
Abstract
Electrodeposition of nickel from slightly acidic gluconate solutions containing chloride or/and sulfate ions was investigated. Electrochemical measurements correlated with bath speciations showed nickel chloride complex and nickel sulfate complexes as crucial species affecting cathodic reactions in a potential range up to −1.3V. At more negative potentials, nickel deposition was governed by a release of nickel cation from nickel-gluconate complex. This was further evidenced by differences in nucleation modes, morphology, and structure of the deposits. Wettability of as-plated and chemically modified nickel layers were determined and correlated with their morphology and corrosion resistance.
Similar content being viewed by others
Introduction
Nickel electroplating technologies have been developed extensively for last 50 years leading currently among electrodeposited metals with upwards of 150,000 tones being deposited worldwide every year [1]. According to Future Market Insights, this segment is expected to hold a little below 31% of the market, in terms of product type, by the end of 2031 [2]. Nickel remains the sought-after metal in the electroplating area as it provides an excellent combination of corrosion and wear resistance, enhanced hardness, superior strength, and ductility as well as bright, luster, or satin finishes [3]. The electrodeposition process offers also superb adhesion properties for subsequent coating layers, usually for chromium. These widespread properties reflect commercial importance of traditional nickel coatings and their versatility in technical and decorative applications. Nickel is used often as a matrix for electrochemical synthesis of composite coatings [4,5,6]. They can contain a variety of hard or soft nano- or microsized particles improving physio-chemical properties of the metallic layers. For the last few years, a new type of nickel deposits has been also developed [7]. They are superhydrophobic coatings characterized by water contact angle above 150o and potentially improved corrosion resistance originated from reduced contact area between the metal and water droplets [8,9,10].
Detailed review [11, 12] of the subject matter showed no data related to the wettability of traditional as-plated nickel. Some accidental results were reported during comparative studies on a fabrication of superhydrophobic coatings post-treated chemically with compounds of low surface energy [7,8,9,10]. This inspired the authors to carry out an investigation on an importance of primary components of the electrolytic bath (i.e., chloride and sulfate anions) and deposition potential in a creation of a special surface morphology of the nickel coatings and, further, their wettability and corrosion resistance. However, this research work involves principally electrochemical studies on the reduction of nickel ions and metal nucleation in a presence of different anions as both phenomena affect further morphology of the electrodeposits and thus their surface properties. Although a role of chloride and sulfate ions in acidic simple salts solutions has been already investigated [13, 14], their effect in more alkaline complex baths is still essentially obscured. The electroreduction of nickel ions from gluconate baths has been not investigated in details yet; therefore, sodium gluconate as environmentally friendly additive was used in this study to complex nickel ions and improve buffer capacity of the electrolytes [15, 16]. These comparative studies can contribute deeper insight into the surface phenomena taking place at a cathode when the electrolyte contains a mixture of anions. The obtained results can give also an answer if there would be a possibility to fabricate hydrophobic nickel layers of improved corrosion resistance with no addition of special crystal modifiers to the bath nor application of uncommon electrolysis modes, or chemical post-treatment of the coatings is necessary to achieve low wettability of the nickel coatings.
Materials and methods
Electrodeposition of nickel was carried out from gluconate solutions due to improved buffer properties, metal complexing ability, non-toxicity of sodium gluconate, and, thus, well applicability of the latter in the nickel plating. The solutions contained 0.25M Ni2+, 0.25M boric acid H3BO3 as buffering component, and 0.25M sodium gluconate C6H11O7Na as buffering-complexing agent. Three baths were used, i.e., chloride (0.5M Cl- as NiCl2), sulfate (0.25M SO42- as NiSO4), and chloride-sulfate (0.25M Cl- and 0.125M SO42- as NiCl2 and NiSO4 in the 1:1 molar ratio). For comparative purposes, blank solutions with potassium salts of the same molar concentrations instead of nickel salts were used. The pH of all baths was 6.0. Ionic speciations of the particular baths were calculated using free available HySS software.
Electrochemical measurements were carried out in a three-electrode cell using a glassy carbon GC working electrode (0.2 cm2), a platinum plate (2 cm2) as a counter electrode and an Ag/AgCl electrode as a reference electrode (all potentials in the further text are referred to this electrode). Cyclic voltammetry CV with a sweep rate of 10 mV/s was initiated from a potential of 0 V vs. Ag/AgCl and continued towards more negative values (i.e., −1.2 V or −1.4 V) during the first scan. The forward scan was finished at the same potential as initial (0 V). Chronoamperometric measurements were performed for 40 s at various potentials. Anodic stripping voltammetry was further performed at the scan rate of 10 mV/s with no removal of the sample from the bath. Nickel layers were deposited at constant potentials for 60 min in the same system, but copper plates (0.44 cm2) as cathode substrates were used. Before each experiment, the glassy carbon electrode with a mirror finish was chemically cleaned, while the copper sheets were chemically polished in a mixture of concentrated acids (HNO3:H3PO4:CH3COOH with a volume ratio of 1:3:1).
Morphology of the nickel coatings was examined using scanning electron microscope (Hitachi). Structure of the deposits was analyzed by X-ray diffractometry (Rikagu diffractometer, CuKα radiation).
Cathodic current efficiency of nickel potentiostatic deposition η was calculated according to the mass of deposit m determined gravimetrically and total electrical charge flowed through circuit during electrolysis Q:
where kNi is the electrochemical equivalent of nickel.
