
Abstract
North temperate tree species such as white spruce (Picea glauca [Moench] Voss) have evolved strategies to protect themselves against abiotic and biotic stresses that trees encounter during the inclement winter months. Chitinases not only play well-documented roles in plant defense but also function during physiological and developmental preparations for overwintering, including growth cessation, cold and desiccation acclimation, and dormancy acquisition. Phylogenetic analysis of 31 white spruce and 52 Norway spruce chitinases identified genes falling into each of the five clusters, which sometimes—but not always—separated the different biochemical classes of chitinases. Digital expression profiling of white spruce and Norway spruce chitinases across multiple conditions revealed a range of spatiotemporal expression patterns. Transcript abundance profiling in buds, needles, stems, and roots by quantitative RT-PCR suggested roles for eight white spruce chitinases during the growth-to-dormancy transition. In silico analyses of these eight sequences suggested that two cluster 2/class I chitinases function as chitinolytic enzymes in the tree’s constitutive defense arsenal during the winter months. A cluster 2/class I, cluster 2/class II, and cluster 1/class IV chitinase each exhibit hallmarks of antifreeze proteins. Additionally, two cluster 2/class I chitinases and a cluster 1/class IV chitinase may serve as vegetative storage proteins. One cluster 3/class II chitinase exhibited attributes suggesting that it is a chitinase-like gene functioning in cell wall synthesis. Taken together, our results imply that dormancy-associated chitinases act in concert to (1) confer protection against freezing injury, pests, and pathogens, (2) store nitrogen, and (3) promote cell maturation that precedes growth cessation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
North temperate forest trees exhibit a number of adaptations that enable these long-lived species to withstand the inclement conditions of winter. These adaptive traits include dormancy acquisition, bud development and the concomitant cessation of growth, as well as acquisition of cold and desiccation tolerance, seasonal nitrogen remobilization, and, in deciduous species, leaf senescence (Cooke et al. 2012). Several genomic studies over the last decade have revealed the large-scale changes that occur to the transcriptome of shoot tips, needles, and stems during the transition from active growth to dormancy of both coniferous and angiosperm forest trees such as Populus spp. (Schrader et al. 2004; Druart et al. 2007; Ruttink et al. 2007; Park et al. 2008; Karlberg et al. 2010; Resman et al. 2010), Pinus spp. (Joosen et al. 2006), and Picea spp. (Holliday et al. 2008; Asante et al. 2011), including white spruce (Picea glauca [Moench] Voss; El Kayal et al. 2011; Galindo-González et al. 2012). Several of these studies have highlighted the large number of stress-associated genes that are upregulated during the transition from active growth to dormancy, such as genes coding for dehydrins, thaumatins, chaperonins, chitinases, glyoxylases, and oxidative stress responses (e.g., Druart et al. 2007; Holliday et al. 2008; Galindo-González et al. 2012). Galindo-González et al. (2012) further demonstrated that a subset of these proteins figure prominently in proteomic profiles of white spruce stems during the transition to dormancy. These authors observed that protein levels of multiple chitinases became particularly abundant in white spruce prior to dormancy acquisition, suggesting that chitinases play roles in biochemical, cellular, and/or physiological processes that are important for tree survival during autumnal acclimation and the ensuing winter season.
Roles for chitinases in plant defense are well-known. Chitinases from many species are strongly induced in plants challenged with fungal or bacterial pathogens (e.g., Lawton et al. 1992; Collinge et al. 1993; Sela-Buurlage et al. 1993; Melchers et al. 1994; Mason and Davis 1997; Wu et al. 1997; Salzer et al. 1997; Büchter et al. 1997; Hong et al. 2000; Hietala et al. 2004). Several chitinases are classified as pathogenesis-related (PR) proteins, making up the PR-3, PR-4, PR-8, and PR-11 groups of these defense-associated proteins (van Loon et al. 2006). Many chitinases have been shown to hydrolyze chitin and effectively inhibit tip growth of fungal hyphae through breakdown of this fungal cell wall component. Some chitinases also exhibit lysozyme-like activity and are active against the peptidoglycan component of bacterial cell walls (Collinge et al. 1993).
Chitinases comprise a large and diverse gene family, however, and characterization of other chitinases from several species has revealed that these proteins can assume functionalities in the plant other than in defense. Chitinases have been implicated in a variety of developmental processes like seed germination (Wu et al. 2001), senescence (Hanfrey et al. 1996), and morphogenesis (Zhang et al. 2004), including somatic embryogenesis (Wiweger et al. 2003; Johnsen et al. 2005). There is increasing evidence that chitinases can mediate developmental processes through biosynthesis, assembly, and/or remodelling of various cell wall components (van Hengel et al. 2001; Zhang et al. 2004; Sánchez-Rodríguez et al. 2012; Wu et al. 2012). Chitinases can also act as antifreeze proteins. Some of the first studies recognizing the potential of chitinases to adsorb to ice-forming surfaces in cold acclimation processes were carried out in winter rye and other cereals (Hon et al. 1995; Griffith et al. 1997; Yeh et al. 2000; Pihakaski-Maunsbach et al. 2001). Chitinases with antifreeze activity have been isolated from apoplastic spaces of needles from overwintering Pseudotsuga menziesii (Mirb.) Franco, Picea abies (L.) Karst., and Picea pungens Engelm., indicating that chitinases play a role in cold hardiness of conifers (Zamani et al. 2003; Jarząbek et al. 2009). Finally, chitinases have also been recognized as vegetative storage proteins (Clendennen et al. 1998; Peumans et al. 2002; Meuriot et al. 2004).
Different chitinase functions are reflected in the primary and secondary structure of the protein. Chitinases are typified by the presence of a glycosyl hydrolase domain which corresponds to the catalytic domain. The chitinase family can be subdivided into two main groups—glycoside hydrolase family 18 (GH 18) and glycoside hydrolase family 19 (GH 19)—based on differences in this domain (Davies and Henrissat 1995; Iseli et al. 1996; Li and Greene 2010). Biochemically, chitinases are grouped into classes I to VII, based on their primary structure and the presence of one or more diagnostic motifs (Collinge et al. 1993; Neuhaus 1999; Grover 2012). More recently, phylogenetic analyses of Arabidopsis and rice chitinases have led to designation of six clusters (Xu et al. 2007). The groupings of chitinases within these phylogenetic clusters do not mirror those of the classical biochemical classification scheme, although there are some commonalities shared between the phylogenetic and biochemical classifications (Xu et al. 2007). With the extensive characterization of many plant chitinases at the gene and protein level that has been carried out to date, in silico analyses of chitinases from other species can reveal considerable insight into their potential functions in planta.
While the accumulation of chitinases in temperate forest tree species—particularly conifers—during preparation for overwintering is well-documented (Ekramoddoullah et al. 1995, 2000; Zamani et al. 2003; Jarząbek et al. 2009), the roles that these chitinases play in the tree during this phase of the annual cycle are less clear. The objective of the present study was to analyze the chitinase gene family in white spruce using both phylogenetic and in silico characterization approaches, with a focus on chitinases that we had shown in previous studies to be differentially expressed during the transition from active growth to dormancy in this north temperate conifer species (El Kayal et al. 2011; Galindo-González et al. 2012). Given that the differentially expressed chitinases belonged to multiple classes, we hypothesized that these proteins assume different functions during the activity-dormancy transition of white spruce. Using available white spruce-expressed gene sequence resources (Rigault et al. 2011), we performed a phylogenetic and structural classification of available white spruce chitinase sequences. We then carried out in silico characterization of dormancy-associated chitinases, combined with qRT-PCR transcript profiling of these chitinases during the short day-induced activity-dormancy transition in white spruce seedlings to gain further insight into the function of these chitinases in tree overwintering.
Materials and methods
Chitinase in silico analyses
White spruce chitinase sequences identified as differentially expressed during the activity-dormancy transition (Galindo-González et al. 2012) as well as chitinases from rice, Arabidopsis, and additional selected plant species (Xu et al. 2007) were used to identify white spruce chitinase transcript sequences representative of unigenes in the white spruce gene catalog (v3.3; Rigault et al. 2011; ftp.gydle.com/pub/arborea), using BLAT (Kent 2002). White spruce cDNA clones representing each of these transcripts were fully sequenced to provide confirmation of the sequence obtained from the EST assembly. Nucleotide sequences, deduced amino acid sequences, unique identifiers, and corresponding NCBI gi numbers are given in Online Resource 1. Norway spruce putative chitinase peptide sequences corresponding to CDS were identified using the same approach from ConGenIE (www.congenie.org), representing the draft Norway spruce genome sequence (Nystedt et al. 2013), and are also included in Online Resource 1. The deduced amino acid sequences were aligned using ClustalW in MEGA 5.1 (Tamura et al. 2011) with sequences from Arabidopsis and selected plant species representing the different chitinase classes found in plants. The parameters used included a gap opening penalty of 10 and a gap extension penalty of 0.1 for the pairwise alignments and a gap opening penalty of 10 and a gap extension penalty of 0.2 for the multiple alignment; the protein weight matrix used was Gonnet; residue-specific and hydrophilic penalties were turned; gap separation distance was set to 4; and the delay for divergence cutoff was set to 30 %. After a preliminary alignment, poorly aligned sequences and sequences covering less than 50 % of the average sequence length were discarded, and a final alignment was built using default parameters. A neighbor-joining tree was built using MEGA 5.1 under the Poisson model, pairwise deletion, and 1,000 bootstrap repetitions (Tamura et al. 2011). An additional tree was built only with white spruce chitinases.
Functional domains were identified using all the algorithms implemented by InterproScan (http://www.ebi.ac.uk/Tools/pfa/iprscan/—accessed February 17, 2015), and matching domain coordinates were translated into each of the white spruce chitinase sequences using VectorNTI® (Invitrogen, Carlsbad, CA, USA). Chitinases were aligned to a class I chitinase from tobacco (Neuhaus et al. 1991) which has most of the structural domains documented in the chitinase gene family, to resolve the presence of canonical domains and critical amino acids.
