
Abstract
Syntheses and biological activities of imidazo-, pyrimido- and diazepino[2,1-f]purinediones containing N-alkyl substituents (with straight, branched or unsaturated chains) are described. Tricyclic derivatives were synthesized by the cyclization of 8-bromo-substituted 7-(2-bromoethyl)-, 7-(3-chloropropyl)- or 7-(4-bromobutyl)-theophylline with primary amines under various conditions. Compound 22 with an ethenyl substituent was synthesized by dehydrohalogenation of 9-(2-bromoethyl)-1,3-dimethyltetrahydropyrimido[2,1-f]purinedione. The obtained derivatives (5–35) were initially evaluated for their affinity at rat A1 and A2A adenosine receptors (AR), showing moderate affinity for both adenosine receptor subtypes. The best ligands were diazepinopurinedione 28 (K i = 0.28 μM) with fivefold A2A selectivity and the non-selective A1/A2A AR ligand pyrimidopurinedione 35 (K i A1 = 0.28 μM and K i A2A = 0.30 μM). The compounds were also evaluated for their affinity at human A1, A2A, A2B and A3 ARs. All of the obtained compounds were docked to the A2A AR X-ray structure in complex with the xanthine-based, potent adenosine receptor antagonist—XAC. The likely interactions of imidazo-, pyrimido- and diazepino[2,1-f]purinediones with the residues forming the A2A binding pocket were discussed. Furthermore, the new compounds were tested in vivo as anticonvulsants in maximal electroshock, subcutaneous pentylenetetrazole (ScMet) and TOX tests in mice (i.p.). Pyrimidopurinediones showed anticonvulsant activity mainly in the ScMet test. The best derivative was compound 11, showing 100 % protection at a dose of 100 mg/kg without symptoms of neurotoxicity. Compounds 6, 7, 8 and 14 with short substituents showed neurotoxicity and caused death. In rat tests (p.o.), 9 was characterized by a high protection index (>13.3). AR affinity did not apparently correlate with the antiepileptic potency of the compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adenosine, a major constituent of nucleic acids, which consists of the purine base adenine linked to the ribose moiety, has important and diverse effects on many biological processes. Some of the physiological actions of adenosine include effects on heart rate and atrial contractility, vascular smooth muscle tone, release of neurotransmitters, lipolysis, renal function and white blood cell functions [1, 2].
Four adenosine receptor (AR) subtypes are known, A1, A2A, A2B and A3, all of which were cloned and pharmacologically characterized. The A2A and A2B receptors are positively coupled to adenylyl cyclase, while A1 and A3 adenosine receptors cause inhibition of cAMP formation. Adenosine acts via these different receptor subtypes, the affinity to which ranges from nanomolar (“high affinity” A1, 3–30 nM; A2A 1–20 nM) to micromolar (“low affinity” A2B, 5–20 μM; A3 > 1 μM) [3, 4]. These receptors belong to the large superfamily of G protein-coupled receptors [2].
Adenosine A1 receptors are ubiquitously expressed, e.g. in the central nervous system (CNS), especially in the brain, with high levels being expressed in many regions. The distribution of adenosine A2A receptors is wide ranging but restricted, including lymphocytes, platelets, brain striatum, vascular smooth muscle and endothelium [2].
The prototypical antagonists of the A1 adenosine receptor are the xanthines: theophylline and caffeine. Natural xanthines are non-specific adenosine antagonists. They are not selective for any of the adenosine receptor subtypes and have low affinity for the A1 receptor. Due to their CNS-stimulating effects, A1 adenosine receptor selective antagonists have been proposed as cognition enhancers for the treatment of dementias, such as Alzheimer’s disease. These receptors have been shown to be involved in sedative, antiseizure and anxiolytic effects. New potential indications are being discovered and investigated: in heart (for the treatment of cardiac arrhythmias and oedemas and as positive inotropic and cardiac protectants), in kidney (for oedemas and nephritis treatment), in lung (for asthma, oedema and lung protection) and in CNS (for depression, stress and coma) diseases. A1 AR antagonists are being investigated as antihypertensives and potassium-saving diuretics with kidney protective effects, for the treatment of depression and asthma and for the prevention of ischemia-induced injuries [5–7].
In the last years, numerous studies have confirmed the ability of A2A adenosine receptor antagonists to prevent neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases, ischemic brain damage and, recently, epilepsy and sensorimotor disorders (restless legs syndrome—RLS) [8–14]. Methylxanthines such as theophylline and caffeine have been known to enhance locomotor activity; however, these compounds are non-selective antagonists and have weak affinity for A2A AR.
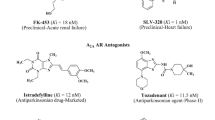
Our efforts were directed towards the development of new selective xanthine adenosine receptor ligands. Our main interest has focussed on the investigation of tricyclic xanthine derivatives [15–19]. The so far most active compounds are shown in Fig. 1. The most potent A1 AR ligands were found among 1,3-dipropyl-substituted benzylpyrimidopurinediones (I, II) [17], while A2A adenosine receptor ligands were 1,3-dimethyl-substituted aryl- (III, IV) [16] cycloalkyl- (V) [19] and phenalkylpyrimidopurinediones (VI) [18] (Fig. 1). Several of the most active ligands at adenosine A2A AR (e.g. III, IV and VI) [20] were demonstrated to exhibit antiparkinsonian effects. Among pyrimidopurinediones, compounds were found which displayed anticonvulsant properties—protective activity in subcutaneous pentylenetetrazole (ScMet) or in maximal electroshock (MES) and ScMet test; however, the mechanism of this action was not clear [16, 19].

Structures and adenosine receptor binding affinities of tricyclic xanthine derivatives (I–VI); Ki values are given in micromolar; h human, r rat
As a continuation of our search for potent adenosine A1 and especially A2A receptor ligands among cycloalkyl annelated xanthines, we have developed a new series of imidazo-, pyrimido- and diazepino[2,1-f]purinedione derivatives possessing aliphatic substituents (open congeners of cycloalkyl derivatives) in the annelated ring, e.g. alkyl, alkynyl and alkenyl chains. The compounds turned out to be selective A1 or A2A AR antagonists with moderate, submicromolar affinity as shown by radioligand binding studies at native rat receptors. Investigated compounds were examined also for A2B and A3 AR affinity at human recombinant AR. Additionally, the most active compounds were evaluated for affinity at human recombinant A1 and A2A AR affinity. Molecular modelling studies were performed to discuss affinity of the compounds through the docking onto the active sites of the A2A adenosine receptor model. Investigated compounds were examined for their anticonvulsant activity as well.
Results and discussion
Chemistry
The synthesis of tricyclic imidazo-, pyrimido- and diazepinopurinediones (Table 1) was accomplished as shown in Fig. 2. As starting material for 1,3-dimethyl-imidazo[2,1-f]purinediones (33, 34), 7-(2-bromoethyl)-8-bromotheophylline (1) was used, the preparation of which had been described by Caccacae [21]. In our laboratory, a modified procedure was developed using a two-phase catalysis method [16]. The other starting compounds, 7-(3-chloropropyl)-8-bromotheophylline (2) for 1,3-dimethyl-pyrimido[2,1-f]purinediones (5–24) and 7-(4-bromobutyl)-8-bromo-theophylline (3) for 1,3-dimethyl-diazepino[2,1-f]purinediones (25–32), were obtained as previously described [22, 23]. 1,3-Dipropyl-7-(3-chloropropyl)-8-bromoxanthine (4) [16, 17] was used as starting material for the synthesis of pyrimido[2,1-f]purinedione (35). The cyclization reaction with amines possessing straight, branched or unsaturated chains was carried out under various conditions (excess of amine, solvent and different reaction time). The synthesis of compounds 8, 9 and 23 was described previously [24], but their structures had been confirmed only by UV spectra, and pharmacological tests had not been performed. Unsubstituted compounds 5, 25 and 33 were previously synthesized in our group [25, 26] and were now subjected to pharmacological tests to compare them with substituted derivatives. Compound 22 with an ethenyl moiety was obtained by dehydrohalogenation of 9-(2-bromoethyl)-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purinedione (21) [27] (formed from the appropriate hydroxy ethyl derivative 20 [28]) with ethanolic potassium hydroxide (Fig. 2). The structures of the synthesized compounds were confirmed by UV, IR and 1H NMR spectra: UV spectra showed a bathochromic shift typical for 8-aminoxanthine derivatives with λ max of about 300 nm [29]. The IR absorption bands were typical of xanthine derivatives [30], and in the 1H NMR spectra, the expected chemical shifts were observed. All compounds were purified by recrystallization.

Synthesis of substituted imidazo-, pyrimido- and diazepino[2,1-f]purinediones. Reagents and conditions: i appropriate primary amines, solvent (EtOH, n-PrOH, n-BuOH, DMF or without solvent), reflux; ii aminoethanol, reflux; iii PBr3, CHCl3, reflux; iv KOH, EtOH, reflux
X-ray structure analysis
Among derivatives with various alkyl substituents at N(9) or (10) (Table 1), monocrystals of three of them—10, 11 and 30—suitable for X-ray structure analysis could be selected. The structures represent pyrimido- (10 and 11) and diazepino- (30) purinediones with N1,N3-dimethyl substituents at the xanthine nitrogen atoms. They feature linear (pentyl and hexyl) or branched (iso-pentyl) substituents at N(9) or (10). The description and discussion of 10, 11 and 30 spatial properties are attached in the supplementary section.