Selected nickel samples were chemically post-treated with 0.2-M ethanol solutions (anhydrous) of three saturated organic acids: lauric C12H24O2, palmitic C16H32O2, and stearic C18H36O2. The coatings were immersed in the solutions for 10 min at 60 oC and then remained to dry at ambient temperature.
Water contact angles WCA of stationary water droplets were measured with a goniometer (Krűss DSA25). A scaler installed at a deionized water container injector was used to control the volume of the water droplet. The water contact angle tester contained a needle that could hold very small droplet (2 μl), which could drop on the sample under gravity. It was followed by taking a series of snapshots accompanied by automatic measurements of WCAs until stable values were obtained.
Corrosion resistance of the coatings in 0.5 M NaCl and 1 M H2SO4 aqueous solutions was determined from potentiodynamic electrochemical experiments. Linear voltammetry with a sweep rate of 10 mV/s was realized always towards positive potentials in a potential range from −1.4V to −0.1V in H2SO4 and from −1.6V to −0.1V in NaCl.
All electrochemical experiments were performed at ambient temperature (22 ± 1 oC) using Autolab potentiostat/galvanostat (PGSTAT30) and non-agitated solutions with a volume of 20 cm3. Individual measurements reported in this paper were repeated three times and average values are shown.
Results
Bath speciation
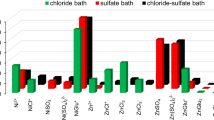
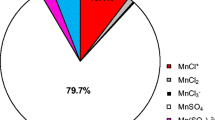
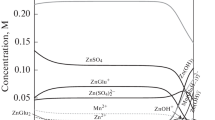
Nickel cations in aqueous electrolytes can exist as various species due to their tendency to combine with water molecules or chloride, sulfate, and gluconate anions. As result, cationic, neutral, or anionic complexes of different stability are formed (Table 1). Figure 1 shows the equilibrium distribution of the nickel species in the electrolytes. It was found that simple metal cation represents 13–19% (40 ± 6 mM) of total nickel in the solutions, while remaining portions exist as different complexes. The latter is mainly gluconate complex showing 68–75% (180 ± 10 mM) of all nickel forms. In turn, chloride (max 15 mM) or sulfate (max 40 mM) nickel complexes show lesser fractions. Hydroxide or borate complexes can be omitted in the considerations due to their negligible percentages (i.e., 10-3 mM).

Distribution of nickel species in gluconate baths at pH = 6.0
The existence of particular nickel species in the solutions determines the mechanism of the metal electrodeposition [8]. However, it must be emphasized that free anions like chloride, sulfate, and gluconate or boric acid also play important roles, since they affect a buffer capacity of the baths [15, 19, 20] and morphology of the deposits [15]. It should be also noted that during electrolysis a composition of the electrolyte adjacent to the cathode can change due to depletion of metal ions or hydrogen coevolution. In the latter case, the equilibrium speciation for higher pH may be recommended for taking account.
Voltammetric studies
Figure 2 shows the CV curves registered for two switching potentials. Generally, one cathodic branch C and single anodic peak A were observed. However, the anodic response was absent in the sulfate bath if the switch potential was negative not enough. This indicates an inhibiting effect of sulfate anions on the nickel electrodeposition. Such hindering was not evidenced in the chloride-sulfate solution, where the inhibiting action of SO42- was depressed by activation effect of chloride species. The analysis of the cathodic branches indicates that nickel starts to deposit at the potentials of −1.04V, −1.07V, and −1.20V in the chloride, chloride-sulfate, and sulfate bath, respectively. In all cases, the cathodic branches involve two electroreduction processes, since nickel deposition is indistinguishable from hydrogen evolution reaction.

Cyclic voltammetric curves registered in various baths for two switching potentials: a) -1.2 V, b) -1.4 V
During the reverse scans, two cross points between the negative and positive sweeps were observed. First cross-point corresponding to nucleation potential En was at the loop typical for a nucleation and growth of a new phase. Such crossover, associated with electrocatalytic points, was about −1.2V and −1.3V for both chloride-containing baths and sulfate bath, respectively. The En values were practically independent on the switch potential in both chloride-containing solutions, while En stabilized at −1.2V in the sulfate bath when the reverse potential was −1.6V (not shown). Second crossover potential Ec appeared at more positive values, i.e., about −0.9V in the chloride-containing baths and −0.4V in the sulfate bath (Fig. 2b, inset). Such point is often related to an equilibrium potential of a M/Mz+ electrode. However, this was not the case, since all recorded values were different from a formal potential of the nickel electrode calculated according to the Nernst equation for the concentration of simple nickel cations, i.e., −0.52V vs. Ag/AgCl for 40±6 mM Ni2+. This could be attributed to complex occurrences at the cathode during potentiodynamic measurement.
The cathodic currents recorded between En and Ec were always higher during the reverse than during the forward sweep. It is obvious, since energy required for the nickel overpotential deposition on the foreign substrate (glassy carbon) is higher than that for the metal deposition on the native layer (nickel) produced during the previous scan [21]. However, the backward CV part was dependent on the bath indicating differences in a behavior of the nickel species at the electrode surface.