3D models were built using the SWISS-MODEL homology modeling tool under default parameters (Arnold et al. 2006) for the white spruce chitinases used in the phylogenetic analysis. Generated models were visualized using Raswin-Rasmol (Sayle and Milner-White 1995). To obtain root mean square deviation values (RMSDs), all predicted structures were compared to each other using the FAST algorithm (Zhu and Weng 2005) implemented in the ProCKSI-Server (Barthel et al. 2007; http://www.procksi.net—accessed February 12, 2013) for multiple comparisons. The RMSD distance matrix of all pairwise comparisons was standardized, and the values were used to create clusters and a tree topology. The tree was visualized using ProCKSI_Viz (http://www.procksi.net), and the Newick tree file was exported to MEGA 5.1 (Tamura et al. 2011) to modify the tree for publication. Each of the white spruce chitinase sequences was also analyzed for intrinsically disordered regions (IDRs) using PONDR-FIT (Xue et al. 2010).
Prediction of ice-binding surfaces was done using AFPredictor (Doxey et al. 2006) (http://doxey.uwaterloo.ca/cgi-bin/AFPredictor.cgi—accessed February 18, 2013) with a solvent accessibility (SAS) threshold (t) = 15, maximum length of allowed vectors (r) = 7.55 Å, minimum length of allowed vectors (q) = 4.5 Å, vector matching cutoff (v) = 1.09 Å, distance threshold for clustering ordered surface carbons (OSCs) (c) = 7.66 Å, polar atom distance cutoff (p) = 4 Å, and SAS threshold for polar atoms = 0 Å2.
Putative active site residues in protein models were predicted based on structural alignments to the top five enzyme homologs using COFACTOR (Roy et al. 2012) (http://zhanglab.ccmb.med.umich.edu/COFACTOR/—accessed March 4, 2013). When an amino acid was predicted in the spruce protein as constituting part of the active site in at least four of the five protein alignments, it was designated a putative active site residue.
Plant material
Experiments with 2-year-old white spruce seedlings derived from Quebec provenances were conducted as described in El Kayal et al. (2011) and Galindo-González et al. (2012). Following bud burst, seedlings were grown in a complete randomized block design in controlled environment growth chambers under long days (LD; 16 h day/8 h night, 20 °C, 50 to 60 % relative humidity (RH)) for ca. 8 weeks. Shortly before seedlings were to begin bud formation, the photoperiod was changed to short-day conditions (SD; 8 h day/16 h night, 20 °C, 50 to 60 % RH) to induce synchronous bud formation. Developing buds, needles, lignified stems representing current year growth, and roots were sampled at 0, 3, 7, 14, and 70 days SD and flash frozen in liquid nitrogen. Remaining plants were maintained in SD for additional 8–15 weeks and then transferred to low temperatures (LT, 2–4 °C) for 3 to 4 weeks with continuing SD prior to harvest of buds, needles, and lignified whole stems.
RNA extraction and quantitative RT-PCR
RNA was extracted according to Pavy et al. (2008) and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). Primer design, cDNA synthesis, and qRT-PCR using SYBR Green were carried out as previously described (El Kayal et al. 2011). Translation initiation factor 5A (TIF5A, GenBank DR448953) was used as the reference gene, since this gene has been used successfully in previous studies for identical material (El Kayal et al. 2011; Galindo-González et al. 2012), and did not show statistically significant differences in transcript abundance in tissues over the time course (P = 0.305 for shoot tips, P = 0.247 for stems, P = 0.361 for needles, and P = 0.967 for roots). Use of a single reference gene with empirically demonstrated invariant expression ensured that the reference could be assayed with the target gene on each and every plate, thereby reducing experimental variability. Although actin and α-tubulin are commonly used as reference genes, our previous studies showed that genes encoding both actin and α-tubulin were unsuitable, as transcript abundance varied over the time course of growth cessation (Galindo-González et al. 2012; data not shown). Gene-specific primers for the target and reference genes are included in (Online Resource 2). Four biological replicates, each with three technical replicates, were analyzed per sample. Reactions were carried out with 20 ng of cDNA in a volume of 10 μL on an ABI PRISM 7900HT Sequence Detection System (Life Technologies, Burlington, ON). Quantification of both target and reference genes was carried out using standard curves.
Statistical analyses were performed in SAS v9.1 (SAS Institute, Cary, NC). To meet assumptions of normality and heterogeneity of variance, data were log10 transformed prior to analysis using general linear models (PROC GLM). Significant differences were determined by Tukey’s HSD test with overall α = 0.05. In two instances, data could not be successfully transformed to meet the assumptions of GLM. In these instances, non-parametric analyses were carried out using kernel density estimation with Gaussian density as the kernel (PROC KDE), followed by pairwise comparisons by t test.
Results
Spruce chitinase genes, phylogenetic relationships, and predicted protein structures
Based on sequence similarity and the presence of one or more characteristic motifs (described in more detail below), 32 expressed sequences encoding partial or full-length putative chitinases were identified in the white spruce gene catalog that comprises more than 27,000 unique expressed genes reported by Rigault et al. (2011) and Online Resources 1 and 3. Seventeen of these have a corresponding partial or full-length genomic DNA sequence (Online Resource 1) obtained by sequence capture of PG-29 (Sena et al. 2014), the genotype used for the draft genome sequence (Birol et al. 2013). Using the same criteria, 80 partial or full-length gene models were identified in the P. abies 1.0 assembly (Nystedt et al. 2013). One and four additional distinct chitinase sequences were identified in NCBI for white spruce and Norway spruce, respectively.
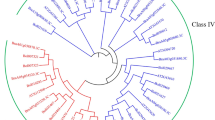
After filtering sequences that were too short for alignment, deduced amino acid sequences corresponding to 31 white spruce and 52 Norway spruce sequences were used to construct a phylogenetic tree, together with 58 previously characterized chitinases of Arabidopsis thaliana and other selected plant species (Fig. 1). The neighbor-joining tree with 1,000 bootstrap replicates yielded five well-supported clusters. A clear division between GH families 18 and 19 could be seen (Davies and Henrissat 1995; Iseli et al. 1996). The GH 19 family comprised clusters 1 through 3, with these chitinases representing structural classes, I, II, IV, and VII. The GH 18 family contained clusters 4 and 5 and comprised members of chitinase classes III and V, respectively. The spruce putative chitinase sequences were distributed among each of the five clusters, with 22 white spruce and 37 Norway spruce sequences found in clusters 1, 2, and 3 of GH 19. While some clusters clearly distinguished just one type of structural class of chitinases, others represented more than one class (Fig. 1). Two subclusters of conifer chitinases could be readily distinguished within clusters 1 and 2. The topology of the tree allowed for identification of several putative orthologous pairs of white spruce–Norway spruce chitinase sequences (Online Resource 3).

Neighbor-joining tree showing the relationships of chitinases from white spruce, Norway spruce, and selected other plant species. The first two letters in each sequence identifier represent the species, followed by the GenBank gene ID (gi) for sequences with known functions, the full-length insert cDNA (FLIC) ID for the white spruce sequences, or the v1.0 gene model ID for the Norway spruce sequences. At, Arabidopsis thaliana; Zm, Zea mays; St, Solanum tuberosum; Ca, Capsicum annum; Os, Oryza sativa; Rg, Rehmannia glutinosa; Gh, Gossypium hirsitum; Ta, Triticum aestivum; Vv, Vitis vinifera; Nt, Nicotiana tabacum; La, Lupinus albus; Hv, Hordeum vulgare; Pg, Picea glauca; Cd, Cynodon dactylon; Sc, Secale cereale; Ma, Musa acuminate; Sr, Sesbania rostrata; Ms, Medicago sativa; Pa, Picea abies; Pm, Pseudotsuga menziesii; Vc, Vaccinium corymbosum. For the previously characterized chitinases from other plant species, the reported chitinase structural class is indicated at the end of the sequence identifier in parentheses. Within this tree, the chitinases fall into two main groups according to their major catalytic domain, the glycoside hydrolase family 18 (GH 18) and glycoside hydrolase family 19 (GH 19)
In silico domain analyses were carried out for both white spruce and Norway spruce chitinase sequences (Online Resource 3). These analyses revealed that each of the 31 white spruce and 80 Norway spruce sequences contained a catalytic domain, while 23 white spruce sequences and 40 Norway spruce sequences had a predicted signal peptide. It should be noted that the seven white spruce sequences missing the signal peptide exhibited predicted amino acid sequences not starting with a methionine (Online Resource 1). Therefore, it is likely that these sequences are incomplete at the 5′ end, not permitting identification of a signal peptide in the truncated sequence. This was also the case for some Norway spruce sequences.
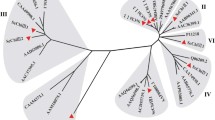
A second tree comprising only white spruce chitinase sequences allowed us to map predicted protein domains in the context of phylogenetic relationships within this gene family (Fig. 2). The topology of this tree was consistent with the larger tree, showing five distinct groups of white spruce genes within the five major clusters, with two subclusters evident within cluster 1.

Predicted structural domains of white spruce chitinases, grouped according to a phylogenetic tree constructed from the full deduced amino acid sequence of each white spruce chitinase. Bootstrap values for the neighbor-joining tree are given for the major branches on the phylogenetic tree. The Arabic numbers to the left of the tree correspond to the cluster designations from Fig. 1. The predicted structural domains for each of the white spruce sequences in the phylogenetic analysis are also indicated. The different structural domains corresponding to each sequence are depicted in the figure legend: arrow signal peptide domain, thick bar CBD, thin bar linker domain, rounded rectangle catalytic domain
White spruce and Norway spruce chitinases were assigned to classes using both the position within the tree and the presence of diagnostic domains according to Neuhaus (1999). The classification scheme is outlined in Online Resource 3. Cluster 1 contained several well-characterized class IV chitinases from other species. All full-length white spruce and Norway spruce sequences from cluster 1 that exhibited a chitin binding domain (CBD, Fig. 2, Online Resource 3) are likely to be class IV chitinases, based on the presence of the CBD, linker sequence, and greatest sequence similarity in the catalytic domain to class IV chitinases of other species (Neuhaus 1999). These grouped together into one subcluster of cluster 1 (Fig. 1). Several white spruce and Norway spruce sequences falling into the other subcluster of cluster 1 showed high sequence similarity within the catalytic domain to class IV chitinases but lacked a CBD. In older chitinase nomenclature schemes, these would have been designated class II chitinases, on the basis of the absent CBD (Hamel et al. 1997). However, according to the newer naming convention put forth by the Chitinase Nomenclature Commission, chitinases showing homology to class IV catalytic domains but lacking a CBD should be classified as class VII (Neuhaus 1999). Accordingly, these white spruce and Norway spruce sequences which would be otherwise classified as class II by similarity to other plant chitinases previously classified as class II (see Fig. 1) are likely better designated as class VII chitinases.