Pharmacology
All compounds were tested in vitro in radioligand binding assays for affinity to A1 and A2A adenosine receptors in rat cortical membrane and rat striatal membrane preparations, respectively. The results are presented in Table 2. The non-selective AR ligand caffeine, selective A2A AR antagonist Preladenant (SCH420814) [31] and selective A1 AR antagonist PSB-36 [31, 32] were included for comparison. Examined compounds were additionally tested for affinity at human recombinant A1, A2A, A2B and A3 receptors stably expressed in Chinese hamster ovary (CHO) cells (Table 3). The following radioligands were used: [3H]2-chloro-N6-cyclopentyladenosine ([3H]CCPA) for A1, [3H]1-propargyl-3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)xanthine ([3H]MSX-2) [33, 34] for A2A, [3H]PSB-603 [3H]8-(4-(4-(4-chlorophenyl)piperazine-1-sulfonyl)phenyl)-1-propylxanthine [35] for A2B and [3H]2-phenyl-8-ethyl-4-methyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purine-5-one ([3H]PSB-11) [36] for A3 binding studies.
Tricyclic xanthine derivatives, previously obtained in our laboratory, showed anticonvulsant activity [15–18]. Therefore, compounds 5–35 were evaluated in vivo as potential anticonvulsants by the ADP (Antiepileptic Drug Development Program of the National Institute of Neurological Disorders and Stroke NINDS) according to the Antiepileptic Screening Project. Compounds were injected intraperitoneally as a suspension in 0.5 % methylcellulose into the mice and evaluated in the preliminary screenings with at least three dose levels (30, 100 and 300 mg/kg at 0.5- and 4-h time periods). Phase I of the evaluation included three tests: MES, ScMet seizure tests and the rotorod test for neurological toxicity (TOX). The tests were described in detail by Stables and Kupferberg [37–39].
The MES test is a model for generalized tonic–clonic seizures and identifies compounds which prevent seizure spread. The ScMet is a model to test compounds that raise seizure threshold. The minimal motor impairment was measured by the rotorod test. The results are given in Table 4.
Some compounds (9, 11, 13, 15, 16, 22 and 30) were also administered orally to rats and examined in the MES, ScMet screen and TOX test (Table 5). Compound 9 was tested also in the hippocampal binding model in rats to evaluate its ability to prevent or modify both the expression and acquisition of focal seizures [39] (Table 6). For two compounds (9 and 13), a quantitative test in mice (i.p.) was made (ED50 and TD50 determination). The results of these experiments compared with literature data for valproate [40] are collected in Table 7.
In vitro tests
Synthesized tricyclic xanthine derivatives displayed affinity towards both A1 and A2A ARs in radioligand binding studies performed at rat brain membranes (Table 2). The most active but not selective A1 AR ligand was 1,3-dipropylpyrimidopurinedione (35). In the group of diazepinopurinediones were found compounds which displayed submicromolar affinity towards adenosine A2A receptors. Diazepinopurinedione 28 (K i = 0.28 μM) showed moderate A2A AR selectivity (about sixfold). A2A AR ligands with submicromolar affinity were also found among pyrimidopurinediones: compounds 11 and 13 displayed K i values of 0.82 and 0.87 μM, respectively. N-substituted derivatives displayed higher affinity than unsubstituted analogues (5, 25 and 33). Elongation of the straight N-alkyl substituents led to an increase in both A1 and A2A AR affinities in the group of diazepinopurinediones. For pyrimidopurinediones, this tendency was not so obvious; it was only observed at A1 AR. In the case of A2A AR, the most active ones were compounds with 6 (comp. 11) and 3 (comp. 8) atom chains. Branched N-alkyl, especially α-branched substituents, generally were not favourable for AR affinity. In case of A1 AR ligands, such modifications led to a loss of affinity (compounds 12, 14, 15, 16 and 18) or its decrease (compound 19). A2A AR ligands were also sensitive to these modifications; however, compounds with branched N-alkyl substituents still showed micromolar affinity, and β-branched derivative 13 was the most active one with submicromolar affinity. Enlargement of the annelated ring was favourable for both A1 and A2A AR activities. Diazepinopurinediones with the same N-alkyl substituents as pyrimidopurinediones were up to sixfold more potent (compounds 9 and 28, and 8 and 27). An exception here was the N-isopropyl derivative 34 which possesses a five-membered annelated ring; however, there are only two members of this group for comparison. Introduction of N1,N3-dipropyl substituents into the xanthine core definitely has favourable influence on A1 AR affinity of N-alkylpyrimidopurinediones: compound 35 was 15-fold more potent than its N1,N3-dimethyl analogue 9. Ligands with unsaturated N-substituents (22–24) displayed only moderate A1 and A2A AR affinity.
Selected compounds are mainly those that were the most active ones at the A2A AR (11, 13, 22, 27, 28 and 35) and were also tested for affinity to human A1 and A2A receptors (Table 3). Generally, the affinity at human recombinant A1 as well as A2A receptors was worse than that for native rat receptors (in the range of 1.5—threefold lower affinity). Only in the case of isobutylpyrimidopurinedione 13, the affinity for the human A1 AR was better than that for the rat receptor and almost equal for A2A AR.
Affinity to the human A2B and A3 receptors of all of the investigated compounds was very weak; only the dipropyl derivative 35 showed micromolar affinity at A2B AR and submicromolar affinity to the human A3 AR. Only one compound with a double-branched, long chain (19) from the group of dimethyl derivatives showed affinity for A3 AR in the micromolar range.
Two of the most potent rat and human A2A receptor ligands (compounds 28 and 35) were investigated for their functional properties using cAMP accumulation assay. They were investigated for their potency to inhibit NECA-induced cAMP accumulation in CHO cells expressing the human A2A receptor (Fig. 3). The compounds clearly behaved as competitive antagonists as the concentration–response curve of NECA was shifted to the right in a parallel fashion in their presence. K b values determined in living CHO cells expressing the human adenosine A2A receptor were well in accordance with K i values determined in radioligand binding studies at membrane preparations of the same cell line. Owing to the structural similarity of all compounds in this series, we suppose that they are all antagonists.

cAMP accumulation studies in CHO cells expressing the human adenosine A2A receptor. The dose–response curves for the NECA-induced stimulation of cAMP accumulation were generated with NECA in the absence or in the presence of two different concentrations of 28 (a) or 35 (b). Graphs from two independent experiments performed in duplicates with mean values ± SEM are shown. Both investigated compounds shifted the concentration–response curve for NECA in a parallel manner to the right, indicating competitive antagonism. Apparent K b values were as follows: 1,510 ± 20 nM (28) and 1,210 ± 130 nM (35)
Molecular modelling studies
In our previous molecular modelling studies on A1 and A2A adenosine receptors, the comparison of rhodopsin- and β2-adrenergic-based homology models through the docking studies was performed [41]. Since that time, a few X-ray structures of A2A AR in complex with various ligands have been reported in the Protein Data Bank [42, 43], among which those co-crystallized with xanthines: XAC (PDB code: 3REY) and caffeine (PDB code: 3RFM) are of great importance for our research. Analysis of the ligand binding mode, observed in the crystals 3REY and 3RFM, compared to the binding mode of an inverse agonist ZM241385 bound to A2A AR (PDB codes: 3PWH [42] and 3EML [43]) indicates flexibility of some amino acid side chains within the receptor binding cleft. In particular, the side chain of Asn253 (6.55), described as a crucial residue for ligand binding [44–46], in 3REY is rotated relative to other structures, e.g. 3RFM with caffeine as a ligand. Nevertheless, in both crystal structures of A2A AR co-crystallized with xanthines, the terminal amino group of Asn253 (6.55) forms the hydrogen bond with the same carbonyl group present in purinedione core of ligands.
Due to the structure and the size of synthesized molecules, A2A AR-XAC (3REY) crystal seems to be the best choice as a template for docking. To validate the utilized molecular docking methods, XAC ligand was redocked to its X-ray structure of A2A receptor. In case of simulation without any constraints, the obtained highest ranked pose was distinct from the one in the crystal; superposition of both conformations gave a high RMSD value of 9.68 Å. In this pose, an imidazol nitrogen atom of the ligand plays a role of an H-bond acceptor involved in the contact with the distal amino group of Asn253 (6.55), while a phenyl ring is situated in the proximity of TM6 and ECL2. The docking was then repeated, setting the H-bond interaction between Asn253 (6.55) and one out of carbonyl groups of the ligand as a constraint. This time, the overlay with the experimental binding mode was better (Fig. 4, RMSD = 4.53 Å). The superposition of phenylpurinedione cores was almost perfect, giving an RMSD value of 0.42 Å, but calculated and experimental poses differ with orientation of a polar tail of XAC. However, in the crystal structure, the position of this flexible chain is not strictly fixed, as its electron density is not complete [42]. The protocol including constrained H-bond between a ligand and Asn253 (6.55) was chosen for further docking simulations.

Superposition of the XAC ligand from 3REY crystal structure (purple) and redocked pose of XAC (orange)
As an additional validation to the tested series of xanthines, the structure of caffeine was added and docked together with the rest of the compounds. Docking simulations of the set of all 30 compounds to both templates gave results that can be grouped in three clusters according to the calculated docking score values.
In the two first, highest ranked clusters, the obtained poses adopt a reflected or rotated orientation of the heterocyclic core compared to the XAC conformation from the crystal. In this position, in both cases, the carbonyl group C2 = O2 of the purinedione interacts with a side chain amino group of Asn253 (6.55), while C4 = O4 either corresponds to the 2-oxo fragment of XAC or points to the top of the receptor, between side chains of Glu169 and Leu267 (7.32).
However, it can be noticed that in both cases of published X-ray structures co-crystallized with xanthines, purinedione core interacts with Asn253 (6.55) in a similar way, with the same carbonyl oxygen atom (C4 = O4) of the ligand, even if Asn253 in this crystal acid adopts two different conformations of the side chain. Taking into account positions of both XAC and caffeine in the binding site, we can presume that the synthesized tricyclic derivatives of xanthine would bind into the A2A receptor in a similar mode (shown in Fig. 5a, b) despite lower docking scores. For this reason, the conformations belonging to the first two clusters were rejected.