Details of the negative and positive scans registered on the GC electrode in the absence and the presence of nickel ions are shown in Figure 3. The forward scans demonstrated enhanced initiation of the cathodic processes in both chloride-containing nickel baths (Figs. 3a, c) associated with easier formation of nickel nuclei and easier hydrogen coevolution on the metal than on glassy carbon [22]. A comparison of the curves showed their overlapping from the potential of about −0.9 V indicating similar electrochemical behavior when chloride ions were present. The cathodic reactions were much more inhibited in the nickel sulfate solution, where the currents started to rise at more negative potential of −1.1V (Fig. 3b). Interestingly, in both sulfate-containing nickel solutions, flat regions iL,F on the curves were found. The length of plateau was proportional to the sulfate concentration. These limiting currents iL,F could be attributed to mass transfer limitation during two-step hydrogen evolution [23] or a reduction of dissolved oxygen. However, the limiting currents (~ 5 mA/dm2) cannot be ascribed solely to these processes since similar plateau should be found in all electrolytes having the same pH and the same air saturation (the electrolytes were not deaerated before experiments). This suggests that the limiting current iL,F is caused by at least two main processes like a formation of first nickel nuclei and concurrent hydrogen evolution taking place on a new metallic phase. These reactions can be hindered by adsorption of sulfate species and/or slowed release of nickel ions from nickel sulfate complexes. In turn, a lack of the plateau in the chloride bath shows a kind of surface activation by chloride species.

Forward (a-c) and backward (a’-c’) parts of CV curves registered in blank and nickel gluconate solutions: a-a’) chloride, b-b’) sulfate, c-c’) chloride-sulfate
Dissimilarities were also during the reverse scan (Fig. 3 a’-c’). The behavior of both chloride-based systems was quite similar, but a limiting cathodic current iL,B (50 mA/dm2) appeared in the sulfate solution. The limiting current was observed solely if nickel film was previously produced on the cathode surface.
Such differences in the positive scans were further found during anodic sweep voltammetry (Fig.4 a’-c’). Analogous limiting cathodic currents iL,B’ were found only in the sulfate solution and solely if metal was deposited in enough amount during the accumulation stage. For the deposition potentials of -1.1V and -1.15V, a gradual decrease of the cathodic currents with no clear plateau and no anodic peak of the metal dissolution were observed. It is evident that these limiting currents are definitely caused by a process favored by the metal formerly deposited on the electrode during the accumulation step (Fig. 4 b, b’). Moreover, this process cannot be hydrogen coevolution. All observations show that the surface phenomenon is unquestionably correlated with the presence of nickel sulfate complexes in the solution and this cathodic process is enhanced by the nickel surface, but not by the glassy carbon.

Chronoamperometry (a-c) and anodic stripping voltammetry for nickel deposited at various potentials from gluconate solutions: a-a’) chloride, b-b’) sulfate, c-c’) chloride-sulfate
The anodic stripping voltammetry indicated also some differences in location of the anodic peak maximum. The maxima were shifted somewhat towards more negative direction in the sulfate bath in comparison to the chloride bath. In turn, the peak maxima were in the intermediate positions in the chloride-sulfate solution.
The heights of the anodic peaks were also dependent on the deposition potential of the accumulation stage. In the sulfate solution, the deposition potentials up to −1.3V resulted in more enhanced nickel deposition and thus continuous development of the anodic response during dissolution. In turn, the anodic peak started to lessen its height for the deposition potential of −1.3V in the chloride-sulfate bath, while in the chloride bath, this effect was observed already at −1.25V. Such effects indicate existence of the parallel reduction of hydrogen ions responsible for the falling of the cathodic efficiency of metal deposition at more negative potentials. The existence of single anodic peaks in all electrolytes indicates a lack of secondary/intermediate products like Ni(OH)2 incorporated into the metal [24] or nickel passivation effects. This shows that the presence of gluconate having good buffering properties [15] prevents precipitation of hydroxide at the cathode surface even if hydrogen coevolution occurs simultaneously. However, it must be mentioned that conditions at the cathode surface can be changed during prolonged electrolysis times.
To get better insight into the cathodic processes, the parts of the cathodic and anodic branches were plotted in a Tafel coordination system (Fig.5). During cathodic scan, three ranges were observed: I, up to −1.15V was influenced by the anions; II, in a rather narrow potential range near −1.2V was linear and independent on the bath; and III, at the potentials more negative where a concentration polarization appears in all electrolytes. In turn, the rising parts of the anodic peak (Fig.5, inset) were similar in both chloride containing solutions, while in the sulfate bath horizontal part was detected. The linear regions were observed in the narrow potential ranges with average slopes of about −0.11V/dec and 0.12V/dec for cathodic and anodic reaction, respectively. The slopes were in a good agreement with literature data determined at similar solution pH [25]. It shows that nickel can be deposited and dissolved under activation control as one-electron transfer rate-determining step but only at restricted potential ranges (in fact the linear Tafel region on E–log i dependences should be more prolonged for deeper discussion of the mechanism of the electrochemical reaction). However, this was not fully true for the anodic nickel dissolution in the sulfate bath, since the flat part of the curve indicates additional surface phenomena disturbing formation of the metal ions.

Tafel plots of cathodic and anodic (inset) CV branches
The comparison of all voltammetric data clearly evidences different surface phenomena taking place on the glassy carbon and fresh nickel surfaces depending on the presence of chloride or sulfate ions as well as the cathode potential. These anions can form nickel complexes of relatively low stability, which is crucial for the mechanism of nickel electroreduction [11]. It seems that the presence of NiGlu+ of the highest stability affects the metal electroreduction at more negative potentials, i.e., in the III range, while for more positive potentials, the metal deposition was governed by the chloride and sulfate forms.