Similar to cluster 1, cluster 2 contained white spruce chitinases with and without a CBD. This cluster also included a number of well-characterized class I and class II chitinases from other species. A CBD was identified for three white spruce sequences within one of the subclusters of cluster 2. Given the sequence similarity between the known class I chitinases and these chitinases, together with the presence of a CBD, these three chitinases are likely to be class I chitinases. Those chitinases that showed similarity to class I chitinases but lacked the CBD fall into a different subcluster and are more likely to be class II chitinases (Fig. 1, Online Resource 3). No cluster 2 Norway spruce sequences exhibited a CBD, and none were found in this subcluster.
3D protein models were obtained by homology modeling for 30 white spruce predicted proteins. The predicted tertiary structures were aligned to each other using the FAST algorithm (Zhu and Weng 2005) to obtain an RMSD matrix of all pairwise comparisons (Online Resource 4), which were used to cluster the proteins according to their similarities (Fig. 3). The chitinases belonging to the GH 18 family showed a distinct (α/β) eight barrel fold, in contrast to the GH 19 family members, which exhibited structures rich in α-helices and included a distinct catalytic cleft. While the cluster 4 (class III) and cluster 5 (class V) chitinase sequences belonging to GH 18 were clustered similarly according to these predicted tertiary structures as they did for the phylogenetic analysis based on conventional primary sequence alignments (cf. Figs. 2 and 3), GH 19 chitinases exhibited somewhat different groupings in the former than the latter that reflected the presence of common domains, particularly the CBD. A distinctive characteristic of the structural clustering was that putative class I and class IV structures bearing the CBD clustered together, showing that this domain allowed for additional structural matching when performing the alignments (Fig. 3). Most putative class VII chitinases from cluster 1 grouped together in this structure-based tree, while the putative class II chitinases from cluster 2 did not form a distinct group.

Topology of relationships among predicted protein tertiary structures for white spruce chitinases. Structures were compared using the FAST algorithm from the ProCKSI-Server (http://www.procksi.net/). The distance matrix of RMSDs of all pairwise comparisons was standardized and used to cluster sequences and create the tree
Interestingly, the CBD region of each class I and class IV chitinase was predicted to have a high degree of intrinsic disorder (Online Resource 5). A comparable IDR was not detected in sequences lacking this domain. A small number of sequences from clusters 2 and 4 exhibited regions within the C-terminal half of the catalytic region showing lesser degrees of intrinsic disorder. Additionally, all sequences exhibited a short IDR at the C-terminal, while all full-length sequences exhibited a short IDR at the N-terminal that corresponded to the predicted signal peptide domain.
Characterization of autumnal acclimation-associated white spruce chitinases
Classification
The eight chitinases previously identified as differentially expressed in white spruce stems during the activity-dormancy transition (Galindo-González et al. 2012) fall into cluster 1 (Pg_GQ03206_D15.1, Pg_GQ03803_J20.1, Pg_GQ0207_E09.1), cluster 2 (PgGQ0183_A06.2, Pg_GQ03904_P01.1, Pg_GQ03703_O09.1, Pg_GQ03902_B01.1), and cluster 3 (Pg_GQ0046_J03.1) of Fig. 1. No genes identified as differentially expressed by Galindo-González et al. (2012) belonged to the GH 18 clusters 4 or 5. The clustering of these genes with previously characterized chitinases from other species (Online Resource 6) and the presence or absence of a CBD suggests that Pg_GQ03206_D15.1 and Pg_GQ03803_J20.1 (cluster 1) are class IV chitinases; Pg_GQ0207_E09.1 (cluster 1) is a class VII chitinase; PgGQ0183_A06.2 and Pg_GQ03904_P01.1 (cluster 2) are class I chitinases; Pg_GQ03703_O09.1 and Pg_GQ03902_B01.1 (cluster 2) are class II chitinases; and Pg_GQ0046_J03.1 (cluster 3) is a class II chitinase or chitinase-like (CTL) sequence (sensu Zhang et al. 2004; Sánchez-Rodríguez et al. 2012).
To further resolve the classification of these dormancy-associated chitinases, the sequences were examined in more detail by alignment to a class I tobacco chitinase (Neuhaus et al. 1991) that exhibits all of the canonical chitinase motifs in order to identify motifs important to their classification (Fig. 4). We used the structural classification characters outlined by Hamel et al. (1997) and Neuhaus (1999) to guide the classification. The alignment showed that Pg_GQ0183_A06.2 and Pg_GQ03904_P01.1—which showed similar domain features—shared more sequence similarity to the tobacco chitinase than the others (62 and 66 %, respectively, identity while the remaining six sequences ranged from 33 to 58 %; Online Resource 7), providing additional evidence that these two sequences are class I chitinases. In particular, the full-length Pg_GQ0183_A06.2 exhibits the N-terminal CBD separated from the catalytic domain by a linker sequence, characteristic of class I chitinases (Hamel et al. 1997; Neuhaus 1999). Pg_GQ03904_P01.1, missing the 5′ region of the sequence, does not exhibit these motifs (probably due to an incomplete sequence) but shows high sequence similarity within the catalytic domain. Pg_GQ03703_O09.1 and Pg_GQ03902_B01.1 (Fig. 4) likely belong to class II because they each lack a CBD and carboxy-terminal extension (CTE), and they exhibit a deletion in loop 2 of the catalytic domain of 17 and 14 amino acids, respectively (Hamel et al. 1997). Chitinases Pg_GQ3206_D15.1 and Pg_GQ3803_J20.1 did not exhibit any deletions in loop 2. Instead, these sequences exhibit a 13 amino acid deletion in loop 1 of the catalytic domain and 5 and 13 amino acid deletions within and flanking loop 3 of the catalytic domain. Loss of loop 1 is characteristic of class IV chitinases (Neuhaus 1999). Additionally, Pg_GQ3206_D15.1 and Pg_GQ3803_J20.1 had no identifiable CTE, but each exhibited a CBD, which is also characteristic of class IV chitinases. Pg_GQ0207_E09.1 did not show evidence of a CBD or CTE but is more similar to class IV than class I chitinases. This is consistent with its classification as a class VII chitinase based on the scheme outlined by Neuhaus (1999). Furthermore, Pg_GQ0207_E09.1 exhibited the identical deletions associated with loop 2 and loop 3 as the class IV chitinases Pg_GQ3206_D15.1 and Pg_GQ3803_J20.1. Pg_GQ0046_J03.1 did not exhibit deletions within the catalytic domain, although there was a six amino acid insertion in loop 1. Pg_GQ0046_J03.1 further displayed no evidence of a CBD or CTE, and showed a low sequence identity when compared to the other white spruce sequences (Online Resource 7). This sequence showed the most similarity to the A. thaliana sequences encoding CHITINASE-LIKE 1 (CTL1) and CHITINASE-LIKE 2 (CTL2), which are designated class II chitinases, although they lack chitinase activity (Zhang et al. 2004; Hermans et al. 2010; Sánchez-Rodríguez et al. 2012). Both AtCTL1 and AtCTL2 lack the highly conserved histidine-glutamic acid-threonine-threonine (HETT) motif within loop 1 that is essential for chitinase activity (Passarinho and de Vries 2002). Pg_GQ0046_J03.1 also lacks this HETT motif, which is replaced by threonine-glutamic acid-threonine-serine (TETS). Thus, Pg_GQ0046_J03.1 could be considered a class II chitinase or CTL.

Motif identification in dormancy-associated white spruce chitinases. The white spruce chitinase sequences were aligned to a class I chitinase from tobacco, since this sequence has all features of a traditional class I chitinase, plus a carboxy terminal extension which has been linked to vacuolar localization (Neuhaus et al. 1991). Identical residues in all sequences are highlighted in a darker shade, while conserved and similar residues are demarcated in the corresponding lighter shade. Shaded blocks represent domains: signal peptide (green), CBD (blue), catalytic domain (red), and carboxy terminal extension (yellow). Significant deletions are boxed
These more detailed sequence analyses provide further support for the class designations made in Figs. 1 and 2 and Online Resource 3, including the distinct clustering of class IV and class VII (presently called class II) genes within cluster 1, and distinct clustering of class I and class II genes within cluster 2.
qRT-PCR expression profiling
Quantitative RT-PCR was used to generate transcript profiles for each of these eight sequences in shoot tips, needles, stems, and roots during the short day-induced transition from active growth to dormancy (Fig. 5). Based upon the analyses conducted in El Kayal et al. (2011) and Galindo-González et al. (2012), bud formation was nearly complete after 70-day short day treatment and was completed before the trees were transferred to 4 °C. Neither short days nor cold temperatures are necessary to achieve cold tolerance or transition to dormancy in these seedlings, although short days accelerate these processes (Hamilton, El Kayal, and Cooke, unpublished). Six of the eight sequences showed statistically significant changes in transcript abundance in stems, the tissue in which they were originally identified as differentially expressed by microarray or proteomics analysis (Galindo-González et al. 2012). Each of these six sequences also showed statistically significant differences in transcript abundance in at least one other tissue. Most of these genes showed upregulation during the latter stages of autumnal acclimation. A notable exception was Pg_GQ0046_J03.1, which showed significant reductions in transcript abundance in all tissues over time. While some closely related genes exhibited very similar transcript profiles, e.g., the cluster 2/class I Pg_GQ0183_A06.2 and Pg_GQ03904_P01.1, other closely related genes, did not exhibit similar patterns of transcript abundance. For example, while the cluster 1/class IV chitinases Pg_GQ3206_D15.1 and Pg_GQ3803_J20.1 were closely related (Figs. 1 and 2), Pg_GQ3206_D15.1 was much more highly expressed than Pg_GQ3803_J20.1, and only the former showed significant changes over time in shoot tips, needles, and stems (Fig. 5). Similarly, the cluster 2/class II Pg_GQ03703_O09.1 showed strong upregulation in all tissues over time, while the closely related cluster 2/class II Pg_GQ03902_B01.1 showed very low transcript abundance with no significant differences in any tissue over time.