a The model of compound 9 (orange) docked to 3REY crystal structure (chosen residues in green and XAC ligand in purple). b The model of compound 35 (orange) docked to 3REY crystal structure (chosen residues in green and XAC ligand in purple). c Superposition of XAC ligand (purple) and compound 9 (orange) in the binding site of A2A receptor
The third cluster is created by poses predicted for most of the compounds including caffeine (Fig. 5a, b). Although docking scores are lower than those for the two first sets, this pose is in agreement with the position of the ligand XAC in the crystal structure. The carbonyl group C4 = O4 interacts here with the terminal amino group of Asn253 (6.55), and the whole purinedione core of the modelled xanthines overlies the heterocyclic part of XAC with very good RMSD values (from 0.39 to 0.93 Å, for caffeine 0.58 Å), making π–π stacking with the aromatic part of Phe168. Two N-methyl (or N-propyl) substituents point towards the bottom of the binding site, likewise in case of XAC. The third annelated ring is situated between Phe168 and Glu169 of ECL2 from one side and Ile274 (7.39) and Met270 (7.35) from the other. The alkyl chain is buried in a similar pocket of the binding site to the phenoxy tail of XAC, in the long narrow cleft limited by residues from top fragments of TM2 (Ala63 (2.61), Ile66 (2.64) and Ser67 (2.65)) and TM7 (Leu267 (7.32) and Tyr271 (7.36)) as well as Tyr9 (1.35) (Fig. 5c).
The compounds 25 and 33, unsubstituted in the annelated ring, were not successfully docked in this position, while long alkyl chains in this position, branched or not, are easily adapted in the groove created by TM2 and TM7. Similarly, longer propyl fragments at 1- and 3-positions of 35 make hydrophobic contacts with residues Leu85 (3.33), Leu249 (6.51) and Met177 (5.38) from one side and Ile66 (2.64), Ala81 (3.29) and Val84 (3.32) from the other, increasing affinity to the receptor relative to short methyl substituents (35 vs. 9). Compounds 24 and 30 were not found among poses from this cluster either—the most probable explanation is that the linear structure of the triple bond in 24 or a double-branched chain of 30 causes a steric clash with residues forming the cleft.
The performed docking experiments did not explain in detail the correlation between structure and A2A AR affinity of the obtained xanthines. Nevertheless, it can be stated that both the third annelated ring and alkyl chains as N-substituents fit well to the A2A binding pocket, forming additional interactions, and their presence has a big influence on the affinity to the adenosine receptors compared to caffeine—non-selective weak A2A AR antagonist. The similar effect is observed for two N-propyl substituents and the phenyl ring of XAC, very potent adenosine receptor antagonist (35, Fig. 5b).
In vivo tests
Unsubstituted pyrimido- (5) and diazepinopurinedione (25) did not show protective activity in both electric and chemical seizures (Table 4). Introduction of N-alkyl substituents resulted in anticonvulsant activity in the pyrimidopurinediones, whereas these modifications did not affect in the same way the group of diazepinopurinediones as only compounds 31 and 32 showed protective activity in a dose of 300 mg/kg, but after 4 h caused death of tested animals. Probably, enlargement of the annelated ring in the xanthine derivatives is not favourable for anticonvulsant activity. Unsaturated substituents introduced into the tricyclic xanthine derivatives (23 and 24) were also disadvantageous. Generally, N-alkyl derivatives showed good protective activity in mice (i.p.), especially in ScMet test in short time (0.5 h). The best compound was N-hexylpyrimidopurinedione 11, with 100 % protection at a dose of 100 mg/kg with no symptoms of neurotoxicity. The other two compounds with long lipophilic chains 18 and 19 also showed good protection: derivative 19 was active in both tests, and ligand 18 displayed protection in ScMet test. Both compounds did not show neurotoxicity. The N-butyl and N-pentyl derivatives 9 and 10 displayed protective properties in MES and ScMet tests but at the same time showed neurotoxicity. Compounds with short chains (6, 7, 8 and 14) caused death of test animals. The most toxic substance was N-propargylpyrimidopurinedione 24, causing death following clonic seizures and tonic extension.
The most active compounds in mice were also examined for activity in rats after oral administration, showing higher seizure protection (Table 5). The best compound was the butyl derivative 9, showing 100 % protection in ScMet test in the dose of 50 mg/kg in short time (0.25 and 0.5 h) and 50 % protection in dose of 10 mg/kg after 1 2 and 4 h. Ligand 16 was active in MES test at 30 mg/kg and in ScMet test at 50 mg/kg. No symptoms of neurotoxicity were observed in both tested compounds. Ligand 9 was also tested in a hippocampal kindled seizure screen (Table 6). Results suggest the ability of 9 to prevent or modify fully kindled seizures. The N-butyl and N-isobutyl derivatives 9 and 13 were advanced for phase II of evaluation for quantification of activities (ED50 and TD50) against MES- and ScMet-induced seizures in mice (i.p.) (Table 7). These pharmacological parameters were compared with data for valproate [40, 47]. Both tested substances were characterized by a PIScMet higher than that of valproate, but a lower PIMES.
The anticonvulsant activity of the examined compounds was analysed for correlation with AR affinity. Some coincidence of adenosine A2A affinity and anticonvulsant activity was observed. Compounds showing anticonvulsant activity in ScMet test (9, 11, 13, 14, 15, 16 and 18) were among the pyrimidinepurinedione derivatives showing affinity towards A2A AR. However, there were also compounds showing anticonvulsant activity but were devoid of A2A AR affinity. The most active A2A AR ligand, the diazepine derivative 28 and the dipropyl derivative 35 were not active as anticonvulsants. Comparing in vitro and in vivo activities of the studied tricyclic xanthine derivatives, there is no clear correlation between A1/A2A AR affinity and anticonvulsant activity of the investigated ligands. So our previous observation that A2A selectivity of AR antagonists may be important for anticonvulsant activity has not been confirmed in the present study [19].
Conclusions
Tricyclic pyrimido- and diazepinopurinediones are moderately potent A1 and A2A AR antagonists. Enlargement of the annelated ring caused an increase in affinity but not selectivity. Pyrimidopurinodiones showed anticonvulsant activity in mice (i.p.) and rats (p.o.), but lower homologs were toxic. Enlargement of the annelated ring was unprofitable for anticonvulsant activity and led practically to a lack of the activity. Although there is no apparent correlation between anticonvulsant activity and adenosine receptor affinity, it seems that a lipophilic substituent is necessary for both activities.
Abbreviations
- ADP:
-
Antiepileptic Drug Development Program
- ARs:
-
Adenosine receptors
- CHO:
-
Chinese hamster ovary
- CNS:
-
Central nervous system
- DMF:
-
Dimethylformamide
- DMSO:
-
Dimethylsulfoxide
- ([3H]CCPA):
-
[3H]2-chloro-N6-cyclopentyladenosine
- ([3H]PSB-603):
-
[3H]8-(4-(4-(4-chlorophenyl)piperazine-1-sulfonyl)phenyl)-1-propylxanthine
- ([3H]PSB-11):
-
[3H]2-phenyl-8-ethyl-4-methyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purine-5-one
- ([3H]MSX-2):
-
[3H]1-propargyl-3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)xanthine)
- IR:
-
Infrared
- MES:
-
Maximal electroshock
- NINDS:
-
National Institute of Neurological Disorders and Stroke
- NMR:
-
Nuclear magnetic resonance
- PI:
-
Protection index
- PDB:
-
Protein data bank
- PSB-36:
-
1-Butyl-3-hydroxypropyl-8-noradamantylxanthine
- RMSD:
-
Root mean square deviation
- ScMet:
-
Subcutaneous pentylenetetrazol
- TLC:
-
Thin layer chromatography
- UV:
-
Ultra-violet
- XAC:
-
Xanthine amine congener
References
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63:1–34
Müller CE, Jacobson KA (2011) Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta 1808:1290–1308
Trincavelli ML, Daniele S, Martini C (2010) Adenosine receptors: what we know and what we are learning. Curr Top Med Chem 10:860–877
Yuzlenko O, Kieć-Kononowicz K (2006) Potent adenosine A1 and A2A receptors antagonists: recent developments. Curr Med Chem 13:3609–3625
Müller CE (2002) P2-pyrimidinergic receptors and their ligands. Curr Pharm Des 8:2353–2369
Brunschweiger A, Müller CE (2006) P2 receptors activated by uracil nucleotides—an update. Curr Med Chem 13:289–312
Schenone S, Brullo C, Musumeci F, Bruno O, Botta M (2010) A1 receptors ligands: past, present and future trends. Curr Top Med Chem 10:878–901
Cristalli G, Müller CE, Volpini R (2009) Recent developments in adenosine A2A receptor ligands. Handb Exp Pharmacol 193:59–98
Manera C, Saccomanni G (2010) A2A receptor ligands: past, present and future trends. Curr Top Med Chem 10:902–922
Yu L, Shen H-Y, Coelho JE, Araújo IM, Huang Q-Y, Day Y-J, Rebola N, Canas PM, Rapp EK, Ferrara J, Taylor D, Müller CE, Linden J, Cunha RA, Chen J-F (2008) Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol 63:338–346
Salamone JD, Betz AJ, Ishiwari K, Felsted J, Madson L, Mirante B, Clark K, Font L, Korbey S, Sager TN, Hockemeyer J, Müller CE (2008) Tremorolytic effects of adenosine A2A antagonists: implications for parkinsonism. Front Biosci 13:3594–3605
Ishiwari K, Madson LJ, Farrar AM, Mingote SM, Valenta JP, DiGianvittorio MD, Frank LE, Correa M, Hockemeyer J, Müller CE, Salamone JD (2007) Injections of the selective adenosine A2A antagonist MSX-3 into the nucleus accumbens core attenuate the locomotor suppression induced by haloperidol in rats. Behav Brain Res 178:190–199
Müller CE, Ferré S (2007) Blocking striatal adenosine A2A receptors: a new strategy for basal ganglia disorders. Recent Pat CNS Drug Discov 2:1–21
Müller CE, Jacobson KA (2011) Xanthines as adenosine receptor antagonists. In: Fredholm BB (ed) Methylxanthines handbook of experimental pharmacology. Springer, Heidelberg, pp 151–199