Electrodeposition and characterization of deposits
Nickel was deposited at constant potentials selected according to the potentiodynamic results. Figure 6 shows the influence of the deposition potential on the amount of deposited metal and current efficiency. Comparable amounts of nickel were produced independently on the type of anion, but the key parameter revealed the deposition potential. The courses of the curves in Fig. 6a reflect the conclusions drawn from the Tafel plots and clear ranges of the cathodic process can be distinguished showing different rate-determining steps. The most favorable conditions for the plating were in a narrow range of the potentials, i.e., between −1.1V and −1.2V, where deposition run under activation control. This was followed by a decrease in the nickel deposition caused by mass transfer limitation and plateau of the limiting currents below −1.4V. The latter appeared in the same potential range signifying similar rate-determining step in all baths. Finally, in both sulfate-containing solutions, the amounts of the metal increased a little from the potential −1.8V, despite that the cathodic current efficiency remained practically unchanged (Fig. 6b).

Influence of deposition potential and bath composition on: a) amount of deposited nickel, b) cathodic current efficiency
Such ranges can be then correlated with the surface morphology of the nickel layers (Fig. 7). High similarity of the nickel surfaces produced in the chloride-containing baths is evident, while other morphology was observed in the sulfate electrolyte.

Surface morphology of nickel deposited from gluconate baths: a) chloride, b) sulfate, c) chloride-sulfate
Three types of the deposits depending on the deposition potential were found in particular baths. In each case two-level roughness of the surface was visible at low and high magnifications. In the chloride bath, compact layers with cauliflower-like surface were produced at the potentials from −1.0 to −1.2V. Such specific morphology consisted of clearly visible grains changing from nodular (−1.0V) to star-like or needle-like/platelets (−1.1V and −1.2V) in shape. For more negative potentials, thin smooth deposits were obtained. These reproduced roughness of the copper substrate in a center of the sample. At the most negative potentials, porous deposits with inorganic precipitates at the edges of the sample were formed. Similar morphologies were observed in the chloride-sulfate solution, except the most negative potentials. In the latter cases, thicker, cracked, and rough nickel layers were formed. Such effects were more prevented in the sulfate bath. At the less-negative potentials gradual change from nodular (−1.0V) to polyhedral big grains at (−1.3V) were observed. At −1.4V and −1.5V thin layers of similar morphology like in other baths were produced. The surface began to change with more negative potentials. The smooth layer started to crack (−1.6V) and ripple (−1.7V) with an initiation of columnar growth at the edges of the cracks (−1.8V and below). A development of the columnar structures was more favored at the edges of the sample, but no inorganic precipitates were observed.
SEM observations evidenced precipitation of secondary nickel oxide/hydroxide products on the surface of the samples produced mainly in the chloride-containing baths, at the most positive and negative potentials. At the more positive potentials (−1V), reduction of hydrogen ions proceed electroreduction of nickel ions due to thermodynamic reasons, but at the more negative potentials hydrogen evolution is favored due to mass transfer limitation of nickel species. This resulted in the alkalization of the electrolyte at the cathode surface and enhanced hydrolysis of the nickel salts. Such effects were prevented in the sulfate bath confirming former hypothesis on the inhibiting effects of the sulfate species.
X-ray diffraction patterns indicated a face centered cubic structure of the polycrystalline deposits (Fig. 8). Nickel diffractograms could not be obtained for all thin layers due to refining nickel crystallite sizes and thus strong signals of the copper substrate. Therefore, some general deductions could be drawn for only few samples. Three peaks located at 44.6o, 52.1o, and 76.5o were indexed to nickel (111), (200), and (220) planes, respectively. Relative intensity of the peaks seemed to be dependent on the anion type in the electrolyte (Table 2), despite that the preferential orientation of the close-packed plane (111) is dominant in all cases. However, it is clearly seen that intensity of the (111) reflexion is dependent on the bath (thickness of the deposits is comparable, Fig.6a). It was found that chloride ions impede formation of the (111) and (200) planes, enhancing development of the (220) plane. In turn, sulfate ions stabilize development of the (111) and (200) planes. The influence of particular anions is reflected in the crystalographic structure of the nickel deposits produced from the mixed-anion bath. Preferential growth modes of the nickel deposits is correlated with the grain shapes shown above since the final form of the crystals obviously depends on the relative development of the planes. However, if the metal crystals are produced under different conditions, they can exhibit the same crystallographic structure but different external shapes and facets [26].

XRD of nickel deposits produced at -1.2V
The average nickel crystallite sizes Dc were estimated from XRD patterns for the (111) reflection using the classical Scherrer’s formula:
where λ is the wavelength of X-rays (0.154 nm), β is a full width at the half-maximum intensity (rad), θ is the Bragg diffraction angle (rad), and K is a dimensionless shape factor assumed to be 0.9.
It was found that the nickel crystalline domains were nanometric in size having about 14, 24, and 21 nm for the chloride, sulfate, and chloride-sulfate bath, respectively.