Quantitative RT-PCR transcript abundance of eight white spruce chitinases previously demonstrated to be differentially expressed in stems during the during SD-induced activity-dormancy transition (Galindo-González et al. 2012). The cluster and putative biochemical class is indicated above the sequence identifier. Expression was determined relative to TIF5A; both the target and reference genes were quantified using standard curves. Values represent the mean ± SE, n = 4. For data analyzed by GLM, data were first log transformed to meet the assumptions of normality and homogeneity of variance. For each gene, different letters denote statistical difference within a tissue at P < 0.05. In the cases of stem tissue data for Pg_GQ0183_A06.2 and Pg_GQ03703_O09.1, transformation was insufficient to meet assumptions of normality and homogeneity of variance required for parametric tests. Here, data were analyzed using a non-parametric test as described in “Materials and methods,” and significant differences within a tissue at P < 0.05 are indicated with asterisks
In silico analyses
We performed an in silico prediction of putative active site residues using COFACTOR. For this analysis, we used the five top closest enzyme homologs determined by structural alignment to determine the most likely residues involved in the active site (Online Resource 8). If the residue was part of the active site in four of the five alignments for each of the spruce chitinases, it was classified as a predicted active site residue (Fig. 6). When the modeled sections of the proteins were aligned, it was shown that the predicted active site amino acids were conserved (Holm and Sander 1994; Hart et al. 1995). For each of the proteins, two glutamic acids were predicted to constitute part of the active site (Fig. 6), but a serine that was predicted to be part of the active site was only present in five of the eight sequences: Pg_GQ03904_P01.1, PgGQ0183_A06.2, Pg_GQ0207_E09.1, Pg_GQ03803_J20.1, and Pg_GQ03206_D15.1. Three amino acids (Gln, Tyr, and Asn) important for substrate binding (Verburg et al. 1993; Hart et al. 1995) were conserved in five sequences, while Pg_GQ03703_O09.1 exhibited a Ser in place of the Tyr, Pg_GQ0207_E09.1 exhibited a Phe in place of the Tyr, and Pg_GQ0046_J03.1 exhibited a Pro in place of the Gln. In summary, four sequences exhibited all six active site residues: Pg_GQ03206_D15.1 (cluster 1/class IV), Pg_GQ03803_J20.1 (cluster 1/class IV), Pg_GQ0183_A06.2 (cluster 2/class I), and Pg_GQ03904_P01.1 (cluster 2/class I). However, only the two cluster 2/class I sequences exhibit the HETT motif found to be essential for activity (Passarinho and de Vries 2002), whereas the two cluster 1/class IV sequences exhibit a substitution in this motif.

Depiction of predicted active site amino acids in dormancy-associated white spruce chitinases. Black circles below sequences show predicted active site amino acids by structural alignment of white spruce sequences based on predicted 3D models to the top five closest enzyme homologs using COFACTOR (http://zhanglab.ccmb.med.umich.edu/COFACTOR/). An open circle is used to indicate the predicted glutamic acid that was only present in two of the five top enzyme homologs (see Online Resource 8). Plus signs indicate amino acids important in substrate binding (Hart et al. 1995)
We also carried out in silico analyses of protein properties consistent with an antifreeze function. Prediction of putative ice binding residues using AFPredictor (Doxey et al. 2006) was performed for the eight dormancy-associated white spruce chitinases and four plant chitinases from other species known to be involved in cold acclimation (Table 1). The analysis predicted that two of the four chitinases involved in cold acclimation (one from Vaccinium corymbosum and one from Secale cereale) contain OSCs, indicative of ice-binding surfaces (Doxey et al. 2006). Likewise, six out of the eight white spruce chitinases also presented OSCs. The total surface area (TSA) of ice binding was higher in five of the six white spruce proteins relative to the previously characterized proteins from blueberry and rye, and two white spruce proteins had TSA values over 200 Å. The fraction of surface area (FSA) was likewise higher in the white spruce proteins. The two white spruce proteins with the higher TSA—Pg_GQ03803_J20.1 and Pg_GQ03902_B01.1—also exhibited the higher FSAs (0.021 and 0.025). When the white spruce sequences from the predicted 3D models bearing OSCs were aligned (Online Resource 9), it was evident that many of the amino acids which made up the predicted ice-binding surfaces were conserved between sequences. For Pg_GQ03206_D15.1 and Pg_GQ03803_J20.1, the amino acids Cys, Gly, Ala, and Ser were conserved, while for Pg_GQ03703_O09.1, Pg_GQ03902_B01.1, and Pg_GQ03904_P01.1, the amino acids Thr, Ala, Gly, and Val/Ala were also conserved among the three sequences.
Digital expression profiling of spruce chitinases
We used transcriptome data derived from ca. 6.6 million Roche 454 reads representing seven different white spruce tissue libraries—vegetative buds, current year foliage, mature tree xylem, juvenile tree xylem, juvenile tree bark, megagametophytes, and somatic embryogenic tissue—to further characterize gene expression for the eight dormancy-associated white spruce chitinases (Online Resource 10). These libraries are described in detail in Raherison et al. (2012). Five of the eight genes—GQ03803_J20, GQ0183_A06, GQ03904_P01, GQ_03902_B01, and GQ03703_O09—were expressed in nearly all tissues. These five genes and GQ0046_J03 were all found to be expressed in vegetative buds, consistent with our data. GQ03206_D15 was not found to be expressed in vegetative buds in this transcriptomic dataset. Several other chitinases representing all classes and clusters were also found to be expressed in vegetative buds.
Transcripts for GQ0207_E09 were only noted in the somatic embryogenic tissue. Two thirds of the chitinases were expressed in embryogenic tissue, more than in any other tissue that was surveyed. Chitinases tended to be more highly expressed in embryogenic tissue than other tissues.
We also carried out digital expression profiling in ConGenIE of RNASeq data for 22 libraries (Nystedt et al. 2013). Fifty-six of the 76 Norway spruce sequences were represented in this dataset. At least one chitinase showed high transcript abundance for 18 of the 22 libraries (Online Resource 11). Clustering revealed co-regulated groups of chitinases, although there was no clear relationship between co-regulation and biochemical class or phylogenetic cluster. Every sequence exhibited high transcript abundance under at least one condition, but few sequences exhibited high transcript abundance under multiple conditions. Contrary to expectation, diseased or wounded samples did not show the highest proportion of highly expressed chitinases.
Three of the eight white spruce chitinases associated with autumnal acclimation had a putative Norway spruce orthologue with corresponding RNA-Seq data. GQ03803_J20.1 (class IV) and GQ3902_B01.1 (class II) were putatively orthologous to MA_159244g0020 and MA_10313114g0010, respectively. Both of these genes showed very high transcript abundance in early season developing buds. The former was not expressed more than average in any other tissue, while the latter showed moderately elevated transcript abundance in needles from girdled twigs and needles from vegetative shoots produced in 2010. GQ0207_E09.1 was putatively orthologous to MA_10427109g0010. This sequence showed high transcript abundance in late wood and moderately elevated transcript abundance in early morning needles and pineapple galls.
Twelve additional Norway spruce chitinases, mostly cluster 1/class VII, showed high transcript abundance in tissues associated with the activity-dormancy transition, namely early season buds, late season buds, and late wood. Three of these had a putative white spruce orthologue: Pa_MA_10431378g0020–GQ03811_E13.2, Pa_MA_18470g0010–WS00749_F20.1, and Pa_MA_10262473g0010–GQ03407_A15.1. Pa_MA_10431378g0020 was highly expressed in late buds, while Pa_MA_18470g0010 and Pa_MA_10262473g0010 were highly expressed in late wood.
Discussion
The spruce chitinase gene family
A total of 31 unique white spruce chitinase sequences were identified in this study (Rigault et al. 2011). Eighty distinct partial or full-length Norway spruce chitinases were identified in the v1.0 draft genome sequence (Nystedt et al. 2013), of which 53 were supported by corresponding transcripts. Given the incomplete status of the Norway spruce genome assembly, there is a high likelihood that (a) true genes were undetected, and (b) some sequences represent the same locus, although great care was taken to remove redundant sequences. As such, the number of predicted Norway spruce genes reported here should be viewed as an estimate at best and subject to revision with future versions of the Norway spruce genome. The Norway spruce results imply that the white spruce sequences identified in this study represent only a subset of the total number of chitinase genes in the white spruce genome. Mining of the v1.0 draft of the white spruce genome sequence (Birol et al. 2013) yielded fewer putative genes than expressed sequences (J. Cooke and K. Muirhead, unpublished), but this will undoubtedly improve with future versions. Notwithstanding, the available evidence from these white spruce and Norway spruce genomic resources suggest that the chitinase family in Picea species is expanded relative to that of Arabidopsis and rice, which comprise 24 and 37 members, respectively (Xu et al. 2007).