Kieć-Kononowicz K, Drabczyńska A, Pękala E, Michalak B, Müller CE, Schumacher B, Karolak-Wojciechowska J, Duddeck H, Rockitt S, Wartchow R (2001) New developments in A1 and A2 adenosine receptor antagonists. Pure Appl Chem 73:1411–1420
Drabczyńska A, Müller CE, Lacher SK, Schumacher B, Karolak-Wojciechowska J, Nasal A, Kawczak P, Yuzlenko O, Kieć-Kononowicz K (2006) Synthesis and biological activity of tricyclic aryloimidazo-, pyrimido-, and diazepinopurinediones. Bioorg Med Chem 14:7258–7281
Drabczyńska A, Müller CE, Karolak-Wojciechowska J, Schumacher B, Schiedel A, Yuzlenko O, Kieć-Kononowicz K (2007) N9-benzyl-substituted 1,3-dimethyl- and 1,3-dipropyl-pyrimido[2,1-f]purinediones: synthesis and structure–activity relationships at adenosine A1 and A2A receptors. Bioorg Med Chem 15:5003–5017
Drabczyńska A, Müller CE, Schiedel A, Schumacher B, Karolak-Wojciechowska J, Fruziński A, Zobnina W, Yuzlenko O, Kieć-Kononowicz K (2007) Phenylethyl-substituted pyrimido[2,1-f]purinediones and related compounds: structure–activity relationships as adenosine A1 and A2A receptor ligands. Bioorg Med Chem 15:6956–6974
Drabczyńska A, Yuzlenko O, Köse M, Paskaleva M, Schiedel AC, Karolak-Wojciechowska J, Handzlik J, Karcz T, Kuder K, Müller CE, Kieć-Kononowicz K (2011) Synthesis and biological activity of tricyclic cycloalkylimidazo-, pyrimido- and diazepinopurinediones. Eur J Med Chem 46:3590–3607
Drabczyńska A, Zygmunt M, Sapa J, Filipek B, Müller CE, Kieć-Kononowicz K (2011) Antiparkinsonian effects of novel adenosine A2A receptor antagonists. Arch Pharm 1:20–27
Cacace F, Masironi R (1956) Derivati della 8-mercapto-teofillina: sintesi di una 2′,3′-tiazolidino-7,8-teofillina. Ann Chim (Roma) 46:806–812
Rockitt S, Wartchow R, Duddeck H, Drabczyńska A, Kieć-Kononowicz K (2001) Modes of xanthine complexation to dirhodium tetrakis [(R)-a-methoxy-a-(trifluoromethyl)-phenylacetate] in solution and in the solid state. Z Naturforsch 56b:319–324
Pawłowski M, Drabczyńska A, Gorczyca M, Malec D, Modzelewski J (1994) Synthesis and pharmacological screening of novel 10-substituted diazepino-[2,1-f]-purines. Acta Pol Pharm 51:385–391
Eckstein M, Łosoń W (1968) A search of new drugs in the group of xanthine derivatives. Dissert Pharm Pharmaceut 20:35–41
Eckstein M, Drabczyńska A (1973) A search for new drugs in the group of xanthine derivatives. XXXV. A new method of synthesis of 6,7,8,9-tetrahydropyrimido-[2,1-f]-purine-2,4-(1H,3H)-dione system. Pol J Pharmacol Pharm 25:171–173
Eckstein M, Drabczyńska A (1979) A new method for the anellation of 5-, 6-, and 7-membered rings containing two N-atoms at the 7,8 position of the xanthine system. Synthesis 8:581–583
Pawłowski M, Drabczyńska A, Gorczyca M, Malec D, Modzelewski J (1991) Synthesis and preliminary pharmacological assessment of novel 9-substituted pyrimidino-[2,1-f]-purines. Pol J Pharmacol Pharm 43:61–70
Eckstein M (1962) A search for new drugs in the group of xanthine derivatives. XVI. Reactions of 7-halogenalkylderivatives of 8-bromo- and 8-chloro-theophylline with amines. Dissert Pharm 14:435–441
Bergmann F, Dickstein S (1955) The relationship between spectral shifts and structural changes in uric acids and related compounds. J Am Chem Soc 77:691–694
Rybár A, Antoš K (1970) 1,3,7-Trisubstituted 8-isothiocyanatomethylxanthines. Coll Czech Chem Commun 35:1415–1419
Jacobson KA, Balasubramanian R, Deflorian F, Gao Z-G (2012) G-protein-coupled adenosine (P1) and P2Y receptors: ligand design and receptor interactions. Purinergic Signal 8:419–436
Weyler S, Fülle F, Diekmann M, Schumacher B, Hinz S, Klotz K-N, Müller CE E (2006) Improving potency, selectivity, and water-solubility of adenosine A1 receptor antagonists: xanthines modified at position 3 and related pyrimido[1,2,3-cd]purinediones. ChemMedChem 1:891–902
Müller CE, Maurinsh J, Sauer R (2000) Binding of [3H]MSX-2 (3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine) to rat striatal membranes—a new, selective antagonist radioligand for A2A adenosine receptors. Eur J Pharm Sci 10:259–265
Klotz K-N, Lohse MJ, Schwabe U, Cristalli G, Vittori S, Grifantini M (1989) 2-Chloro-N6-[3H]cyclopentyladenosine ([3H]CCPA)—a high affinity agonist radioligand for A1 adenosine receptors. Naunyn Schmiedeberg’s Arch Pharmacol 340:679–683
Borrmann T, Hinz S, Bertarelli DCG, Li W, Florin NC, Scheiff AB, Müller CE (2009) 1-Alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J Med Chem 52:3994–4006
Müller CE, Diekmann M, Thorand M, Ozola V (2002) [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 adenosine receptors. Bioorg Med Chem Lett 12:501–503
Porter RJ, Cereghino JJ, Gladding GD, Kupferberg HJ, Scoville B, White BG (1984) Antiepileptic Drug Development Program. Clev Clin 51:293–305
Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA (1978) Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia 19:409–428
Stables JP, Kupferberg KJ (1997) In: Avanzini G, Tanganelli P, Avoli M (eds) Molecular and cellular targets for antiepileptic drugs. John Libbey, London, pp 191–198
Mulzac D, Scott KR (1993) Profile of anticonvulsant activity and minimal toxicity of methyl 4-[(p-chlorophenyl)amino]-6-methyl-2-oxo-cyclohex-3-en-1-oate and some prototype antiepileptic drugs in mice and rats. Epilepsia 34:1141–1149
Yuzlenko O, Kieć-Kononowicz K (2008) Molecular modeling of A1 and A2A adenosine receptors: comparison of rhodopsin- and β2-adrenergic-based homology models through the docking studies. J Comput Chem 30:14–32
Dore AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH (2011) Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19:1283–1293
Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217
Kim SK, Gao ZG, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, Jacobson KA (2003) Modeling the adenosine receptors: comparison of the binding domains of A2A agonists and antagonists. J Med Chem 46:4847–4859
Ivanov AA, Barak D, Jacobson KA (2009) Evaluation of homology modeling of G-protein-coupled receptors in light of the A2A adenosine receptor crystallographic structure. J Med Chem 52:3284–3292
Dal Ben D, Lambertucci C, Marucci G, Volpini R, Cristalli G (2010) Adenosine receptor modeling: what does the A2A crystal structure tell us? Curr Top Med Chem 10:993–1018
White HS, Johnson M, Wolf HH, Kupferberg HJ (1995) The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital J Sci 16:73–77
Hockemeyer J, Burbiel JC, Müller CE (2004) Multigram-scale syntheses, stability, and photoreactions of A2A adenosine receptor antagonists with 8-styrylxanthine structure: potential drugs for Parkinson’s disease. J Org Chem 69:3308–3318
Ozola V, Thorand M, Diekmann M, Qurishi R, Schumacher B, Jacobson KA, Müller CE (2003) 2-Phenylimidazo[2,1-i]purin-5-ones: structure–activity relationships and characterization of potent and selective inverse agonists at human A3 adenosine receptors. Bioorg Med Chem 11:347–356
Bulicz J, Bertarelli DCG, Baumert D, Fülle F, Müller CE, Heber D (2006) Synthesis and pharmacology of pyrido[2,3-d]pyrimidinediones bearing polar substituents as adenosine receptor antagonists. Bioorg Med Chem 14:2837–2849
Klotz K-N, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ (1998) Comparative pharmacology of human adenosine receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn Schmiedeberg’s Arch Pharmacol 357:1–9
Yan L, Bertarelli DCG, Hayallah AM, Meyer H, Klotz K-N, Müller CE (2006) A new synthesis of sulfonamides by aminolysis of p-nitrophenylsulfonates yielding potent and selective adenosine A2B receptor antagonists. J Med Chem 49:4384–4391
Bertarelli DCG, Diekmann M, Hayallah AM, Rüsing D, Iqbal J, Preiss B, Verspohl EJ, Müller CE (2006) Characterization of human and rodent native and recombinant adenosine A(2B) receptors by radioligand binding studies. Purinergic Signal 2:559–571
Hayallah AM, Sandoval-Ramìrez J, Reith U, Schobert U, Preiss B, Schumacher B, Daly JW, Müller CE (2002) 1,8-Disubstituted xanthine derivatives: synthesis of potent A2B-selective adenosine receptor antagonists. J Med Chem 45:1500–1510
Sauer R, Maurinsh J, Reith U, Fülle F, Klotz K-N, Müller CE (2000) Water-soluble phosphate prodrugs of 1-propargyl-8-styrylxanthine derivatives, A2A-selective adenosine receptor antagonists. J Med Chem 43:440–448
Maestro v.9.0, MacroModel v.9.7, Glide v.5.5; Schrödinger, LLC (2009) New York
PyMOL Molecular Graphics System v.0.99, DeLano Scientific
Acknowledgments
The authors wish to acknowledge Professor James P. Stables for providing pharmacological data through the Antiepileptic Drug Development Program National Institutes of Health. Financial support of this research by the Ministry of Science and Higher Education and National Science Centre, Poland (grant No. 4 P05F 014 18, grant No. NN 405 297 836 and grant DEC-2012/04/M/NZ4/00219) and by the BMBF, Germany (01EW0911) in the frame of ERA-NET NEURON is gratefully acknowledged. We would like to thank Mrs. Inge Renner and Katharina Thiemke for the technical assistance in the biological experiments.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Experimental section
Chemistry
Melting points were determined on a MEL-TEMP II apparatus, and they are uncorrected. IR spectra were taken as KBr discs on an FT Jasco IR 410 spectrometer. 1H-NMR spectra (in CDCl3) for compounds 10, 13, 14 and 22 were obtained at ambient temperature with a Bruker VM 250, for other compounds with a Varian Mercury 300-MHz spectrometer, using signal of undeuterated solvent as an internal standard. UV spectra were recorded on a Jasco UV/Vis V-530 apparatus in 10−5 mole/L in methanol. Elemental analyses (CHN) were performed on an Elementar Vario-EL III apparatus and agreed with theoretical values within ±0.4 %. TLC data were obtained with Merck silica gel 60 F254 aluminium sheets with benzene: acetone (7:3) as developing system. Spots were detected under UV light. Syntheses of compounds 5, 25 and 33 were described elsewhere [25, 26].