A possibility of the creation of different surface morphologies can affect surface properties of the coatings like wettability and then corrosion resistance. Wettability is seriously dependent on the roughness due to possibility to maintain water droplets on tops of “hills” if regularly distributed on the surface [7]. Obviously, the best situation to obtain superhydrophobicity is hierarchical morphology in a micro- and nanoscale helping to trap a large amount of the air that reduces attractive interactions between the solid surface and the liquid. Second option is an application of chemical compounds of low surface energy if no specific two-tier surface can be obtain. The microscopic observations of the nickel surfaces produced in this research showed that hierarchical surface roughness was not succeeded. Therefore, a few selected samples were chemically treated with three saturated organic acids: lauric C12H24O2, palmitic C16H32O2 and stearic C18H36O2. The wettability of as-plated and modified nickel layers was determined. The results are presented in Figure 9. Water contact angles WCA changed from weakly hydrophilic to weakly hydrophobic (but not superhydrophobic), as seems, dependently on the deposit thickness and, hence, the surface roughness. This effect was mostly evidenced for less negative deposition potentials where gradual changes in the nickel morphology were identified. At more negative potentials, surface composition and morphology changed due to precipitation of inorganic hydrolysis products combined with a serious increase in the surface roughness. Modification of the nickel coatings with organic acid decreased wettability of the surfaces. Such effect is related to the adsorption of the molecules with polar group –COOH at the metal surface and non-polar hydrocarbon chain directed outside. In general, lauric acid with the shortest chains appeared to be the most effective, probably due to denser arrangement of the molecules attached to the metal substrate. However, no superhydrophobic properties were obtained. This shows that surface modification with the compounds of simple structure is not sufficient to obtain high WCA if no special metal surface morphology exists.

Influence of deposition potential on wettability of as-plated (a-c) and modified (a’-b’) nickel deposits produced from the baths: a-a’) chloride, b-b’) sulfate, c-c’) chloride-sulfate
Corrosion properties of the coatings were investigated in neutral and acid environments. However, no correlations among corrosion currents, corrosion potentials, and wettability of nickel were found. In general, the corrosion currents and corrosion potentials were (2.6 ± 0.5)∙10-2 mA/cm2 and −0.70 ± 0.05V in NaCl solution, whereas 0.9 ± 0.2 mA/cm2 and −0.30 ± 0.05V in H2SO4 solution. These values indicate that conventional nickel coatings with no specific surface architecture are not able to achieve special properties.
Nickel nucleation
Early stages of the metal formation affect often morphology of the coatings. It is traditionally agreed that metal electrodeposition on a foreign substrate includes the reduction of metal ions to adatoms, whose aggregation can successively generate nuclei at active sites randomly distributed over the substrate These particles can further grow by diffusion of metal ions and charge transfer reaction at nuclei surface. The competition between nucleation of the metal and its growth determines the granularity and morphology of the deposit.
The nucleation stage is investigated usually by analysis of the chronoamperometric curves registered at constant potentials. The i-t transients shown in Figure 4a-c exhibit the deposition behavior with the characteristic peak corresponding to the nuclei overlapping followed by a tendency of the curves to merge into a common curve for longer times typically for a diffusion controlled growth of the nuclei. It is seen that the initial decay in the current density caused by double-layer charging does not occur completely. This may be caused by concurrent hydrogen evolution or rapid replacement of consumed ions. The i-t curves demonstrate also induction periods tind which were the longest in the sulfate bath. This delayed nucleation was less emphasized in the chloride-containing solutions. Such induction period is considered as the time needed to reach a steady-state distribution of subcritical clusters associated with adsorption-desorption of electroactive ions or the time needed for the appearance of an active site on the electrode surface [26]. In this stage no significant nucleation can occur; the small clusters may also dissolve into the solution. Prolongation of this stage indicates some hindrance of the active sites creation by adsorbed non-electroactive species or hydrogen.
Initiation of the metal layer deposition is associated with a formation of 2D or 3D nuclei. Deposition is continued through layer-by-layer of adatoms on the depositing plane in the case of 2D growth, whereas random incorporation of isolated supercritical nuclei on the substrate occurs in the case of 3D growth. The nucleation stage and subsequent growth can take place by two different mechanisms called instantaneous and progressive. Instantaneous nucleation IN occurs when the rate of nucleation is faster than subsequent growth rate of nuclei. In this case, all nuclei are created within very short time at early stage of electrodeposition. In progressive nucleation PN, the rates of the nucleation and the successive growth are comparable, while the nucleation process is continued simultaneously with growing nuclei formerly created. Kinetics of the early stages of the electrochemical deposition is investigated by analyzing the current-time transients (Fig.4a-c) in terms of dimensionless dependences characteristic for particular nucleation modes. These are plotted as i/imax or (i/imax)2 against t/tmax, where imax is potentiostatic current maximum and tmax is the corresponding time in the chronoamerometric curve. The equations for the individual nucleation models were proposed by Bewick et al. [27] and Scharifker and Hills [28] for the analysis of 2D and 3D nucleation, respectively. The 2D nucleation model undertakes the incorporation of atoms or molecules at the expanding periphery of the centers as the rate-determining step in the electrocrystallization, while the growth of 3D nuclei is assumed as diffusion-limited. The following relationships were given for the nucleation with nuclei overlap [26,27,28,29]:
These expressions describe both the rising and falling portions of the transients.