The phylogenetic tree constructed from (a) deduced amino acid sequences of white spruce, Norway spruce, and selected angiosperm species or from (b) deduced amino acid sequences of white spruce showed five well-supported clusters. Xu et al. (2007) identified six distinct clusters for a phylogeny based mainly on the Arabidopsis and rice chitinases. However, one of the clusters identified by Xu et al. comprised only three rice chitinases, which were not included in this present analysis. The study by Xu et al. (2007) showed that of the GH 19 chitinases, Arabidopsis sequences of class I and II made up one cluster and those of class IV formed a separate sister group, similar to our findings. In our study, class I and most class II chitinases fall into one cluster, while class IV and class VII (Neuhaus 1999) chitinases fall into another cluster. Earlier chitinase classification schemes did not include classes VI and VII (e.g., Hamel et al. 1997). Under the convention of these schemes, chitinases that we have designated as class VII would have been classified as class II. Hamel et al. (1997) argued that class II chitinases behave as an artificial group because (a) the domain structure is inconsistent among this group, and (b) they share several critical amino acids with both class I and class IV chitinases (Hamel et al. 1997; Tyler et al. 2010; Islam et al. 2010). Class VII was designated to accommodate this artificiality (Neuhaus 1999). Consistent with our classification, P. glauca × engelmannii and P. contorta chitinases classified by Kolosova et al. (2014) as class VII fall into cluster 1, show high sequence similarity within the catalytic domain to class IV chitinases, and lack a CBD. These sequences reported by Kolosova et al. (2014) showed high sequence similarity to Pg03803_J20.1 and Pg_GQ0207_E09.1, which we have designated as class VII. Furthermore, although an additional class VII was proposed for cotton chitinases (Li and Liu 2003), and we see that several other sequences form a distinctive cluster with the cotton chitinases (cluster 3), we believe that the classification of these enzymes is doubtful. Li and Liu (2003) proposed that the class VII differ from class II in the presence of a NYNYG domain. We found that this domain is present in several class II chitinases which lie outside this cluster (data not shown). Therefore, while these may be a distinct class of chitinases (or chitinase-like proteins), the same process of domain evolution would allow classification of these sequences as class II and not class VII.
Our study supported the notion that cluster 2/class I and cluster 2/class II chitinases likely have a common ancestor, whereas cluster 1/class VII and cluster 1/class IV chitinases share a different common ancestor (Araki and Torikata 1995; Neuhaus 1999). One of the major discriminating features between class I and class II chitinases is the absence of a CBD in the latter; similarly, the absence of a CBD discriminates class VII from class IV chitinases (Neuhaus 1999). Neuhaus (1999) suggested that the CBD has been lost multiple times during the evolutionary history of the GH 19 family; this notion is supported by the fact that other ancestral lysozymes exhibit the CBD (Hamel et al. 1997). The phylogenetic analysis presented in this study is consistent with this hypothesis. The analysis further suggests that the loss of the CBD in the class VII and class II conifer chitinases occurred following the split between angiosperms and gymnosperms, since the class IV conifer sequences are more closely related to the class VII conifer sequences than to the class IV angiosperm sequences, and the conifer class I and class II chitinases are more closely related to each other than each is to the analogous class of angiosperm chitinases.
We demonstrated that the CBD represents an IDR within the chitinases bearing this domain. IDRs are structurally pliable regions that do not assume a single, stable 3D structure but rather undergo a disorder-to-order transition upon binding of a partner molecule (Tompa 2012; Sun et al. 2013). We expect that the CBD of CBD-containing chitinases with chitinolytic activity recognizes chitin. In contrast, we predict that the CBD of CBD-containing chitinases without chitinolytic activity has other binding partners. IDRs often have the ability to interact with multiple partners, undergoing binding-induced folding to facilitate binding of these different partners (Sun et al. 2013). This raises the intriguing possibility that chitinase CBDs may interact with more than one target substrate or interacts with a given substrate in different configurations.
Putative functions of dormancy-associated white spruce chitinases
Defense against pathogens and pests
Expression of defense-associated genes during overwintering preparation including chitinases has been well-documented in conifers (Ekramoddoullah et al. 1995, 2000; Zamani et al. 2003; Galindo-González et al. 2012). Pathogen-responsive chitinases often belong to classes I and IV (e.g., Sela-Buurlage et al. 1993; Davis et al. 2002; Passarinho and de Vries 2002; Hietala et al. 2004; Islam et al. 2010; Kolosova et al. 2014) and generally exhibit diagnostic active site residues (Holm and Sander 1994; Hart et al. 1995), CBDs (Iseli et al. 1993), and a signal peptide targeting the proteins to the secretory pathway (Nielsen et al. 1997).
Two cluster 2/class I chitinases (Pg_GQ0183_A06.2 and Pg_GQ03904_P01.1) and two cluster 1/class IV chitinases (Pg_GQ03206_D15.1 and Pg_GQ03803_J20.1) exhibited hallmarks of functional chitinases (Table 2). Each of these sequences exhibited three amino acids—Glu, Glu, and Ser (Fig. 6)—demonstrated to be important for catalytic function (Holm and Sander 1994; Hart et al. 1995). However, only Pg_GQ0183_A06.2 and Pg_GQ03904_P01.1 exhibited the characteristic HETT motif of functional chitinases (Passarinho and de Vries 2002), while Pg_GQ03206_D15.1 and Pg_GQ03803_J20.1 exhibited HETG in place of HETT. Recently, Kolosova et al. (2014) demonstrated that a P. glauca × engelmannii (interior spruce) class I chitinase cDNA nearly identical to Pg_GQ0183_A06.2 and very similar to Pg_GQ03904_P01.1 encoded a protein with chitinolytic activity. In the same study, proteins encoded by P. glauca × engelmannii class II and class IV chitinase cDNAs nearly identical to Pg_GQ03904_P01.1 and Pg_GQ03206_D15.1, respectively, as well as several cluster 1/class VII P. glauca × engelmannii and Pinus contorta (lodgepole pine) chitinase cDNAs did not display chitinolytic activity (Kolosova et al. 2014). Davis et al. (2002) also reported class II Pinus elliottii (slash pine) and Pinus taeda (loblolly pine) chitinases that lacked chitinolytic activity.
Kolosova et al. (2014) demonstrated that while only class I chitinases exhibited measureable chitinolytic activity, class I, class II, and class IV interior spruce chitinases were upregulated in response to fungal pathogens (Leptographium abietinum) and weevils (Pissodes strobi). As such, we infer that the three white spruce chitinases showing the highest similarity to these interior spruce chitinases—Pg_GQ0183_A06.2, Pg_GQ03904_P01.1, and Pg_GQ03206_D15.1—could also play a role in tree defense (Table 2). Each of these three genes was substantially upregulated at the level of transcript abundance during the latter stages of the transition to dormancy, suggesting that these chitinases constitute an important component of the tree’s protective arsenal against pests and pathogens that continue to pose a threat to the tree in the autumn and early spring.
Pg_GQ0183_A06.2 and Pg_GQ03904_P01.1 exhibited the CTE shown to be necessary and sufficient for targeting of a tobacco chitinase and a barley lectin to the vacuole (Neuhaus et al. 1991; Dombrowski et al. 1993). We speculate that vacuolar-localized chitinases with chitinolytic activity may act later in the response of the plant to invasion by fungal pathogens or herbivorous insects, when the cell has been disrupted and the contents of the vacuole make contact with the pest or pathogen (Wang and Dong 2011). This model has been proposed for bean, where accumulation of chitinase in the vacuole of leaf cells will have the most effect on the invading fungus when the cell is broken (Mauch and Staehelin 2013). In contrast, it is less clear what functional role the putatively non-chitinolytic Pg_GQ03206_D15.1 plays in defense.
Wiweger et al. (2003) and Hietala et al. (2004) provided evidence that class IV chitinases are involved in developmental events such as programmed cell death and suggested that class IV chitinases with enzymatic activity that are localized to the apoplastic space may also be involved in signalling through generation of N-acetylglucosamine and/or oligonucleotides that in turn trigger changes in gene expression. If this is the case, it is conceivable that the dormancy-associated class IV chitinases identified in this study could be involved in mediating developmental events associated with the activity-dormancy transition.
Freezing acclimation
Freezing tolerance for forest trees and other perennials increases over the course of bud formation and the transition to dormancy (Hawkins and Shewan 2000; Colombo et al. 2003). Chitinases from several species have been shown to play a role in cold and freezing acclimation, with some of these having demonstrated antifreeze activity (e.g., Griffith et al. 1997; Yeh et al. 2000; Pihakaski-Maunsbach et al. 2001; Kikuchi and Masuda 2009; Jarząbek et al. 2009). Chitinases with demonstrated antifreeze activity from V. corymbosum (class II) and S. cereale (class I and II) are present in cluster 2 (Yeh et al. 2000; Kikuchi and Masuda 2009). Cluster 2 also contains PgGQ0183_A06.2 (class I), Pg_GQ03904_P01.1 (class I), Pg_GQ03703_O09.1 (class II), and Pg_GQ03902_B01.1 (class II). Several lines of evidence suggest that some of these cluster 2 white spruce chitinases play a role in freezing acclimation. Putative ice-binding surfaces were predicted in Pg_GQ03902_B01.1, Pg_GQ03904_P01.1, and Pg_GQ03703_O09.1. Pg_GQ03904_P01.1 and Pg_GQ03703_O09.1 showed strong upregulation during the latter stages of transition to dormancy, particularly following completion of bud formation and the subsequent transfer to low temperatures (4 °C). It is likely that Pg_GQ03902_B01.1 and Pg_GQ03703_O09.1 do not exhibit chitinolytic activity, as neither contained a CBD nor the full complement of putative active site and substrate binding amino acids (Table 2). Pg_GQ03904_P01.1 was truncated at the 5′ end, so the presence of a CBD cannot be determined, but the sequence did possess all of the putative active site and substrate binding amino acids. Taken together, these data suggest that Pg_GQ03904_P01.1 (cluster 2/class I) and Pg_GQ03703_O09.1 (cluster 2/class II) may contribute to freezing acclimation in shoot tips, needles, stems, and roots through antifreeze activity.
While most previously characterized chitinases from cluster 1/class IV have been implicated in embryogenesis (Dong and Dunstan 1997; Passarinho et al. 2001; Wiweger et al. 2003) and pest or pathogen responses (Stein et al. 2006; Zheng et al. 2006; Little et al. 2007; Pré et al. 2008; Islam et al. 2010), some cluster 1/class IV chitinases have been implicated in abiotic stress responses, including cold acclimation (Gong et al. 2001; Oono et al. 2006; Swindell 2006). The cluster 1/class IV chitinase Pg_GQ03206_D15.1, which was strongly upregulated during the latter stages of the activity-dormancy transition, exhibited potential ice-binding surfaces, suggesting that it could function as an antifreeze protein (Table 2).
