic current up to a maximum for all potentials (at shorter times for more negative potentials) with their final part falling to the plateau. The course of such i–t transients is typical for the response of a 3D nucleation and growth process occurring under the mass transfer control [27]. At very short times, sudden decrease in the current was observed due to the charging of the double layer. Further rise of the current in the i–t plots corresponds to the increase in the electroactive area due to the growth of the individually isolated metal nuclei and/or the increase of the number of nuclei. At this step, transport of tin species to the growing centers occurs through hemispherical diffusion zones formed around each isolated nucleus. Radii of the hemispherical zones increase with time, and the diffusion zones start to overlap forming finally a planar diffusion layer. The maximum of the cathodic current is related to the maximal electroactive area, i.e., the moment at which hemispherical metal nuclei are on the point of the collision. The current then falls, and the transients are characterized by the Cottrell equation [25], since the electrochemical reaction run under linear diffusion. At potentials of −1.0 V or more negative, maxima were not observed in the current–time transients and reduction of Sn(II) ions occurred at the diffusion controlled rate. For longer times, transients overlapped and then plateau corresponding to the effect of natural convection was observed.

Potentiostatic current–time transients and analysis of the chronoamperometric curves for tin deposition (GC electrode) in various baths: a chloride–gluconate, b chloride–sulfate–gluconate
For a planar electrode, the Cottrell equation:
shows that cathodic current density i is a linear function of t −1/2. The plot should pass through the origin, and the diffusion coefficient D of the reduced species can be found from the slope of the straight lines [25]. Data presented in the insets in Fig. 11 show that such dependencies can be obtained for the falling parts of the transients registered at the potentials −1.0 V or higher, while at more negative potentials linear functions were accompanied by various residual cathodic currents and similar slope values for different plating potentials. The latter can be ascribed to the course of the parallel reaction of the hydrogen evolution at this potential range. Applying above tests for the diffusion control of metal deposition, average diffusion coefficients of tin(II) ions were calculated. Obtained values were (7.55 ± 0.01)·10−6 cm2 s−1 and (1.15 ± 0.02)·10−6 cm2 s−1 for chloride–gluconate and chloride–sulfate–gluconate baths, respectively. These seem to be reasonable when compared with the data presented in the literature [23] for the metallic ions in aqueous solutions at the room temperature. Diffusion coefficients determined in this study are comparable to that obtained for tin ions in sulfate [8], sulfate–gluconate [6], sulfate–tartrate [10], and chloride–citrate [12] solutions. However, it should be noted that speciation of the baths is rather complicated (Fig. 1); hence, resulting diffusion coefficients cannot correspond strictly to the diffusion coefficients of free Sn2+ ions. Therefore, it was accepted that obtained values (calculated for total Sn2+ concentrations) are rather effective diffusion coefficients, i.e., average diffusion coefficients of various tin species with consideration for their molar parts in the solutions [6].
Theory of the potentiostatic current transients for 3D multiple nucleation of tin with diffusion controlled growth was discussed by various authors [7, 8, 10, 11]. Mathematical model for instantaneous and progressive nucleation developed by Sharifker and Hills [28] has been often used to analyze the current transients of metals deposition [10, 29]. A classification of the nucleation type is possible by comparison of the experimental results with theoretical curves plotted in a dimensionless form. In the case of instantaneous nucleation, all nuclei are generated simultaneously and they grow further with the same rate. Under these conditions, the following diagnostic equation is satisfied [28]:
During progressive nucleation, the nuclei are formed constantly and they grow to different sizes and/or at different rates; hence, in this case, corresponding expression is as follows:
Figure 11 (right side) shows a comparison of non-dimensional plots for instantaneous and progressive nucleation models with the experimental data. At t/t max < 1, the transients showed a behavior closely related to the progressive nucleation, while at t/t max > 1 a deviation from the predicted curves appeared and all experimental curves were located between both theoretical ones. It is supposed that deviation of the experimental curves from the model can result from the additional step affecting the kinetics of the metal deposition, i.e., release of tin ions from the complex species in the baths, which can appear with the plating time. Comparison of the literature data [12] and the results of this study shows that chloride ions enable progressive nucleation of tin, while in the sulfate system instantaneous nucleation mode can be observed [7, 10].