To identify the mode of the nickel nucleation a comparison of the theoretical curves with the experimental data was performed. Figure 10a shows the results plotted in the t/tmax-(i/imax)2 coordination system (Eq. (3)-(4) were recalculated) for the deposition potentials at which the formation of metal should be least disturbed by concurrent processes. It was observed that experimental points for both chloride-containing baths are closely following two theoretical curves (2D IN and 3D PN) up to the maximum current peak, but then partially cover the diagnostic curve of 3D instantaneous nucleation. The latter model seems to be characteristic for early stages of the nickel deposition from the sulfate bath. A transition between modes observed for tmax < t (i.e., when nuclei overlap with each other) suggests that the nucleation type can change if new nickel nuclei are formed on previously created thin metal layer. This could be confirmed by second weak maximum marked on the transient at shorter times for the chloride containing baths if deposition potential was more negative.

To distinguish nucleation behavior at very early times (on glassy carbon), the rising parts of the chronoamperometric curves were plotted in a logarithmic coordination system (Fig.10b). Linear dependence with a general formula:
describes generally i-tn functions developed for different kinds of nucleation and growth stages at early stages of the electrocrystallization [26, 29]. Therefore, the values n can serve as the diagnostic criterion for the type of the process. The rising parts of the transients for the identified nucleation modes are described by the following equations:
where z is the number of electrons, F the Faraday constant, h the height of the deposited layer, k the rate of incorporation, No and N the number densities of isolated nuclei, M the molar mass of the metal, ρ the metal density, D the diffusion coefficient of the electroactive species, and c the concentration of metal ions.
For both chloride containing solutions, the slopes of the linear parts of the rising portions of the transients for −1.2V were 0.9 indicating 2D instantaneous nucleation (n = 1). However, at more negative potential (-1.25V) the slope was more reduced if sulfate ions were present. In the sulfate bath, the slope confirms 3D instantaneous nucleation with diffusion limited growth (n = 0.5), but the change in the potential again affects the results. All of it confirms that nickel nucleation is affected by the presence of sulfate species.
Comparison of the identified nucleation models with SEM micrographs (Fig. 11) shows that along the time, instability in the nickel growth takes place and a transition from one to other growth mode takes place. This is indicated not only by the analysis of the transients but also evolution of the surface morphology with the deposition potential. Samples produced at the potential of -1V seem to consist of grains of narrow size distribution. It may confirm 2D or 3D instantaneous nucleation. The morphologies changed when thickness of the deposits increased. In both chloride-containing baths, thin platelet-type star-like grains of quite uniform size were observed. This appeared to support transition from 2D to 3D instantaneous nucleation on the tops of the 2D nuclei. In turn, polyhedral grains with broad grain size distribution were formed in the sulfate bath indicating 3D progressive nucleation type. Therefore, the transition from 3D instantaneous to 3D progressive nucleation can appear in this case. Obviously, it should be in mind that the nucleation mode can be affected by the substrate type (glassy carbon or nickel), while the further growth of the metal crystals can be modified by adsorbed species [26].

Surface morphology of nickel deposited at -1.2 V from gluconate solutions: a) chloride, b) sulfate, c) chloride-sulfate
Discussion
The electrochemical measurements and morphological investigations clearly show significant role of anions in the nickel electrodeposition from the gluconate baths. This is additionally complicated by the deposition potential used, which determine the rate-controlling step of the electroreduction.
It is generally accepted that nickel electrodeposition from slightly acid baths involves two consecutive one-electron charge transfers with adsorption of intermediary nickel cationic complex [12, 24, 26, 30]:
The anion X- can be Cl- in chloride or chloride-sulfate baths or OH- in sulfate electrolytes. The rate-determining step is first electron transfer, i.e., reaction (11). This corresponds to the Tafel slope of -0.12V/dec.
A significance of hydrogen in the nickel electrodeposition cannot be omitted; hence a sequence of reaction can also occur [31]:
leading to the hydrogen incorporation into the nickel layer [32].
Taking above and the experimental results of the current study into account, it is seen that the differences in the electrochemical behavior of the electrolytes is evidently correlated with the bath speciations.
In the chloride solution and in the deposition ranges I and II, the course of the cathodic reactions involves concurrent adsorption and reduction of H+ and NiCl+. The NiCl+ complex as the positively charged species can be easily attracted to the cathode initiating the nickel deposition at lower overpotentials, i.e. at more positive potentials than observed in the sulfate electrolyte. Moreover, the adsorbed NiCl+ can disturb reduction of hydrogen ions resulting in shorter induction times during nucleation [33] and higher current efficiencies during prolonged electrolysis. It is not also excluded that this inhibited reduction of hydrogen ions results in lesser incorporation of Hinc into the metal and formation of α-Ni (a solid solution of H in Ni; a H/Ni atomic ratio approx. 0.03) [32]. This supposition can be reflected by shifting the anodic peaks to more positive values.
