
Abstract
Mitochondrial respiratory chain deficiencies are a group of more than 100 disorders of adults and children, with highly variable phenotypes. The high prevalence of mitochondrial disorders (MIDs) urges the clinician to diagnose these disorders accurately, which is difficult in the light of highly variable and overlapping phenotypes, transmission patterns and molecular backgrounds. Fibroblast growth factor 21 (FGF-21) is an important endocrine and paracrine regulator of metabolic homeostasis. The FGF-21 transcript is reported to be abundantly expressed in liver, but little is known about the regulation of FGF-21 expression in other tissues. FGF-21 could play a role in the metabolic alterations that are often associated with mitochondrial diseases. The aim of this study was to show the association of the FGF-21 biomarker with human primary MIDs and secondary MIDs in suspected patients in Iran. Serum FGF-21 levels were determined using ELISA in 47 mitochondrial patients, including 32 with primary MIDs, 15 patients with Friedreich ataxia as a secondary MID and 30 control subjects. Serum FGF-21 levels were significantly higher in subjects with the primary MIDs (p < 0.05), compared to subjects without MIDs. However, serum FGF-21 levels did not show significant increase in subjects with FA as a secondary MID. There is an association between increasing concentrations of FGF-21 with mitochondrial diseases, suggesting FGF-21 as a biomarker for diagnosis of primary MIDs in humans. However, this biomarker is not appropriate for the diagnosis of FA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human fibroblast growth factor (FGF) family includes at least 22 members with diverse biological functions, such as cell growth, cell differentiation, and wound healing [1]. Recent data has shown that this family may play important roles in defining and regulating the functions of some endocrine-relevant tissues and organs, as well as modulating various metabolic processes [2].
FGF-21 has been identified as a potent metabolic regulator [3]. Although the physiological role of FGF-21 in humans is not understood, higher circulating levels are strongly associated with certain metabolic states, where fat is accumulated in the liver, triglycerides are high and HDL cholesterol is low (all features associated with hepatosteatosis and dyslipidemia) [4]. FGF-21 is a recently identified player in carbohydrate and lipid metabolism [5–7]. Mounting evidence from animal-based studies suggests FGF-21 as a potent metabolic regulator with multiple beneficial effects on obesity and diabetes [8, 9]. Serum levels of FGF-21, which has been suggested as a potential candidate for the treatment of diabetes, are increased in obesity. In addition, there is a positive association of serum FGF-21 levels with age, several parameters of adiposity including BMI, waist circumference, waist-to-hip ratio, fat percentage, insulin resistance, and adverse lipid profiles [10].
FGF-21 is detected in fat [11] and muscle [12], and the concept of FGF-21 as a novel hepatokine, adipokine and even a myokine were suggested. However, FGF-21 is also present in the exocrine pancreas and b-cells at drastically higher levels than in the liver, adipose tissue and muscle [13–15]. In skeletal muscles, FGF-21 protein expression is essentially comparable to that of fasted liver. Hepatic FGF-21 transcription is upregulated by fasting [16, 17], thus skeletal muscle can be an important source of FGF-21 [12].
In this study, we measured and investigated the correlation of FGF-21 concentrations in the sera of patients with primary and secondary MIDs to assess whether it is a feasible biomarker for human RCD.
Materials and methods
Patients with mitochondrial diseases were excluded by the fact that these defects were the result of primary MIDs. The genetic defect was established in mtDNA extracted from muscle and blood using established techniques. Confirmation of the mtDNA defect in each patient was sought but was not required for inclusion, assuming the clinical features were consistent and a pathogenic mutation had been confirmed within the pedigree. This approach was validated by a near-100 % rate of positive genetic tests where samples were available.
The suspected mitochondrial diseases were referred to the Medical Genetics Department at the Special Medical Center in Iran for investigation. Those with pathogenic mtDNA mutations were identified. Some FA patients were recruited as candidates for secondary MIDs because of the link between FA and MID. A total of 30 control subjects with no family history of MIDs were randomly selected.
Human FGF-21 enzyme-linked immunosorbent assay (ELISA) kits were obtained from BioVendor Laboratory Medicine (Modrice, Czech Republic). For the measurement of FGF-21. Serum samples were diluted 1:3 before the assay. Then, 100 μl of diluted sera, calibrators and quality controls were added to 96-well microtiter plates coated with an affinity-purified polyclonal anti-human FGF-21 antibody. The assay was conducted according to the manufacturer’s protocol.
A calibration curve was constructed by plotting the absorbance values at 450 nm versus the FGF-21 concentrations of the calibrators, and concentrations of unknown samples were determined using this calibration curve.