Current step method
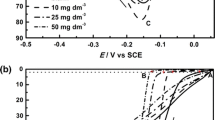
Electrodeposition of tin was studied also in galvanostatic experiments. Figure 12 shows cathodic polarization curves registered in both solutions. It was found that reduction of Sn(II) ions on the glassy carbon substrate requires relatively high overvoltage. Inhibiting effect of sulfate ions on the electrode reaction occurred, similarly to the earlier voltammetric experiments. Reactions run at low rate (below 0.3 A dm−2) at the potentials from −0.6 V to −1.4 to –1.3 V, but the increase in the cathodic current up to 4 A dm−2 corresponds to the potential range of −1.7 to −1.3 V. Cathodic polarization curves plotted in the Tafel type coordination system show that reduction of tin species occurs under limiting current (0.19 A dm−2 and 0.10 A dm−2 for the chloride and chloride–sulfate bath, respectively), but at currents higher than 0.3 A dm−2 (i.e., below −1.4 V), the process is accompanied by intensive hydrogen coevolution. It may suggest that mass transfer is a rate determining step of tin deposition. However, in the complex baths, characteristic limiting current can be also associated with the release of the electroactive metal ions from the complexes as a rate determining step. This is in accordance with the voltammetric and potentiostatic data obtained in the previous experiments.

Galvanostatic cathodic polarization curves for tin deposition (GC electrode) (current step 0.1 mA/s)
Measurements in the galvanostatic mode are relatively seldom used for the investigations of the metal deposition [8, 12, 30]. In this type of experiment, the current flowing in the cell is instantaneously stepped from zero to some fixed value and the potential of the working electrode is monitored as a function of time [25]. Figure 13 shows chronopotentiometric curves registered for the chloride and chloride–sulfate solutions of the same Sn(II) total concentration. As the current pulse was applied, there was an initial fairly sharp decrease in the potential due to charging of the double layer, and then a potential, at which reduction occurs on the cathode, was reached. There was then slow decrease in the potential until concentration of tin species at the electrode surface may eventually reach zero. It was visible especially for low current densities accompanied by the reduction of tin ions. At higher current densities, a sharp drop of the potential corresponded to the reduction of hydrogen ions as a main cathodic reaction running at potentials below −1.4 V, since the flux of Sn(II) ions to the working surface by natural convection was no longer sufficient to maintain the applied current.

Chronopotentiometric curves registered at various current densities (GC electrode) for tin deposition from 0.1 M Sn(II)–0.2 M gluconate baths: a chloride–gluconate, b chloride–sulfate–gluconate
Jump of the electrode potential caused by the depletion of the solution at the electrode surface with the electroactive metal ions is associated with so-called transition time τ d given by the Sand equation [25]:
Transition time can be determined from the plot, but it should be noted that double layer charging effects can disturb the precise determination of the time period. At fixed total concentration of tin ions, the product of i . τ d 1/2 should be constant for all current densities applied. It was realized for current densities up to 2 A dm−2 and the obtained average values were 0.76 ± 0.07 and 0.34 ± 0.01 A s1/2 dm−2 for chloride–gluconate and chloride–sulfate–gluconate solutions, respectively. Hence, these were constants as expected for diffusion-controlled process. Linear dependencies of the cathodic current density on τ d −1/2 allowed to determine the average effective diffusion coefficients from the slopes of the straight lines (Fig. 14). The obtained values were 0.7·10−6 and 3.0·10−6 cm2 s−1 for Sn–Cl–Glu and Sn–Cl–S–Glu systems, respectively. These were close to that determined in former experiments mainly for chloride–sulfate bath.

Dependence of the cathodic current density on the inverse of the transition time square root for 0.1 M Sn(II)–0.2 M gluconate baths
Electrodeposition of tin from chloride–gluconate solutions was studied more detailed, and the obtained results will be described in the next paper.
Conclusions
Electrodeposition of tin from acidic baths of various compositions was investigated. Distribution of tin(II) species in chloride–gluconate and chloride–sulfate–gluconate solutions was presented. Cyclic voltammetric, potentiostatic, and galvanostatic measurements showed that the cathodic process can run under limiting current determined probably by the release of the electroactive tin species from the complexes. Simultaneously, the presence of sulfate ions inhibited cathodic process and affected the morphology of tin deposits. Anodic response was found as dependent on the bath composition and experimental conditions. Chronoamperometric studies suggested nucleation of tin according to the progressive mechanism. Experimental methods used in this study allowed to estimate effective diffusion coefficients of tin(II) species.