The role of free chloride ions in the electrolyte (0.48M) cannot be omitted in the metal phase formation, since they show specific adsorption forming often chemical bonds with the metal surface [34]. These adsorbed anions by blocking reaction sites can inhibit 3D nuclei formation on the glassy carbon leading to instantaneous creation of 2D metal nuclei at early stages of electrodeposition. However, when first metal layers are formed this specific adsorption occurs on some preferential planes enabling growth of 3D nuclei [26]. Such performance is further reflected by the fine-grained crystal structure of the deposit indicating preferred adsorption of the species on the (200) plane and also weaker on the (111) plane. Such modification of the nickel crystallographic structure cannot be attributed solely to the adsorption of H+ads, H2 or even NiOH+ads [14] if generated at prolonged time (here: 1mM NiOH+ at pH of 8), since the relative ratio of the diffraction reflections is characteristic for the chloride bath. Moreover, the specific nickel morphology (star-like grains) was obtained solely in the chloride-containing baths. Thus, the special behavior of Cl-ads species is evident as arising from their inhibition of certain three-dimensional growth facets as the faces with the largest adsorption have the slowest growth rate [35]. It seems also that even easier precipitation of secondary products in both chloride-containing baths during prolonged electrolysis was observed they do not appear to interfere with the metal deposition.
A behavior of the sulfate system in the I and II ranges was much more complicated and more influenced by the substrate surface (glassy carbon, nickel). It is known that free SO42- anions can adsorb on the cathode surface and desorb as the cathode potential becomes more negative. The desorption of sulfate anions from nickel surface was observed in sodium sulfate solution at −0.85V vs. Ag/AgCl at pH 3, but such desorption peak was difficult to distinguish at pH 6 due to convolution with the background current [36]. This sulfate adsorption can disturb the nickel deposition leading to higher cathode polarization due to competitive occupation of adsorption sites on the electrode surface, but hydrogen coevolution seems not disturbed as previously. Therefore, it is assumed that large sulfate anions are rather steric hindrances than being bound by chemical bonds. This is evidenced by longer induction times, formation of 3D nuclei, lower current efficiencies, and the anodic peaks shifted towards more negative potentials. The latter can be correlated with the anodic dissolution of β-Ni (an interstitial hydrogen alloy of nickel, a H/Ni atomic ratio above 0.6) [32]. However, these possibilities do not explain the cathodic limiting currents observed if nickel film existed on the electrode surface. Hence, it is accepted that nickel surface can catalyze reduction of Ni(SO4)22- to NiSO4-. The latter can then adsorb on the metal surface and can be reduced to the metallic form:
This NiSO4-ads adsorbate is weakly bonded to the metallic nickel resulting in insignificant disturbance of the nickel crystals formation, but the reaction (18) can be responsible for longer induction times.
The concurrent action of chloride and sulfate species is evidenced in the mixed-anion system. Strong bonding of the chloride species to the metal surface is weakened by steric obstacle of the sulfate species and reverse. This is revealed by short plateau of limiting current on CV curves followed by their overlapping below −0.9V. This complex electrochemical behaviour is reflected by the morphology and structure of the deposits.
The nickel electrodeposition in the III range is practically the same in all baths. This implies that release of nickel ions from the same complex NiGlu+ is the rate-determining step. It led to formation of very fine-grained deposits of the same morphology.
Interesting effect was observed in both sulfate-containing baths at the most negative potentials. Increased mass of the deposit was correlated with the growth of columnar structures at the edges of cracks in the metal layer. These cracks are places of preferential growth due to locally increased current density (micro-edge effect). This was not observed in the chloride bath, where secondary precipitation of hydrolysis product was more favored due to less buffering properties of the bath.
Conclusions
Electrodeposition of nickel from slightly acidic gluconate solutions containing sulfate and/or chloride ions was investigated and the following conclusions have been drawn:
-
Electrochemical measurements correlated with bath speciations showed chloride and sulfate complexes of nickel or as crucial species affecting the cathodic reactions in a potential range up to −1.3V. At more negative potentials the nickel deposition was governed by a release of nickel cation from nickel-gluconate complex as a rate-determining stage.
-
The actions of particular species were supported by differences in the nucleation mode, morphology and structure of the deposits. Strong bonding of chloride species resulted in the formation of fine-grained deposits and inhibited growth of (200) and (111) crystal planes caused by Cl-. Fast reduction of NiCl+ favored 2D-3D instantaneous nucleation. In turn, large sulfate species revealed as stearic hindrances weakly adsorbed on the cathode surface, resulting in 3D instantaneous nucleation stage followed by transition to 3D progressive nucleation on the first nickel layers. It resulted in a formation of larger polyhedral nickel crystals. The role of NiGlu+ was clarified below −1.4V, where thin and very fine-grained deposits were produced
-