While the cluster 2/class II Pg_GQ03902_B01.1 and the cluster 1/class IV chitinase Pg_GQ03803_J20.1 have attributes such as putative ice-binding surfaces, the lower and relatively constitutive level of expression of these genes suggests that the proteins play lesser roles in freezing acclimation. It is possible that Pg_GQ03902_B01.1 and Pg_GQ03803_J20.1 require sub-zero temperatures for upregulation and that these chitinases act in complementary timeframes during winter (Hon et al. 1995; Yeh et al. 2000).
It is notable that predicted amino acids bearing the OSCs involved in ice binding map to three distinct regions of the aligned protein sequences (Online Resource 9). It would be interesting to investigate if mutagenesis of such amino acids can influence ice-binding surfaces, once the antifreeze properties have been tested for these chitinases.
Nitrogen storage and seasonal nitrogen cycling
Perennials undergo seasonal nitrogen cycling, whereby nitrogen resources such as vegetative storage proteins (VSPs) are accumulated in perennating tissues as the growing season ends, and remobilized as needed to sustain metabolic and biosynthetic activities during the winter months and the initial phases of regrowth the following spring (Cooke and Weih 2005). VSP accumulation during the late summer and early autumn has been characterized for a number of perennials, including forest trees (Wetzel et al. 1989; Langheinrich and Tischner 1991; Avice et al. 2003). Proteins from diverse families have been recruited to function as VSPs, including chitinases. Although most chitinases characterized as VSPs are GH 19 cluster 4/class III chitinases (Peumans et al. 2002; Meuriot et al. 2004; Rao and Gowda 2008), a storage function for chitinases belonging to other classes cannot be discounted. There is also the possibility of multi-functional chitinases, such as chitinolytic chitinases that serve as VSPs (Meuriot et al. 2004) or chitinolytic chitinases with additional VSP and cold acclimation functions (Avice et al. 2003). Chitinolytic chitinases could conceivably even contribute to seasonal N cycling by releasing nitrogen from N-acetylglucosamine-containing endogenous substrates, although this is speculative.
Roberts et al. (1991) and Binnie et al. (1994) demonstrated that P. glauca × Picea engelmannii accumulated proteins during fall acclimation and that these proteins likely functioned as VSPs. The identity of these proteins was not established, however. The protein corresponding to cluster 2/class I Pg_GQ0183_A06.2 (cluster 2/class I) showed the largest accumulation of all proteins during the activity-dormancy transition (Galindo-González et al. 2012) and thus is also a candidate VSP. The pattern of transcript abundance for this sequence is in agreement with the protein accumulation pattern. Three other autumn-associated chitinases, Pg_GQ03703_O09.1 (cluster 2/class II), Pg_GQ3904_P01.1 (cluster 2/class I), and Pg_GQ03206_D15.1 (cluster 1/class IV), also showed substantial increases in transcript abundance during the activity-dormancy transition.
VSPs are often localized to vacuoles for sequestration (Wetzel et al. 1991; Sauter and van Cleve 1994). Galindo-González et al. (2012) showed that white spruce stems accumulate considerable protein in storage vacuole-like bodies in the phloem parenchyma and ray cells during the transition to dormancy, similar to VSP accumulation in other species. The CTE present on the cluster 2/class I chitinases Pg_GQ03904_P01.1 and Pg_GQ0183_A06.2 suggests that these proteins could be targeted to the vacuole, providing further evidence that these chitinases are VSP candidates. Further experiments to determine the annual pattern of protein abundance and subcellular localization are necessary to ascertain if these chitinases function as VSPs.
Interestingly, the three class III white spruce chitinases identified in this study all showed greatest expression in megagametophytes and embryogenic tissue, raising the possibility that they serve a storage role in these tissues even though they are not classical seed storage proteins.
Cell wall modification
Pg_GQ0046_J03.1 grouped with class II Arabidopsis CTLs shown to be involved in cell wall modification (Zhang et al. 2004; Sánchez-Rodríguez et al. 2012) in cluster 3. Sánchez-Rodríguez et al. (2012) demonstrated that AtCTL1 and AtCTL2 affect cellulose biosynthesis, possibly by promoting interactions between cellulose microfibrils and matrix glycans. Pg_GQ0046_J03.1 exhibited a signal peptide but lacked a CTE, suggesting that it is targeted to the cell wall (Table 2). Pg_GQ0046_J03.1, which was shown by digital expression profiling to be very highly expressed in all tissues with extensive cell wall biosynthesis activity, was the only one of the eight dormancy-associated chitinases that was downregulated during the activity-dormancy transition. The timing of downregulation mirrored growth cessation and the concomitant completion of cell maturation and showed similarities to transcript profiles of other known cell wall biosynthetic genes (El Kayal et al. 2011; Galindo-González et al. 2012). Furthermore, Pg_GQ0046_J03.1 lacked one of the conserved substrate binding amino acids and did not have a CBD, hinting that the preferred substrate for this protein is not chitin but perhaps another carbohydrate such as the glucan polymers that are bound by AtCTL1 and AtCTL2. Preliminary experiments indicate that, indeed, the protein coded for by Pg_GQ0046_J03.1 does not hydrolyze chitin in in vitro assays (El Kayal, Morris, and Cooke, unpublished).
Conclusion
In this study, we have demonstrated that a diverse array of chitinases participate in the transition of white spruce from active growth to dormancy, playing putative roles in defense, cold acclimation, seasonal nitrogen cycling, and cell wall modification. In silico analyses suggest that some of these dormancy-associated chitinases are multifunctional. No two chitinases appear to be redundant in their spatiotemporal expression pattern and inferred function. In some cases, closely related chitinases exhibit amino acid differences that lead to different functions, suggestive of neofunctionalization. In other cases, closely related chitinases with very similar sequences exhibit different patterns of transcript abundance across tissues over time, suggestive of subfunctionalization (Duarte et al. 2006; Lynch and Conery 2012). Tyler et al. (2010) hypothesized that because of a higher rate of mutation in the catalytic domain of GH 19 members than that of GH 18 members, the former exhibit adaptive functional modifications that emerge as a result of pathogen pressure and therefore increased genetic variability that could result in functional diversity. In support of this notion, Liu et al. (2011) recently demonstrated that nucleotide variation in a class IV chitinase gene was significantly associated with Pinus monticola resistance against the pathogen Cronartium ribicola. SNPs have been identified and validated for most of the white spruce chitinases reported in this study (Pavy et al. 2013). It will be of great interest to determine if non-synonymous SNPs or indels observed within populations can be connected to altered chitinase functional attributes and if there is any relationship between these altered functional attributes and signatures of selection.
In summary, our results imply that through a non-redundant complement of spatiotemporal expression patterns and functions, this suite of chitinases contribute to the tree’s ability to withstand the rigors of winter and resume growth the following spring. In-depth functional studies of these chitinases are now underway to determine whether the in vitro and in vivo functions of these proteins are consistent with the functions predicted by in silico analysis. Availability of the draft white spruce genome sequence (Birol et al. 2013) facilitates analysis of the regulation of these genes through promoter studies.
References
Araki T, Torikata T (1995) Structural classification of plant chitinases: two subclasses in class I and class II chitinases. Biosci Biotechnol Biochem 59:336–338
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi:10.1093/bioinformatics/bti770
Asante DKA, Yakovlev IA, Fossdal CG et al (2011) Gene expression changes during short day induced terminal bud formation in Norway spruce. Plant Cell Environ 34:332–346. doi:10.1111/j.1365-3040.2010.02247.x
Avice J, Le Dily F, Goulas E et al (2003) Vegetative storage proteins in overwintering storage organs of forage legumes: roles and regulation. Can J Bot 81:1198–1212. doi:10.1139/B03-122
Barthel D, Hirst JD, Błazewicz J et al (2007) ProCKSI: a decision support system for protein (structure) comparison, knowledge, similarity and information. BMC Bioinf 8:416. doi:10.1186/1471-2105-8-416
Binnie S, Grossnickle SC, Roberts DR (1994) Fall acclimatation patterns of interior spruce seedlings and their relationship to changes in vegetative storage proteins. Tree Physiol 14:1107–1120
Birol I, Raymond A, Jackman SD et al (2013) Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 29:1492–1497. doi:10.1093/bioinformatics/btt178
Büchter R, Strömberg A, Schmelzer E, Kombrink E (1997) Primary structure and expression of acidic (class II) chitinase in potato. Plant Mol Biol 35:749–761. doi:10.1023/A:1005830706507
Clendennen SK, López-Gómez R, Gómez-Lim M et al (1998) The abundant 31-kilodalton banana pulp protein is homologous to class-III acidic chitinases. Phytochemistry 47:613–619
Collinge DB, Kragh KM, Mikkelsen JD et al (1993) Plant chitinases. Plant J 3:31–40
Colombo SJ, Glerum C, Webb DP (2003) Daylength, temperature and fertilization effects on desiccation resistance, cold hardiness and root growth potential of Picea mariana seedlings. Ann For Sci 60:307–317. doi:10.1051/forest