Synthetic procedures and analytical data of tricyclic alkylimidazo-, pyrimido- and diazepinopurinediones
General procedure for the synthesis of 9-substituted 1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (6–19, 23, 24). A mixture of (0.66 g, 2 mmol) of 7-(3-chloropropyl)-8-bromotheophylline (2) and amine derivative (4–66 mmol) was heated under reflux in the appropriate solvent (ethanol, propanol, butanol and DMF) or without solvent for 5 to 15 h. After cooling, the precipitate was separated. If the crude compound included a little amount of unreacted 7-(3-chloropropyl)-8-bromotheophylline (monitoring by TLC), it was purified by dissolving in 10 % HCl, alkalizing to pH 8 with 10 % NaOH and washing with water. If DMF was used as a reaction medium, the product was precipitated by adding water to the reaction mixture. Compound 17 did not crystallize after cooling and was obtained by removing the excess of 2-propynylamine by steam distillation and cooling the water solution. All compounds were purified by crystallization.
1,3,9-Trimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (6)
Reaction medium, ethanol; time of heating under reflux, 15 h; excess of amine, 33. Crystallization with ethanol gave pure compound 6. Yield 47 %, mp 251–252 °C; TLC: R f: 0.31; Anal. Calcd for C11H15O2N5 (6): C, 53.00; H, 6.07; N, 28.10. Found: C, 52.77; H, 5.91; N, 27.82. 1H-NMR δ (ppm): 2.12–2.20 (m, 2H, N5CH2CH2CH2N9), 3.12 (s, 3H, CH3), 3.29–3.33 (m, 3H + 2H, N3CH3 + CH2N9), 3.48 (s, 3H, N1CH3), 4.19 (t, J = 6.04Hz, 2H, N5CH2). IR KBr (cm−1): 2,950–2,871—CH2, 1,699—CO (2), 1,655 (4), 744—CH2. UV λ max, logε: 300.5, 4.15.
9-Ethyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (7)
Reaction medium, ethanol; time of heating under reflux, 5 h; excess of amine, 29. Crystallization with ethanol (30 %) gave pure compound 7. Yield 34 %, mp 215–216 °C; TLC: R f: 0.49; Anal. Calcd for C12H17O2N5 (7): C, 54.74; H, 6.52; N, 26.60. Found: C, 55.00; H, 6.52; N, 26.25. 1H-NMR δ (ppm): 21.22 (t, J = 7.14 Hz, 3H, CH2CH3), 2.13–2.20 (m, 2H, N5CH2CH2CH2N9), 3.34–3.37 (m, 3H + 2H, N3CH3 + CH2N9), 3.50 (s, 3H, N1CH3), 3.54–3.63 (m, 2H, CH2CH3), 4.20 (t, J = 6.04 Hz, 2H, N5CH2). IR KBr (cm−1): 2,967–2,872—CH2, 1,697—CO (2), 1,666 (4), 784—CH2. UV λ max, logε: 301.5, 4.28.
1,3-Dimethyl-9-propyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (8)
Reaction medium, ethanol; time of heating under reflux, 5 h; excess of amine, 12. Crystallization with ethanol (30 %) gave pure compound 8. Yield 60 %, mp 188–190 °C; TLC: R f: 0.52; Anal. Calcd for C13H19O2N5 (8): C, 56.31; H, 6.91; N, 21.25. Found: C, 56.25; H, 6.97; N, 24.96. 1H-NMR δ (ppm): 0.95 (t, J = 7.42 Hz, 3H, CH2CH 3), 1.60–1.83 (m, 2H, CH2CH2CH3), 2.11–2.19 (m, 2H, N5CH2CH2CH2N9), 3.34–3.46 (m, 3H + 2H, N3CH3 + CH2N9), 3.49–3.59 (m, 2H + 3H, CH2CH3 + N1CH3), 4.21 (t, J = 5.91 Hz, 2H, N5CH2). IR KBr (cm−1): 2,958–2,871—CH2, 1,697—CO (2), 1,664 (4), 784—CH2. UV λ max, logε: 302, 4.34.
9-Butyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (9)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 15. Crystallization with ethanol gave pure compound 9. Yield 81 %, mp 174–176 °C; TLC: R f: 0.37; Anal. Calcd for C14H21O2N5 (9): C, 57.71; H, 7.27; N, 24.04. Found: C, 57.63; H, 7.08; N, 23.84. 1H-NMR δ (ppm): 0.95 (t, J = 7.28 Hz, 3H, (CH2)3CH3),1.31–1.38 (m, 2H, CH 2CH3), 1.54–1.62 (m, 2H, CH2CH2CH3), 2.09–2.17 (m, 2H, N5CH2CH2CH2N9), 3.31–3.34 (m, 3H + 2H, N3CH3 + CH2N9), 3.48–3.53 (m, 2H + 3H, CH2CH3 + N1CH3), 4.19 (t, J = 6.04 Hz, 2H, N5CH2). IR KBr (cm−1): 2,958–2,873—CH2, 1,702—CO (2), 1,666 (4), 784—CH2. UV λ max, logε: 302, 4.25.
1,3-Dimethyl-9-pentyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (10)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 20. Crystallization with ethanol (80 %) gave pure compound 10. Yield 59 %, mp 183–185 °C; TLC: R f: 0.43; Anal. Calcd for C15H23O2N5 (10): C, 58.99; H, 7.60; N, 22.93. Found: C, 58.76; H, 7.26; N, 23.08. 1H-NMR δ (ppm): 0.92 (t, J = 6.25 Hz, 3H, (CH2)4CH3), 1.27–1.65 (m, 6H, (CH2)3CH3), 2.10–2.19 (m, 2H, N-CH2CH2CH2N9), 3.33–3.37 (m, 3H + 2H, N3CH3 + CH2N9), 3.46–3.54 (m, 2H + 3H, CH2CH3 + N1CH3), 4.24 (t, J = 6.25 Hz, 2H, N5CH2). IR KBr (cm−1): 2,952–2,869—CH2, 1,699—CO (2), 1,662 (4), 784—CH2. UV λ max, logε: 301, 4.34.
9-Hexyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (11)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 12. Crystallization with ethanol gave pure compound 11. Yield 57 %, mp 149 °C; TLC: R f: 0.55; Anal. Calcd for C16H25O2N5 (11): C, 60.16; H, 7.89; N, 21.93. Found: C, 60.13; H, 3.94; N, 22.04. 1H-NMR δ (ppm): 0.90 (t, J = 5.77 Hz, 3H, (CH2)5CH3), 1.33–1.64 (m, 8H, (CH2)4CH3), 2.11–2.18 (m, 3H + 2H, N3CH3 + CH2N9), 3.49–3.53 (m, 2H + 3H, CH2CH3 + N1CH3), 4.21 (t, J = 6,04 Hz, 2H, N5CH2). IR KBr (cm−1): 2,952–2869—CH2, 1,698—CO (2), 1,664 (4), 742—CH2. UV λ max, logε: 301.5, 4.32.
9-Isopropyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (12)
Reaction medium, propanol; time of heating under reflux, 5 h; excess of amine, 12. Crystallization with ethanol (30 %) gave pure compound 12. Yield 65 %, mp 241–242 °C; TLC: R f: 0.48; Anal. Calcd for C13H19O2N5 (12): C, 56.31; H, 6.91; N, 25.25. Found: C, 56.10; H, 6.58; N, 25.05. 1H-NMR δ (ppm): 1.20 (d, J = 6.86 Hz, 6H, CH(CH3)2), 2.05–2.13 (m, 2H, N5CH2CH2CH2N9), 3.26 (s, 3H, N3CH3), 3.48 (s, 3H, N1CH3), 4.17 (t, J = 6.04 Hz, 3H, N1CH3), 4.62–4.71 (m, 1H, N9CH). IR KBr (cm−1): 2,967–2,848—CH2, 1,704—CO (2), 1,662 (4), 742—CH2. UV λ max, logε: 302, 4.29.