References
Hirsch S (1995) Tin–lead, lead and tin plating. Met Fin 93(1):298–313
Schlesinger M, Paunovic M (2010) Modern electroplating. Wiley, Hoboken, pp 139–204
Medvedev GI, Makrushin NA (2002) A study of the kinetics of tin electrodeposition from sulfate electrolyte with organic additives. Russ J Appl Chem 75(8):1234–1236
Guaus E, Torrent-Burgués J (2006) Voltammetric study of Sn(II) reduction on a glassy carbon electrode from sulfate–tartrate baths. Russ J Electrochem 42(2):141–146
He A, Liu Q, Ivey DG (2008) Electrodeposition of tin: a simple approach. J Mater Sci Electron 19:553–562
Survila A, Mockus Z, Kanapeckaite S, Stalnionis G (2012) Kinetics of Sn(II) reduction in acid sulphate solutions containing gluconic acid. J Electroanal Chem 667:59–65
Gomez E, Guaus E, Sanz F, Vallés E (1999) Tin electrodeposition on carbon electrodes. From nuclei to microcrystallites. J Electroanal Chem 465:63–71
Barry FJ, Cunnane VJ (2002) Synergistic effect of organic additives on the discharge, nucleation and growth mechanisms of tin at polycrystalline copper electrodes. J Electroanal Chem 537:151–163
Wen S, Szpunar J (2005) Nucleation and growth of tin on low carbon steel. Electrochim Acta 50:2393–2399
Torrent-Burgués J, Guaus E (2005) Electrodeposition of tin from tartrate solutions. Portug Electrochim Acta 23:471–479
Torrent-Burgués J, Guaus E, Sanz F (2002) Initial stages of tin electrodeposition from sulfate baths in the presence of gluconate. J Appl Electrochem 32:225–230
Han C, Liu Q, Ivey DG (2009) Nucleation of Sn and Sn–Cu alloys on Pt during electrodeposition from Sn–citrate and Sn–Cu–citrate solutions. Electrochim Acta 54:3419–3427
Tachikawa N, Serizawa N, Katayama Y, Miura T (2008) Electrochemistry of Sn(II)/Sn in a hydrophobic room-temperature ionic liquid. Electrochim Acta 53:6530–6534
Gomez E, Guaus E, Torrent J, Alcobe X, Valles E (2001) Tin–cobalt electrodeposition from sulfate–gluconate baths. J Appl Electrochem 31:349–354
Guaus E, Torrent-Burgues J (2003) Tin–zinc electrodeposition from sulphate–gluconate baths. J Electroanal Chem 549:25–36
Chrisholm CU, El-Sharif M, Kuzmann E, Stichleutner S, Homonnay Z, Vértes A (2010) Electrodeposition of Sn–Fe alloys using gluconate electrolytes and X-ray diffractometry and mössbauer studies of the deposits. Mat Chem Phys 120:558–565
Chrisholm CU, Kuzmann E, El-Sharif M, Doyle O, Stichleutner S, Solymos K, Homonnay Z, Vértes A (2007) Preparation and characterization of electrodeposited amorphous Sn–Co–Fe ternary alloys. Appl Surf Sci 253:4348–4355
Chen K, Wilcox GD (2006) Tin–manganese alloy electrodeposits. From nuclei to microcrystallites. J Electrochem Soc 153(9):C634–C640
Cigala RM, Crea F, De Stefano C, Lando G, Milea D, Sammartano S (2012) The inorganic speciation of tin (II) in aqueous solution. Geochim Cosmochim Acta 87:1–20
Pettine M, Millero FJ, Macchi G (1981) Hydrolysis of tin(II) in aqueous solutions. Anal Chem 53:1039–1043
Müller B, Seward TM (2001) Spectrophotometric determination of the stability of tin (II) chloride complexes in aqueous solution up to 300 °C. Geochim Cosmochim Acta 65(22):4187–4199
Sillen LG, Martell AE (1971) Stability constants of metal-ion complexes. Suppl. No. 1 Alden Press Oxford
Kalendarz chemiczny (1954) PWN Warszawa (in Polish)
Sawyer DT, Sobkowiak A, Roberts JL (1995) Electrochemistry for Chemists. Wiley, New York
Greef R, Peat R, Peter LM, Pletcher D, Robinson J (1985) Instrumental methods in electrochemistry. Ellis Horwood Ltd., Chichester
Begum ASN, Basha A, Muralidharan VS, Lee CW (2012) Electrochemical behaviour of tin in alkali solutions containing halides. Mat Chem Phys 132:1048–1052
Budevski E, Staikov G, Lorenz WJ (1996) Electrochemical phase formation and growth. VCH, Weinhheim
Sharifker B, Hills G (1983) Theoretical and experimental studies of multiple nucleation. Electrochim Acta 28(7):879–889
Hyde ME, Compton RG (2003) A review of the analysis of multiple nucleation with diffusion controlled growth. J Electroanal Chem 549:1–12
Andreazza P, Andreazza-Vignolle C, Kante I, Devers T, Levesque A, Allam L (2005) Buffer layer effect in nanostructured tin electrodeposition on insulating and conducting substrates. Prog Solid State Chem 33:299–308
Acknowledgments
This research work has been financed under research project no. 11.11.180.373. The author thanks to Grzegorz Włoch, Ph.D., from Department of Structure and Mechanics of Solids at the Faculty of Non-Ferrous Metals of AGH University of Science and Technology for his help during SEM observations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Rudnik, E. Effect of anions on the electrodeposition of tin from acidic gluconate baths. Ionics 19, 1047–1059 (2013). https://doi.org/10.1007/s11581-012-0819-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-012-0819-4