Cooke JEK, Weih M (2005) Nitrogen storage and seasonal nitrogen cycling in Populus: bridging molecular physiology and ecophysiology. New Phytol 167:19–30. doi:10.1111/j.1469-8137.2005.01451.x
Cooke JEK, Eriksson ME, Junttila O (2012) The dynamic nature of bud dormancy in trees: environmental control and molecular mechanisms. Plant Cell Environ 35:1707–1728. doi:10.1111/j.1365-3040.2012.02552.x
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859
Davis JM, Wu H, Cooke JEK et al (2002) Pathogen challenge, salicylic acid, and jasmonic acid regulate expression of chitinase gene homologs in pine. Mol Plant-Microbe Interact 15:380–387
Dombrowski JE, Schroeder MR, Bednarek SY, Raikhel NV (1993) Determination of the functional elements within the vacuolar targeting signal of barley lectin. Plant Cell 5:587–596. doi:10.1105/tpc.5.5.587
Dong JZ, Dunstan DI (1997) Endochitinase and beta-1,3-glucanase genes are developmentally regulated during somatic embryogenesis in Picea glauca. Planta 201:189–94
Doxey AC, Yaish MW, Griffith M, McConkey BJ (2006) Ordered surface carbons distinguish antifreeze proteins and their ice-binding regions. Nat Biotechnol 24:852–855. doi:10.1038/nbt1224
Druart N, Johansson A, Baba K et al (2007) Environmental and hormonal regulation of the activity-dormancy cycle in the cambial meristem involves stage-specific modulation of transcriptional and metabolic networks. Plant J 50:557–573. doi:10.1111/j.1365-313X.2007.03077.x
Duarte JM, Cui L, Wall PK et al (2006) Expression pattern shifts following duplication indicative of subfunctionalization and neofunctionalization in regulatory genes of Arabidopsis. Mol Biol Evol 23:469–478. doi:10.1093/molbev/msj051
Ekramoddoullah AK, Hawkins B, Taylor D (1995) Characterization of a fall protein of sugar pine and detection of its homologue associated with frost hardiness of western white pine needles. Can J For Res 25:1137–1147
Ekramoddoullah AKM, Yu X, Sturrock R et al (2000) Detection and seasonal expression pattern of a pathogenesis-related protein (PR-10) in Douglas-fir (Pseudotsuga menziesii) tissues. Physiol Plant 110:240–247. doi:10.1034/j.1399-3054.2000.110214.x
El Kayal W, Allen CCG, Ju CJ-T et al (2011) Molecular events of apical bud formation in white spruce, Picea glauca. Plant Cell Environ 34:480–500. doi:10.1111/j.1365-3040.2010.02257.x
Galindo-González LM, El Kayal W, Ju CJ-T et al (2012) Integrated transcriptomic and proteomic profiling of white spruce stems during the transition from active growth to dormancy. Plant Cell Environ 35:682–701. doi:10.1111/j.1365-3040.2011.02444.x
Gong Z, Koiwa H, Cushman MA et al (2001) Genes that are uniquely stress regulated in salt overly sensitive (sos) mutants. Plant Physiol 126:363–575
Griffith M, Antikainen M, Hon W et al (1997) Antifreeze proteins in winter rye. Physiol Plant 100:327–332
Grover A (2012) Plant chitinases: genetic diversity and physiological roles. Crit Rev Plant Sci 31:57–73. doi:10.1080/07352689.2011.616043
Hamel F, Boivin R, Tremblay C, Bellemare G (1997) Structural and evolutionary relationships among chitinases of flowering plants. J Mol Evol 44:614–624
Hanfrey C, Fife M, Buchanan-Wollaston V (1996) Leaf senescence in Brassica napus: expression of genes encoding pathogenesis-related proteins. Plant Mol Biol 30:597–609
Hart J, Pfluger HD, Monzingo AF et al (1995) The refined crystal structure of an endochitinase. J Mol Biol 248:402–413
Hawkins CDB, Shewan KB (2000) Frost hardiness, height, and dormancy of 15 short-day, nursery-treated interior spruce seed lots. Can J For Res 30:1096–1105
Hermans C, Porco S, Verbruggen N, Bush DR (2010) Chitinase-like protein CTL1 plays a role in altering root system architecture in response to multiple environmental conditions. Plant Physiol 152:904–917. doi:10.1104/pp. 109.149849
Hietala AM, Kvaalen H, Schmidt A et al (2004) Temporal and spatial profiles of chitinase expression by Norway spruce in response to bark colonization by Heterobasidion annosum. Appl Environ Microbiol 70:3948–3953. doi:10.1128/AEM.70.7.3948
Holliday JA, Ralph SG, White R et al (2008) Global monitoring of autumn gene expression within and among phenotypically divergent populations of Sitka spruce (Picea sitchensis). New Phytol 178:103–122. doi:10.1111/j.1469-8137.2007.02346.x
Holm L, Sander C (1994) Structural similarity of plant chitinase and lysozymes from animals and phage. An evolutionary connection. FEBS Lett 340:129–32
Hon WC, Griffith M, Mlynarz A et al (1995) Antifreeze proteins in winter rye are similar to pathogenesis-related proteins. Plant Physiol 109:879–889
Hong J, Jung H, Kim Y, Hwang B (2000) Pepper gene encoding a basic class II chitinase is inducible by pathogen and ethephon. Plant Sci 159:39–49
Iseli B, Boller T, Neuhaus JM (1993) The N-terminal cysteine-rich domain of tobacco class I chitinase is essential for chitin binding but not for catalytic or antifungal activity. Plant Physiol 103:221–226
Iseli B, Armand S, Boller T et al (1996) Plant chitinases use two different hydrolytic mechanisms. FEBS Lett 382:186–188
Islam MA, Sturrock RN, Williams HL, Ekramoddoullah AKM (2010) Identification, characterization, and expression analyses of class II and IV chitinase genes from Douglas-fir seedlings infected by Phellinus sulphurascens. Phytopathology 100:356–366. doi:10.1094/PHYTO-100-4-0356
Jarząbek M, Pukacki PM, Nuc K (2009) Cold-regulated proteins with potent antifreeze and cryoprotective activities in spruces (Picea spp.). Cryobiology 58:268–274. doi:10.1016/j.cryobiol.2009.01.007
Johnsen Ø, Fossdal CG, Nagy N et al (2005) Climatic adaptation in Picea abies progenies is affected by the temperature during zygotic embryogenesis and seed maturation. Plant Cell Environ 28:1090–1103
Joosen RVL, Lammers M, Balk PA et al (2006) Correlating gene expression to physiological parameters and environmental conditions during cold acclimation of Pinus sylvestris, identification of molecular markers using cDNA microarrays. Tree Physiol 26:1297–1313
Karlberg A, Englund M, Petterle A et al (2010) Analysis of global changes in gene expression during activity-dormancy cycle in hybrid aspen apex. Plant Biotechnol 27:1–16. doi:10.5511/plantbiotechnology.27.1
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12:656–664. doi:10.1101/gr.229202
Kikuchi T, Masuda K (2009) Class II chitinase accumulated in the bark tissue involves with the cold hardiness of shoot stems in highbush blueberry (Vaccinium corymbosum L.). Sci Hortic (Amst) 120:230–236. doi:10.1016/j.scienta.2008.11.007
Kolosova N, Breuil C, Bohlmann J (2014) Cloning and characterization of chitinases from interior spruce and lodgepole pine. Phytochemistry 101:32–39. doi:10.1016/j.phytochem.2014.02.006
Langheinrich U, Tischner R (1991) Vegetative storage proteins in poplar. Plant Physiol 97:1017–1025
Lawton K, Ward E, Payne G et al (1992) Acidic and basic class III chitinase mRNA accumulation in response to TMV infection of tobacco. Plant Mol Biol 19:735–743
Li H, Greene LH (2010) Sequence and structural analysis of the chitinase insertion domain reveals two conserved motifs involved in chitin-binding. PLoS One 5:e8654. doi:10.1371/journal.pone.0008654
Li J, Liu J-Y (2003) A novel cotton gene encoding a new class of chitinase. Acta Bot Sin 45:1498–1496
Little D, Gouhier-Darimont C, Bruessow F, Reymond P (2007) Oviposition by pierid butterflies triggers defense responses in Arabidopsis. Plant Physiol 143:784–800. doi:10.1104/pp. 106.090837
Liu J-J, Sniezko RA, Ekramoddoullah AKM (2011) Association of a novel Pinus monticola chitinase gene (PmCh4B) with quantitative resistance to Cronartium ribicola. Phytopathology 101:904–911. doi:10.1094/PHYTO-10-10-0282
Lynch M, Conery JS (2012) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155. doi:10.1126/science.290.5494.1151
Mason ME, Davis JM (1997) Defense response in slash pine: chitosan treatment alters the abundance of specific mRNAs. Mol Plant Microbe Interact 10:135–137. doi:10.1094/MPMI.1997.10.1.135
Mauch F, Staehelin LA (2013) Functional implications of the subcellular localization of ethylene-induced chitinase and β-1,3-glucanase in bean leaves. Plant Cell 1:447–457
Melchers LS, Apotheker-de Groot M, van der Knaap JA et al (1994) A new class of tobacco chitinases homologous to bacterial exo-chitinases displays antifungal activity. Plant J 5:469–480
Meuriot F, Noquet C, Avice J-C et al (2004) Methyl jasmonate alters N partitioning, N reserves accumulation and induces gene expression of a 32-kDa vegetative storage protein that possesses chitinase activity in Medicago sativa taproots. Physiol Plant 120:113–123. doi:10.1111/j.0031-9317.2004.0210.x
Neuhaus J (1999) Plant chitinases (PR-3, PR-4, PR-8, PR-11). Ch 4. In: Datta SK, Muthukrishnan S (eds) Pathogenesis related proteins in plants. CRC Press, Boca Raton, pp 77–105
Neuhaus JM, Sticher L, Meins F, Boller T (1991) A short C-terminal sequence is necessary and sufficient for the targeting of chitinases to the plant vacuole. Proc Natl Acad Sci U S A 88:10362–10366
Nielsen H, Engelbrecht J, Brunak S, von Heijne G (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10:1–6
Nystedt B, Street NR, Wetterbom A et al (2013) The Norway spruce genome sequence and conifer genome evolution. Nature 497:579–584. doi:10.1038/nature12211
Oono Y, Seki M, Satou M et al (2006) Monitoring expression profiles of Arabidopsis genes during cold acclimation and deacclimation using DNA microarrays. Funct Integr Genomics 6:212–234. doi:10.1007/s10142-005-0014-z
Park S, Keathley DE, Han K-H (2008) Transcriptional profiles of the annual growth cycle in Populus deltoides. Tree Physiol 28:321–329
Passarinho PA, de Vries SC (2002) Arabidopsis chitinases: a genomic survey. The Arabidopsis book. The American Society of Plant Biologists, pp 1–25
Passarinho PA, Van Hengel AJ, Fransz PF, de Vries SC (2001) Expression pattern of the Arabidopsis thaliana AtEP3/AtchitIV endochitinase gene. Planta 212:556–567