9-Isobutyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (13)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 25. Crystallization with ethanol (50 %) gave pure compound 13. Yield 68 %, mp 213–215 °C; TLC: R f: 0.44; Anal. Calcd for C14H21O2N5 (13): C, 57.71; H, 7.27; N, 24.04. Found: C, 58.05; H, 6.99; N, 23.79. 1H-NMR δ (ppm): 0.94 (d, J = 6.50 Hz, 6H, CH(CH3)2), 2.01–2.19 (m, 1H + 2H, CH(CH3)2 + N5CH2CH2CH2N9), 3.31–3.37 (m, 2H + 2H, CH2N9CH2), 3.39 (s, 3H, N3CH3), 3.50 (s, 3H, N1CH3), 4.22 (t, J = 6.00 Hz, 2H, N5CH2). IR KBr (cm−1): 2,964–2,844—CH2, 1,698—CO (2), 1,662 (4), 742—CH2. UV λ max, logε: 301.5, 4.34.
9-Sec-butyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (14)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 25. Crystallization with ethanol (50 %) gave pure compound 14. Yield 98 %, mp 188–190 °C; TLC: R f: 0.48; Anal. Calcd for C14H21O2N5 (14): C, 57.71; H, 7.27; N, 24.04. Found: C, 57.90; H, 7.43; N, 23.87. 1H-NMR δ (ppm): 0.90 (t, J = 7.38 Hz, 3H, CH2CH3), 1.19 (d, J = 6.60 Hz, 3H, =CHCH3), 2.06–2.16 (m, 2H, N5CH2CH2CH2N9), 3.22–3.27 (m, 2H, CH2N9), 3.37 (s, 3H, N3CH3), 3.50 (s, 3H, N1CH3), 4.18–4.24 (m, 2H, N5CH2), 4.39–4.48 (m, 1H, N1CH=). IR KBr (cm−1): 2,969–2875—CH2, 1,704—CO(2), 1,652 (4), 744—CH2. UV λ max, logε: 302.5, 4.26.
1,3-Dimethyl-9-(pentan-2-yl)-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (15)
Reaction medium, DMF; time of heating under reflux, 10 h; excess of amine, 4. Crystallization with ethanol/H2O gave pure compound 15. Yield 91 %, mp 143–144 °C; TLC: R f: 0.65; Anal. Calcd for C15H23O2N5 (15): C, 59.00; H, 7.60; N, 22.94. Found: C, 58.90; H, 7.57; N, 22.99. 1H-NMR δ (ppm): 0.93 (t, J = 7.29 Hz, 3H, CH2CH3), 1.17 (d, J = 6.88 Hz, 3H, CH3CH), 1.25–1.59 (m, 4H, CH2CH2CH3), 2.05–2.13 (m, 2H, N5CH2CH2CH2N9), 3.20–3.25 (m, 2H, CH2N9), 3.35 (s, 3H, N3CH3), 3.50 (s, 3H, N1CH3), 4.18–4.24 (m, 2H, N5CH2), 3.49 (s, 3H, N1CH3), 4.14–4.24 (m, 2H, N5CH2), 4.51–4.58 (m, 1H, N1CH=). IR KBr (cm−1): 2,963–2,872—CH2, 1,697—CO (2), 1,666 (4), 743—CH2. UV λ max, logε: 302, 4.28.
1,3-Dimethyl-9-(4-methylpentan-2-yl)-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (16)
Reaction medium, DMF; time of heating under reflux, 5 h; excess of amine, 2. Crystallization with ethanol/H2O gave pure compound 16. Yield 59 %, mp 116–117 °C; TLC: R f: 0.58; Anal. Calcd for C16H25O2N5 (16): C, 60.16; H, 7.89; N, 21.93. Found: C, 60.22; H, 8.01; N, 21.96. 1H-NMR δ (ppm): 0.89–0.94 (m, 6H, CH(CH3)2), 1.15 (d, J = 6.60 Hz, 3H, N9CHCH3), 1.19–1.26 (m, 1H, CH(CH3)2), 1.40–1.58 (m, 2H, CH2CH(CH3)2), 2.03–2.15 (m, 2H, N5-CH2-CH2-CH2-N9), 3.14–3.20 (m, 2H, CH2N9), 3.33 (s, 3H, N3CH3), 3.47 (s, 3H, N1CH3), 4.12–4.22 (m, 2H, N5CH2), 4.57–4.67 (m, 1H, N9CHCH3). IR KBr (cm−1): 2,952–2,867—CH2, 1,699—CO (2), 1,660 (4), 742—CH2. UV λ max, logε: 301.5, 4.21.
1,3-Dimethyl-9-(2-methylhexyl)-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (17)
Reaction medium, DMF; time of heating under reflux, 5 h; excess of amine, 4. Crystallization with ethanol/H2O gave pure compound 17. Yield 81 %, mp 122–124 °C; TLC: R f: 0.63; Anal. Calcd for C17H27O2N5 (17): C, 61.24; H, 8.17; N, 21.01. Found: C, 61.51; H, 8.19; N, 21.12. 1H-NMR δ (ppm): 0.84–0.89 (m, 6H, 2CH3), 1.18–1.59 (m, 8H, CH2 CH(CH2)3 CH2CH3), 1.40–1.58 (m, 2H, CH2CH(CH3)2), 2.06–2.13 (m, 2H, N5CH2CH2CH2N9), 3.17–3.21 (m, 2H, CH2N9), 3.36 (s, 3H, N3CH3), 3.48 (s, 3H, N1CH3), 4.21 (t, J = 6.05 Hz, 2H, N5CH2), 4.29–4.35 (m, 1H, N9CH). IR KBr (cm−1): 2,960–2,861—CH2, 1,699—CO (2), 1,659 (4), 750—CH2. UV λ max, logε: 302, 4.31.
9-(Heptan-2-yl)-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (18)
Reaction medium, DMF; time of heating under reflux, 10 h; excess of amine, 2. Crystallization with ethanol/H2O gave pure compound 18. Yield 57 %, mp 94–96 °C; Anal. Calcd for C17H27O2N5 (18): C, 61.24; H, 8.17; N, 21.01. Found: C, 60.95; H, 7.90; N, 21.05. 1H-NMR δ (ppm): 0.84–0.94 (m, 3H, (CH2)4-CH3), 1.17 (d, J = 6.87 Hz, 3H, CHCH 3), 1.26–1.59 (m, 8H, (CH2 )4CH3), 2.05–2.13 (m, 2H, N5CH2CH2CH2N9), 3.20–3.25 (m, 2H, CH2N9), 3.35(s, 3H, N3CH3), 3.49 (s, 3H, N1CH3), 4.13–4.24 (m, 2H, N5CH2), 4.50–4.56 (m, 1H, N9CH). IR KBr (cm−1): 2,955–2,859—CH2, 1,701—CO (2), 1,656 (4), 745—CH2. UV λ max, logε: 302, 4.30.
1,3-Dimethyl-9-(6-methylheptan-2-yl)-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (19)
Reaction medium, DMF; time of heating under reflux, 5 h; excess of amine, 2. Crystallization with ethanol/H2O gave pure compound 19. Yield 60 %, mp 99–101 °C; TLC: R f: 0.48; Anal. Calcd for C18H29O2N5 (19): C, 61.24; H, 8.17; N, 21.01. Found: C, 61.99; H, 8.21; N, 20.19. 1H-NMR δ (ppm): 0.83–0.86 (m, 6H, CH(CH3)2), 1.18 (d, J = 6.87 Hz, 3H, N9CHCH3), 1.23–1.38 (m 1H CH(CH3)2), 1.43–1.61(m, 6H, (CH2)3), 2.06–2.14 (m, 2H, CH2N9), 3.36 (s, 3H, N3CH3), 3.50 (s, 3H, N1CH3), 4.12–4.27 (m, 2H, N5CH2), 4.46–4.58 (m, 1H, N9CHCH3). IR KBr (cm−1): 2,956–2,858—CH2, 1,708—CO (2), 1,651 (4), 742—CH2. UV λ max, logε: 302.5, 4.25.
9-Allyl-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (23)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 20. Crystallization with ethanol (10 %) gave pure compound 23. Yield 65 %, mp 157–159 °C; TLC: R f: 0.35; Anal. Calcd for C13H17O2N5 (23): C, 56.71; H, 6.22; N, 24.44. Found: C, 56.88; H, 6.48; N, 25.12. 1H-NMR δ (ppm): 2.10–2.18 (m, 2H, N5CH2CH2CH2N9), 3.29 (t, J = 5.65 Hz, 2H, CH2N9), 3.34 (s, 3H, N3CH3), 3.48 (s, 3H, N1CH3), 4.16 (t, J = 6.04 Hz, 2H, N9CH2), 4.20 (t, J = 6.04 Hz, 2H, N5CH2), 5.20–5.26 (m, 2H, CH2 = CH), 5.75–5.88 (m, 1H, CH2 = CH). IR KBr (cm−1): 3,081—CH2-CH = CH2, 2,944–2,902 CH2, 1,695—CO (2), 1,664 (4), 750—CH2. UV λ max, logε: 303.5, 4.30.
1,3-Dimethyl-9-(prop-2-ynyl)-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (24)
Reaction medium, n-butanol; time of heating under reflux, 10 h; excess of amine, 5. Crystallization with ethanol (30 %) gave pure compound 24. Yield 93 %, mp 203–206 °C; TLC: R f: 0.57; Anal. Calcd for C13H15O2N5 (24): C, 57.14; H, 5.54; N, 25.62. Found: C, 57.44; H, 5.66; N, 25.79. 1H-NMR δ (ppm): 2.18–2.29 (m, 3H, N5CH2CH2CH2 + CH≡), 3.35 (s, 3H, N3CH3), 3.35–3.42 (m, 2H, CH2N9), 3.50 (s, 3H, N1CH3), 4.36 (t, J = 6.05 Hz, 2H, N9CH2). IR KBr (cm−1): 3,123—CH2-CH = CH2, 2,952—CH2, 1,701—CO (2), 1,665 (4), 744—CH2. UV λ max, logε: 296, 4.19.