The wettability of as-plated and chemically modified with higher organic acids nickel layers was determined and correlated with the morphology and corrosion resistance of the deposits
References
www.nickelinstitute.org (accessed on 24 February 2021)
https://www.futuremarketinsights.com/reports/electroplating-market (accessed on 24 February 2021)
Rose I, Whittington C (2013) Nickel plating handbook 2014. Nickel Institute, Brussels
Tseluikin VN, Vasilenko EA (2011) Electrodeposition and properties of composite coatings based on nickel. Russ J Appl Chem 84(11):2005–2007
Makarova I, Dobryden I, Kharitonov D, Kasach A, Ryl J, Repo E, Vuorinen E (2019) Nickel-nanodiamond coatings electrodeposited from tartrate electrolyte at ambient temperature. Surf Coat Technol 380:125063
Raghavendra CR, Basavarjappa S, Sogalad I (2018) Electrodeposition of Ni-nano composite coatings: a review. Ing Nano-Met Chem 48(12):583–598
Rudnik E, Chat K (2018) A brief review on bio-inspired superhydrophobic electrodeposited nickel coatings. Trans IMF 96(4):185–192
Khorsand S, Raeissi K, Ashrafizadeh F (2014) Corrosion resistance and long-term durability of superhydrophobic nickel film prepared by electrodeposition process. Appl Surf Sci 305:498–505
Xiang T, Ding S, Li C, Zheng S, Hu W, Wang J, Liu P (2017) Effect of current density on wettability and corrosion resistance of superhydrophobic nickel coating deposited on low carbon steel. Mater Des 114:65–72
Shriram S, Mohan S, Renganathan NG, Venkatachalam R (2000) Electrodeposition of nanocrystalline nickel - a brief review. Trans IMF 78(5):194–197
Oriňáková R, Turonová A, Kladeková D, Gálová M, Smith RM (2006) Recent developments in the electrodeposition of nickel and some nickel-based alloys. J Appl Electrochem 36:957–972
Sadiku-Agboola O, Sadiku ER, Biotidara OF (2012) The properties and the effect of operating parameters on nickel plating (review). Int J Phys Sci 7(3):349–360
Sahari A, Azizi A, Schmerber G, Dinia A (2008) Nucleation, growth and morphological properties of electrodeposited nickel films from different baths. Surf Rev Lett 15(6):717–725
Gómez E, Pollina R, Vallés E (1995) Morphology and structure of nickel nuclei as a function of the conditions of electrodeposition. J Electroanal Chem 397:111–118
Rudnik E, Wojnicki M, Włoch G (2012) Effect of gluconate addition on the electrodeposition of nickel from acidic baths. Surf Coat Technol 207:375–388
Abd El Meguid EA, Abd El Rehim S, Moustafa EM (1999) The electroplating of nickel from aqueous gluconate baths. Trans IMF 77(5):188–191
The IUPAC stability constants data base, Academic Software and IUPAC (1992-2000).
Graff A, Barrez E, Baranek P, Bachet M, Benezeth P (2017) Complexation of nickel ions by boric acid or (poly)borates. J Solut Chem 46:25–43
Ji J, Cooper WC, Dreisinger DB, Peters E (1995) Surface pH measurements during nickel electrodeposition. J Appl Electrochem 25:642–650
Tilak BV, Gendron AS, Mosoiu MA (1977) Borate buffer equilibria in nickel refining electrolytes. J Appl Electrochem 7:495–500
Xu Y, Huang K, Zhu Z, Xia T (2019) Effect of glassy carbon, gold and nickel electrodes on nickel electrocrystallization in an industrial electrolyte. Surf Coat Technol 370:1–10
Rudnik E (2015) Effect of gluconate ions on electroreduction phenomena during manganese deposition on glassy carbon in acidic chloride and sulfate solutions. J Electroanal Chem 741:20–31
de Jesús G-VM, Grozovski V, Kovács N, Broekmann P, Vesztergom S (2020) Full model for the two-step polarization curves of hydrogen evolution, measured on RDEs in dilute acid solutions. J Phys Chem C 124:3988–4000
Cui CQ, Lee JY (1995) Nickel deposition from unbuffered neutral chloride solutions in the presence of oxygen. Electrochim Acta 40(11):1653–1662
Davison W, Harrison JA (1973) The deposition of Ni from aqueous sulphate and sulphamate solutions. Electroanal Chem Interf Electrochem 44:431–443
Nasirpouri F (2017) Electrodeposition of nanostructured materials. Springer, Cham
Bewick A, Fleischmann M, Thirsk HR (1962) Kinetics of the electrocrystallization of thin films of calomel. Trans Faraday Soc 58:2200–2216
Sharifker BR, Hills G (1983) Theoretical and experimental studies of multiple nucleation. Electrochim Acta 28(7):879–889
Pletcher D, Greef R, Peat R, Peter LM, Robinson J (2011) Instrumental methods in electrochemistry. Woodhead Publishing Ltd., Cambridge
Saraby-Reintjes A, Fleischmann M (1984) Kinetics of electrodeposition of nickel from Watts baths. Electrochim Acta 29(4):557–566
Chassaing E, Joussellin M, Wiart R (1983) The kinetics of nickel electrodeposition inhibition by adsorbed hydrogen and anions. J Electroanal Chem 157:75–88
Fleischmann M, Saraby-Reintjes A (1984) The simultaneous deposition of nickel and hydrogen on vitreous carbon. Electrochim Acta 29(1):69–75
Gomez E, Muller C, Proud WG, Valles E (1992) Electrodeposition of nickel on vitreous carbon: influence of potential on deposit morphology. J Appl Electrochem 22:872–876
Magnussen OM (2002) Ordered anion adlayers on metal electrode surfaces. Chem Rev 102:679–725
Bergenstof C, Nielsen W, Horsewell A, Østergard MJL (1997) On texture formation of nickel electrodeposits. J Appl Electrochem 27:839–845
Grujicic D, Pesic B (2006) Electrochemical and AFM study of nickel nucleation mechanisms on vitreous carbon from ammonium sulfate solutions, Electrochim. Acta 51:2678–2690
Acknowledgments
This work was financially supported by the National Science Centre of Poland under a project No. 2018/31/N/ST8/00908.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflicts of interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chat-Wilk, K., Rudnik, E., Włoch, G. et al. Importance of anions in electrodeposition of nickel from gluconate solutions. Ionics 27, 4393–4408 (2021). https://doi.org/10.1007/s11581-021-04166-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-021-04166-y