Pavy N, Boyle B, Nelson C et al (2008) Identification of conserved core xylem gene sets: conifer cDNA microarray development, transcript profiling and computational analyses. New Phytol 180:766–786. doi:10.1111/j.1469-8137.2008.02615.x
Pavy N, Gagnon F, Rigault P et al (2013) Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and Nordic congeners. Mol Ecol Resour 13:324–336
Peumans WJ, Proost P, Swennen RL, Van Damme EJM (2002) The abundant class III chitinase homolog in young developing banana fruits behaves as a transient vegetative storage protein and most probably serves as an important supply of amino acids for the synthesis of ripening-associated proteins. Plant Physiol 130:1063–1072. doi:10.1104/pp. 006551.result
Pihakaski-Maunsbach K, Moffatt B, Testillano P et al (2001) Genes encoding chitinase-antifreeze proteins are regulated by cold and expressed by all cell types in winter rye shoots. Physiol Plant 112:359–371
Pré M, Atallah M, Champion A et al (2008) The AP2/ERF domain transcription factor ORA59 integrates jasmonic acid and ethylene signals in plant defense. Plant Physiol 147:1347–1357. doi:10.1104/pp. 108.117523
Raherison E, Rigault P, Caron S et al (2012) Transcriptome profiling in conifers and the PiceaGenExpress database show patterns of diversification within gene families and interspecific conservation in vascular gene expression. BMC Genomics 13:434
Rao DH, Gowda LR (2008) Abundant class III acidic chitinase homologue in tamarind (Tamarindus indica) seed serves as the major storage protein. J Agric Food Chem 56:2175–2182. doi:10.1021/jf073183i
Resman L, Howe G, Jonsen D et al (2010) Components acting downstream of short day perception regulate differential cessation of cambial activity and associated responses in early and late clones of hybrid poplar. Plant Physiol 154:1294–1303. doi:10.1104/pp. 110.163907
Rigault P, Boyle B, Lepage P et al (2011) A white spruce gene catalog for conifer genome analyses. Plant Physiol 157:14–28. doi:10.1104/pp. 111.179663
Roberts DR, Toivonen P, Mcinnis SM et al (1991) Discrete proteins associated with overwintering of spruce and douglas-fir seedlings. Can J Bot 69:437–441
Roy A, Yang J, Zhang Y (2012) COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 40:W471–477. doi:10.1093/nar/gks372
Ruttink T, Arend M, Morreel K et al (2007) A molecular timetable for apical bud formation and dormancy induction in poplar. Plant Cell 19:2370–2390. doi:10.1105/tpc.107.052811
Salzer P, Hübner B, Sirrenberg A, Hager A (1997) Differential effect of purified spruce chitinases and beta-1,3-glucanases on the activity of elicitors from ectomycorrhizal fungi. Plant Physiol 114:957–968
Sánchez-Rodríguez C, Bauer S, Hématy K et al (2012) Chitinase-like1/pom-pom1 and its homolog CTL2 are glucan-interacting proteins important for cellulose biosynthesis in Arabidopsis. Plant Cell 24:589–607. doi:10.1105/tpc.111.094672
Sauter J, van Cleve B (1994) Storage, mobilization and interrelations of starch, sugars, protein and fat in the ray storage tissue of poplar trees. Trees 8:297–304. doi:10.1007/BF00202674
Sayle RA, Milner-White J (1995) RASMOL: biomolecular graphics for all. Trends Biochem Sci 374–376
Schrader J, Moyle R, Bhalerao R et al (2004) Cambial meristem dormancy in trees involves extensive remodelling of the transcriptome. Plant J 40:173–187. doi:10.1111/j.1365-313X.2004.02199.x
Sela-Buurlage MB, Ponstein AS, Bres-Vloemans SA et al (1993) Only specific tobacco (Nicotiana tabacum) chitinases and [beta]-1,3-glucanases exhibit antifungal activity. Plant Physiol 101:857–863
Sena JS, Giguère I, Boyle B et al (2014) Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol 14:95
Stein M, Dittgen J, Sanchez-Rodriguez C et al (2006) Arabidopsis PEN3/PDR8, an ATP binding cassette transporter, contributes to nonhost resistance to inappropriate pathogens that enter by direct penetration. Plant Cell 18:731–746. doi:10.1105/tpc.105.038372.1
Sun X, Rikkerink EHA, Jones WT, Uversky VN (2013) Multifarious roles of intrinsic disorder in proteins illustrate its broad impact on plant biology. Plant Cell 25:38–55. doi:10.1105/tpc.112.106062
Swindell WR (2006) The association among gene expression responses to nine abiotic stress treatments in Arabidopsis thaliana. Genetics 174:1811–1824. doi:10.1534/genetics.106.061374
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Tompa P (2012) Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci 37:509–516
Tyler L, Bragg JN, Wu J et al (2010) Annotation and comparative analysis of the glycoside hydrolase genes in Brachypodium distachyon. BMC Genomics 11:600. doi:10.1186/1471-2164-11-600
van Hengel AJ, Tadesse Z, Immerzeel P et al (2001) N-acetylglucosamine and glucosamine-containing arabinogalactan proteins control somatic embryogenesis. Plant Physiol 125:1880–1890
van Loon LC, Rep M, Pieterse CMJ (2006) Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44:135–162. doi:10.1146/annurev.phyto.44.070505.143425
Verburg JG, Rangwala SH, Samac DA et al (1993) Examination of the role of tyrosine-174 in the catalytic mechanism of the Arabidosis thaliana chitinase: comparison of variant chitinases generated by site-directed mutagenesis and expressed in insect cells using baculovirus vectors. Arch Biochem Biophys 300:223–230
Wang D, Dong X (2011) A highway for war and peace: the secretory pathway in plant-microbe interactions. Mol Plant 4:581–587. doi:10.1093/mp/ssr053
Wetzel S, Demmers C, Greenwood JS (1989) Seasonally fluctuating bark proteins are a potential form of nitrogen storage in three temperate hardwoods. Planta 178:275–281. doi:10.1007/BF00391854
Wetzel S, Demmers C, Greenwood JS (1991) Protein-storing vacuoles in inner bark and leaves of softwoods. Trees 5:196–202. doi:10.1007/BF00227525
Wiweger M, Farbos I, Ingouff M et al (2003) Expression of Chia4-Pa chitinase genes during somatic and zygotic embryo development in Norway spruce (Picea abies): similarities and differences between gymnosperm and angiosperm class IV chitinases. J Exp Bot 54:2691–2699. doi:10.1093/jxb/erg299
Wu H, Echt CS, Popp MP, Davis JM (1997) Molecular cloning, structure and expression of an elicitor-inducible chitinase gene from pine trees. Plant Mol Biol 33:979–987. doi:10.1023/A:1005708611020
Wu CT, Leubner-Metzger G, Meins F, Bradford KJ (2001) Class I beta-1,3-glucanase and chitinase are expressed in the micropylar endosperm of tomato seeds prior to radicle emergence. Plant Physiol 126:1299–1313
Wu B, Zhang B, Dai Y et al (2012) Brittle culm15 encodes a membrane-associated chitinase-like protein required for cellulose biosynthesis in rice. Plant Physiol 159:1440–1452. doi:10.1104/pp. 112.195529
Xu F, Fan C, He Y (2007) Chitinases in Oryza sativa ssp. japonica and Arabidopsis thaliana. J Genet Genomics 34:138–150
Xue B, DunBrack RL, Williams RW, Dunker AK, Uversky VN (2010) PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804:996–1010. doi:10.1016/j.bbapap.2010.01.011
Yeh S, Moffatt BA, Griffith M et al (2000) Chitinase genes responsive to cold encode antifreeze proteins in winter cereals. Plant Physiol 124:1251–1264
Zamani BA, Sturrock R, Ekramoddoullah AKM, Wiseman SB (2003) Endochitinase activity in the apoplastic fluid of Phellinus weirii-infected Douglas-fir and its association with over wintering and antifreeze activity. For Pathol 33:299–316
Zhang D, Hrmova M, Wan C-H et al (2004) Members of a new group of chitinase-like genes are expressed preferentially in cotton cells with secondary walls. Plant Mol Biol 54:353–372. doi:10.1023/B:PLAN.0000036369.55253.dd
Zheng Z, Qamar SA, Chen Z, Mengiste T (2006) Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J 48:592–605. doi:10.1111/j.1365-313X.2006.02901.x
Zhu J, Weng Z (2005) FAST: a novel protein structure alignment algorithm. Proteins 58:618–627. doi:10.1002/prot.20331
Acknowledgments
We thank Dr. Nathalie Isabel and Marie-Claude Gros-Louis (Natural Resources Canada - Canadian Forest Service, Quebec City) for providing the white spruce seedlings used in this study; Carmen Allen (University of Alberta), Marie Gorda, and Susan Koziel (Alberta Innovates Technology Futures) for assistance with the plant experiments; Adriana Arango-Velez (formerly University of Alberta, currently with the Connecticut Agricultural Experiment Station) for conducting the non-parametric statistical tests; Troy Locke and Dr. Tony Cornish (Molecular Biology Service Unit, University of Alberta) for technical advice and instrument maintenance supporting the qRT-PCR analyses; and André Gagné and Dr. John MacKay (Université Laval) for providing cDNAs used in this study from the Arborea cDNA collection maintained at Université Laval. Funding for this research was provided to JEKC through Genome Canada, Genome Quebec, Genome British Columbia and Genome Alberta for the SMarTForests Project (www.smartforests.ca), and the Natural Sciences and Engineering Research Council Discovery Grants program.
Data archiving statement
NCBI GenBank accession numbers for all white spruce sequences are provided in Online Resource 1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Sederoff
Walid El Kayal and Jeremy S. Morris contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(PDF 427 kb)
Online Resource 2
(PDF 171 kb)
Online Resource 3
(XLSX 29.2 kb)
Online Resource 4
(PDF 726 kb)
Online Resource 5
(PDF 853 kb)
Online Resource 6
(PDF 295 kb)
Online Resource 7
(PDF 123 kb)
Online Resource 8
(PDF 199 kb)
Online Resource 9
(PDF 135 kb)
Online Resource 10
(XLSX 13 kb)
Online Resource 11
(PDF 194 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
González, L.M.G., El Kayal, W., Morris, J.S. et al. Diverse chitinases are invoked during the activity-dormancy transition in spruce. Tree Genetics & Genomes 11, 41 (2015). https://doi.org/10.1007/s11295-015-0871-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-015-0871-0