Synthesis of 9-ethenyl 1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (22)
A mixture of 9-(2-bromoethyl)-1,3-dimethyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H, 3H)-dione (0.68 g, 2 mmol) (21) [27] and KOH (0.11 g, 2 mmol) in 10 ml ethanol was heated at reflux for 20 h. After cooling, the precipitate was separated and washed with water and crystallized from methoxyethanol. Yield 70 %, mp 268–271 °C; TLC: R f: 0.55; Anal. Calcd for C12H15O2N5 (22): C, 55.19; H, 5.79; N, 26.80. Found: C, 55.47; H, 5.55; N, 26.45. 1H-NMR δ (ppm): 2.17–2.28 (m, 2H, N5CH2CH2CH2N9), 3.38 (s, 3H, N3CH3), 3.53 (s, 3H, N1CH3), 3.54–3.59 (m, 2H, N9CH2), 4.26–4.52 (m, 4H, N5CH2 + CH2=), 7.38–7.48 (m, 1H, CH=). IR KBr (cm−1): 3,108–3,089—CH = CH2, 2,983–2883 CH2, 1,701—CO (2), 1,652 (4), 746—CH2. UV λ max, logε: 304, 4.40.
General procedure for the synthesis of 10-substituted 1,3-dimethyl-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-diones (26–32)
A mixture of 7-(4-bromobutyl)-8-bromotheophylline (0.79 g, 2 mmol) (3) [23] and an amine derivative (4–72 mmol) was heated under reflux without solvent or with ethanol, isobutanol or DMF for 5–10 h. Compounds 30 and 31 precipitated by adding water to the reaction mixture; compounds 26 and 27 were obtained by removing the solvent and excess of amine by distillation under reduced pressure and adding water to the residue. Compounds 28, 29 and 32 precipitated by removing the excess of amine by steam distillation and cooling the water solution. All compounds were purified by crystallization from diluted ethanol or by dissolving in ethanol and precipitation with water.
10-Ethyl-1,3-dimethyl-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione (26)
Reaction medium, ethanol; time of heating under reflux, 10 h; excess of amine, 36. Crystallization with ethanol (30 %) gave pure compound 26. Yield 38 %, mp 124–125 °C; TLC: R f: 0.62; Anal. Calcd for C13H19O2N5 (26): C, 56.29; H, 6.91; N, 25.24. Found: C, 56.08; H, 6.64; N, 25.50. 1H-NMR δ (ppm): 1.22–1.26 (t, J = 7.01 Hz, 3H, CH3CH2), 1.27–1.92 (s, 4H, N5CH2(CH2)2CH2), 3.30–3.23 (m, 2H, CH2CH3), 3.37 (s, 3H, N3CH3), 3.51 (s, 3H, N1CH3), 3.54–3.56 (m, 2H, CH2N10), 4.28–4.31 (m, 2H, N5CH2). IR KBr (cm−1): 2,952–2873—CH2, 1,693—CO(2), 1,661 (4), 740—CH2. UV λ max, logε: 300, 4.22.
1,3-Dimethyl-10-propyl-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione (27)
Reaction medium, isobutanol; time of heating under reflux, 10 h; excess of amine, 11. Crystallization with ethanol (30 %) gave pure compound 27. Yield 46 %, mp 153–154 °C; TLC: R f: 0.64; Anal. Calcd for C14H21O2N5 (27): C, 57.71; H, 7.27; N, 24.03. Found: C, 57.76; H, 6.98; N, 23.66. 1H-NMR δ (ppm): 0.91 (t, J = 7.43 Hz, 3H, (CH2)2CH3), 1.57–1.73 (m, 2H, CH2CH2CH3), 1.87–1.91 (m, 4H, N5CH2(CH2)2CH2N10), 3.24 (t, J = 5.45 Hz, 2H, CH2CH2CH3), 3.37 (s, 2H, N3CH3), 3.42–3.47 (s, 2H, CH2N10), 3.51 (s, 3H, N1CH3), 4.31 (t, J = 5.08 Hz, 2H, N5CH2). IR KBr (cm−1): 2,935–2870—CH2, 1,692—CO(2), 1,653 (4), 750—CH2. UV λ max, logε: 300, 4.30.
10-Butyl-1,3-dimethyl-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione (28)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 25. Crystallization with ethanol/H2O gave pure compound 28. Yield 72 %, mp 84–86 °C; TLC: R f: 0.70; Anal. Calcd for C15H23O2N5 (28): C, 58.99; H, 7.60; N, 22.93. Found: C, 58.89; H, 7.22; N, 22.60. 1H-NMR δ (ppm): 0.95 (t, J = 7.28 Hz, 3H, (CH2)3CH3), 1.30–1.42 (m, 2H, CH2CH2CH2CH3), 1.57–1.67 (m, 2H, CH3CH2CH2CH3), 1.87–1.88 (m, 4H N5CH2(CH2)2CH2N10), 3.23 (t, J = 5.01 Hz, 2H N10CH2), 3.37 (s, 3H, N3CH3), 3.48 (t, J = 7.48 Hz, 2H, CH2N10), 3.51 (s, 3H, N1CH3), 4.30 (t, J = 5.09 Hz, 2H, N5CH2). IR KBr (cm−1): 2,957–2,871—CH2, 1,700—CO(2), 1,658 (4), 745—CH2. UV λ max, logε: 300, 4.28.
10-Sec-butyl-1,3-dimethyl-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione) (29)
Reaction medium, solvent-free; time of heating under reflux, 10 h; excess of amine, 10. Crystallization with ethanol (50 %) gave pure compound 29. Yield 57 %, mp 138–140 °C; TLC: R f: 0.53; Anal. Calcd for C15H23O2N5 (29): C, 58.99; H, 7.60; N, 22.93. Found: C, 58.67; H, 7.30; N, 22.75. 1H-NMR δ (ppm): 0.95 (t, J = 1.10 Hz, 3H, CH2CH3), 1.22 (d, J = 7.60 Hz, 3H, CHCH3), 1.45–1.72 (m, 2H, CH3CH2), 1.72–2.00 (m, 4H, N5CH2(CH2)2CH2N10), 2.98–3.28 (m, 2H, CH2 N10), 3.37 (s, 2H, N3CH3), 3.48 (t, J = 7.48 Hz, 2H, CH2N10), 3.51 (s, 3H, N1CH3), 4.01–4.12 (m, 2H, N5CH2), 4.51–4.58 (m, 1H, CHCH3). IR KBr (cm−1): 2,966–2,876—CH2, 1,701—CO(2), 1,660 (4), 745—CH2. UV λ max, logε: 300, 4.20.
1,3-Dimethyl-10-(4-methylpentan-2-yl)-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione (30)
Reaction medium, DMF; time of heating under reflux, 10 h; excess of amine, 2. Crystallization with ethanol/H2O gave pure compound 30. Yield 61 %, mp 96–100 °C; TLC: R f: 0.72; Anal. Calcd for C17H27O2N5 (30): C, 61.22; H, 8.17; N, 21.02. Found: C, 61.52; H, 8.09; N, 20.90. 1H-NMR δ (ppm): 0.92–0.94 (m, 6H, CH(CH3)2), 1.19–1.28 (m, 4H CHCH3 + CH(CH3)2), 1.50–1.67 (m, 2H, CH2CH), 1.73–1.97 (m, 4H, N5CH2(CH2)2CH2N10), 2.99–3.27(m, 2H, CH2N10), 3.36 (s, 3H, N3CH3), 3.50 (s, 3H, N1CH3), 4.02–4.10 (m, 1H, N5CH), 4.27–4.34 (m, 2H, N5CH2), 4.48–4.55 (m, 1H, N10CH). IR KBr (cm−1): 2,946–2,876—CH2, 1,695—CO(2), 1,664 (4), 747—CH2. UV λ max, logε: 300, 4.37.
1,3-Dimethyl-10-(pentan-2-yl)-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione (31)
Reaction medium, DMF; time of heating under reflux, 10 h; excess of amine, 2. Crystallization with ethanol/H2O gave pure compound 31. Yield 78 %, mp 113–115 °C; TLC: R f: 0.70; Anal. Calcd for C16H25O2N5 (31): C, 60.17; H, 7.90; N, 21.93. Found: C, 60.19; H, 7.85; N, 21.78. 1H-NMR δ (ppm): 0.90–0.94 (m, 3H, CH2CH3), 1.21 (d, J = 6.88 Hz, 3H, CHCH3), 1.32–1.64 (m, 4H, (CH2)2CH3), 1.74–1.98 (m, 4H, N5CH2(CH2)2CH2N10), 2.99–3.07 (m, 1H, CH2N10), 3.20–3.28 (m, 1H, CH2N10), 3.50 (s, 3H, N1CH3), 4.01–4.09 (m, 1H, N5CH), 4.17–4.24 (m, 2H, N5CH2), 4.49–4.57 (m, 1H, N10CH). IR KBr (cm−1): 2,949–2,871—CH2, 1,700—CO(2), 1,661 (4), 745—CH2. UV λ max, logε: 300, 4.29.
10-Allyl-1,3-dimethyl-6,7,8,9-tetrahydrodiazepino[2,1-f]purine-2,4(1H,3H)-dione (32)
Reaction medium, solvent-free; time of heating under reflux, 5 h; excess of amine, 20. Crystallization with ethanol/H2O gave pure compound 32. Yield 65 %, mp 228–230 °C; TLC: R f: 0.62; Anal. Calcd for C17H25O2N5 (32): C, 58.11; H, 6.63; N, 24.20. Found: C, 58.23; H, 6.48; N, 24.36. 1H-NMR δ (ppm): 1.83–1.90 (m, 4H, N5CH2(CH2)2CH2N10), 3.15 (t, J = 4.95 Hz, 2H, CH2N10), 3.37 (s, 3H, N3CH3), 3.51 (s, 3H, N1CH3), 4.07 (d, J = 4.95 Hz, 2H, N10CH2), 4.33 (t, J = 4.95 Hz, 2H, N5CH2), 5.21–5.30 (m, 2H, CH2 = CH), 5.89–6.00 (m, 1H, CH2 = CH). IR KBr (cm−1): 3,078–3,010—allyl, 2,936—CH2, 1,696—CO(2), 1,652 (4), 750—CH2. UV λ max, logε: 298, 4.23.
8-Isopropyl-1,3-dimethyl-6,7-dihydro-1H-imidazo[2,1-f]purine-2,4(1H,3H)-dione (34)
A mixture of (0.73 g, 2 mmol) of 7-(2-bromoethyl)-8-bromotheophylline (1) [16, 21] and isopropyl amine (1.42 g, 24 mmol) was heated under reflux for 5 h. After cooling, the precipitate was separated and crystallized from 30 % ethanol to give analytically pure compound 34. Yield 50 %, mp 157–159 °C; Anal. Calcd for C12H17O2N5 (34): C, 54.74; H, 6.50; N, 26.60. Found: C, 54.35; H, 6.50; N, 26.28. 1H-NMR δ (ppm): 1.83–1.90 (m, 4H, N5CH2(CH2)2CH2N10), 3.15 (t, J = 4.95 Hz, 2H, CH2N10), 3.37 (s, 3H, N3CH3), 3.51 (s, 3H, N1CH3), 4.07 (d, J = 4.95 Hz, 2H, N10CH2), 4.33 (t, J = 4.95 Hz, 2H, N5CH2), 5.21–5.30 (m, 2H, CH2 = CH), 5.89–6.00 (m, 1H, CH2 = CH). IR KBr (cm−1): 2,960–2,870—CH2–, 1,696—CO(2), 1,653—CO (4), 749—CH2–. UV λ max, logε: 302, 4.21.
9-Butyl-1,3-dipropyl-6,7,8,9-tetrahydropyrimido[2,1-f]purine-2,4(1H,3H)-dione (35)
A mixture of (0.78 g, 2 mmol) of 1,3-dipropylo-7-(3-chloropropyl)-8-bromo-purine-2,4(3H,6H)-dione (4) [16, 17] and butyl amine (2.20 g, 30 mmol) was heated under reflux for 5 h. After cooling, the precipitate was separated and crystallized from ethanol to give analytically pure compound 35. Yield 45 %, mp 77–79 °C; Anal. Calcd for C18H29O2N5 × H2O (29): C, 59.14; H, 8.42; N, 19.16. Found: C, 58.85; H, 8.81; N, 19.10. 1H-NMR δ (ppm): 0.91–0.98 (m, 8H, CH3CH2CH2 + CH3-CH2CH2CH2), 1.28–1.41 (m, 6H, CH3CH2CH2N1 + CH3CH2CH2CH2), 3.24 (t, J = 5.64 Hz, 2H, N9-CH2 ), 3.52 (t, J = 7.15Hz, 2H, N9-CH2), 3.89–3.98 (m, 4H, N1,N3CH2CH2CH3), 4.20 (t, J = 5.19Hz, 2H, N5CH2). IR KBr (cm−1): 3,518–2,870 H2O, 2,959–2,874—CH2–, 1,689—CO (2), 1,655—CO (4), 754—CH2–. UV λ max, logε: 303, 4.30.
Pharmacology
Adenosine receptor binding studies
[3H]CCPA (58 Ci/mmol) was purchased from NEN Life Sciences; [3H]MSX-2 (84 Ci/mmol), [3H]PSB-11 (28 Ci/mmol) and [3H]PSB-603 (73 Ci/mmol) were obtained from Quotient Biosciences (custom synthesis). The non-radioactive precursors of [3H]MSX-2 (MSX-1) [33, 48], [3H]PSB-11 (PSB-10) [36, 49] and [3H]PSB-603 (PSB-603) [35] were synthesized in our laboratory. Frozen rat brains obtained from Pel Freez®, Rogers, AR, USA, were dissected to obtain cortical membrane preparations for A1 assays and striatal membrane preparations for A2A assays as described [50]. CHO cells stably transfected with the human adenosine A1, A2B and A3 receptors were used for membrane preparations as previously described [50, 51]. For A2A adenosine receptor assays, commercially available membrane preparations containing the human A2A AR were obtained from Perkin Elmer Life Sciences (Boston, MA, USA). Stock solutions of the compounds were prepared in dimethylsulfoxide (DMSO); the final concentration of DMSO in the assays did not exceed 2.5 %. Initial screening was performed at a single concentration of 25 or 10 μM, depending on target receptor. Binding assays were performed as previously described [33, 50–54]. Curves were determined using six to seven different concentrations of test compounds spanning three orders of magnitude. At least three separate experiments were performed, each in triplicate. For non-linear regression analysis, the Cheng–Prusoff equation and KD values of 0.2 nM (rat A1) and 0.6 nM (human A1), respectively, for [3H]CCPA [34]; 8 nM (rat A2A) and 7 nM (human A2A), respectively, for [3H]MSX-2 [33, 55]; 0.41 nM (human A2B) for [3H]PSB-603 (human A2B) [35] and 4.9 nM (human A3) for [3H]PSB-11 [52] were used to calculate K i values from IC50 values.
Anticonvulsant screening
The anticonvulsant evaluation was carried but using reported procedures [39, 40, 47]. Male albino mice (Carworth Farms Nr 1, 18–25 g) and male albino rats (Spraque-Dawley, 100–150 g) were used as experimental animals. A group of three to five mice was used in MES and ScMet tests, and a group of four to eight animals was used in rotorod tests. For the evaluation of activity after oral administration, a group of four rats was used. The test compounds were suspended in 0.5 % methylcellulose water mixture.
In the preliminary screening, each compound was administered as an i.p. injection at three dose levels (30, 100 and 300 mg/kg), with anticonvulsant activity and neurotoxicity assessed at 30-min and 4-h intervals after administration. For some compounds, also intervals of 15 min and 1 h were used. Anticonvulsant efficacy was measured by MES, ScMet and neurological deficit in the rotorod tests, and the data are presented in Table 4. Some selected derivatives were examined for oral activity in the MES and ScMet and neurotoxicity screen (rotorod test) 30 and 50 mg/kg doses. The results are summarized in Table 5.
The pharmacological parameters estimated in the preliminary screening were quantified for compounds 9 and 13 in mice (i.p.) (Table 7) and for comp. 9 also in rats (p.o.) (Table 6).
Anticonvulsant activity was expressed in terms of the median effective dose (ED50) MES and ScMet in mice and ED50 ScMet in rats, and neurotoxicity was expressed as the median toxic dose (TD50). For determination of ED50 and TD50, groups of eight mice and rats were given a range of i.p. or p.o. doses of the test drug until at least three points were established in the range of 10–90 % seizure protection or minimal observed neurotoxicity. From the plot of these data, the respective ED50, TD50 value (95 %) confidence intervals, slope of the regression line and the standard error of the slope were calculated by a computer program based on [38] methods described by Finney. For comp. 9, the hippocampal kindling model of focal seizures was applied (Table 6). In this procedure, the bipolar electrodes were placed surgically in the ventral hippocampus of adult male Spraque-Dawley rats. Stage five behavioural seizures are produced by using a stimulus consisting of a 50-Hz, 10-s train of 1-ms biphasic 200-μA pulses delivered every 30 min for 6 h. A single dose of comp. 9 is then administered intraperitoneally, 15 min following the last stimulation. The anticonvulsant activity of 9 is assessed every 30 min for 2 h, starting 15 min after administering the test material. After each stimulation, seizure scores and after discharge durations were recorded (Table 6).
Molecular modelling
The 3D molecule models of 5–35 and caffeine were built using Schrödinger Maestro molecular modelling environment [56] basing on presented crystal structures of chosen pyrimido- and diazepinopurinediones (10 and 30, respectively). The geometry optimization was performed using the multiple minimization method as implemented in MacroModel with MMFF force field and Truncated Newton Conjugate Gradient options and terminated when the root means square (RMS) of conjugate gradient was below 0.05 kJ mol−1 Å−1. The minimization was carried out for water solvent, with distance-dependent dielectric constant as a way to treat electrostatic interactions.
The template 3REY was prepared to docking using the Protein Preparation Wizard option as a part of Schrodinger package. The ligand binding cavity was confined to a box with the crystal ligand in the center (size of the box set automatically), according to the receptor grid generation procedure. The studies of docking to the rigid receptor binding pocket were performed using Glide program, with standard precision (SP) mode.
For validation, redocking of XAC ligand was done. After initial docking, 10 poses for each ligand, with RMS deviation higher than 0.5 Å, were kept, and post-minimized, RMSD values between predicted and crystallographic positions of the ligand were calculated. Redocking of XAC was performed twice: (1) without any constraints and (2) with the amino hydrogen atom of the Asn253 (6.55) side chain used as a constrained H-bond donor for a carbonyl group of xanthine.
Molecular docking of A2A AR antagonists
Docking simulations of low-energy conformations for all of the synthesized compounds and caffeine were performed with SP mode to both templates with limitation used during redocking: the constrained H-bond between the side chain amino group of Asn253 (6.55) and the ligand. Five poses obtained after docking for each ligand (RMS deviation higher than 0.5 Å) were post-minimized, and final poses were kept and analysed according to the obtained docking score values.
For the graphic presentation of the selected structures with the highest docking scores, representing individual clusters of poses, PyMOL [57] software was used.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Drabczyńska, A., Karcz, T., Szymańska, E. et al. Synthesis, biological activity and molecular modelling studies of tricyclic alkylimidazo-, pyrimido- and diazepinopurinediones. Purinergic Signalling 9, 395–414 (2013). https://doi.org/10.1007/s11302-013-9358-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-013-9358-3