This assay was then used to measure serum FGF-21 levels in 47 Iranian mitochondrial disorders. In our study, 32 patients had primary MIDs and 15 patients had secondary MIDs (patients with FA). Our primary MID patients showed a pathogenic mutation or an abnormally low RC enzyme activity in muscle and had mitochondrial encephalomyopathy, lactic acidosis, myoclonus, epilepsy, ragged-red fibers mutation, point mutation, mtDNA deletions and mtDNA depletion.
Results
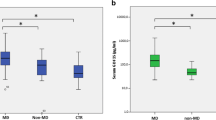
The mean concentrations of FGF-21 in serum are 70 pg/ml (range 15–309 pg/ml) in healthy adults and 114 pg/ml (42–244 pg/ml) in healthy children, respectively [18]. Interestingly, patients with primary MIDs [346.2 pg/ml (17.5–1707); n = 32] had significantly higher serum FGF-21 levels than the controls [87.1 pg/ml (32–269.1); n = 30] (Table 1). No significant difference in serum FGF-21 levels was observed between FA patients and controls. [FA patients (n = 15), 71.6 pg/ml (23.9–93.4) vs. controls [87.1 pg/ml (32–269.1); n = 30] (Table 1).
There were gender differences in serum FGF-21 levels [men (n = 17), median 261.4 pg/ml (interquartile range 17.5–1707] vs. women (n = 15), 442.4 pg/ml (25.2–1,599.1) among patients with primary MIDs, and there were no gender differences in serum FGF-21 levels [men (n = 9), median 69.2 pg/ml (interquartile range 23.9–81.8) vs. women (n = 6), 75.1 pg/ml (36.9–93.4) in FA patients] (Table 1).
Discussion
Mitochondria are vital components of all nucleated cells because of the presence of the respiratory chain (RC), and are the major sites of energy production. Given the complexity of RC genetic inheritance and its function and regulation, a proper oxidative phosphorylation system (OXPHOS) requires the full assembly of functional proteins. Mutations of the nDNA and mtDNA genes encoding the different RC sub-complexes and their regulatory factors can produce a wide range of OXPHOS diseases with extremely heterogeneous clinical manifestations [19]. RC disorders are one of the most common causes of inherited metabolic disorders [20]. Over 100 genes govern the process of oxidative phosphorylation, and mutations in any of these can cause a mitochondrial respiratory chain defect [21].
It has become clear that OXPHOS may be impaired by mutations in many mitochondrial and even non-mitochondrial proteins, or may be disturbed as a secondary effect of other biochemical defects of intracellular metabolism [22]. OXPHOS defects caused by genes encoding non-OXPHOS mitochondrial proteins, such as frataxin, are responsible for FA [23]. FA is an autosomal recessive neurodegenerative disease characterized by progressive ataxia, neuropathy, skeletal abnormalities and cardiomyopathy. In FA, mutations in the nuclear gene encoding frataxin, which is involved in iron homeostasis in mitochondria, result in severe deficiencies of iron–sulphur clusters containing complexes I–III and of the Krebs cycle enzyme aconitase [22]. Defects of intracellular metabolism, particularly excess free radical generation, including nitric oxide or peroxynitrite, may cause secondary damage to the respiratory chain [22]. Other recessive nuclear mutations are known to affect structural subunits or assembly of mitochondrial respiratory chain complexes, and only FA was included in our prevalence figures [23].
Clinical experience in mitochondrial diseases suggest that ~75 % of adult-onset mitochondrial diseases are a consequence of primary mtDNA mutations [23]. The clinical presentation of RCD is highly variable in severity, age of onset, and the combination of organ systems involved. The factors contributing to this variability are poorly understood. Consequently, the diagnosis can be challenging, and there are very limited means of objectively monitoring disease progression [24]. Genetic disorders with impaired OXPHOS are extremely heterogeneous, as their clinical presentation ranges from lesions of single tissues or specialized structures, such as the optic nerve in mitochondrial DNA-associated Leber’s hereditary optic neuropathy and in nuclear DNA-associated dominant optic atrophy, to more widespread pathologies, including myopathies, peripheral neuropathies, encephalomyopathies, cardiopathies, or complex multisystem [25].
Increased concentrations of organic acids (particularly lactic acid and TCA cycle intermediates) in blood, urine, cerebrospinal fluid, as well as in tissues, are frequently encountered in patients with OXPHOS deficiencies, especially in children and in those with the most severe biochemical defects [26]. Serum biomarkers for MIDs include lactate, pyruvate, amino acids, creatine kinase, and possibly serum creatine [18, 27]. The ratio of lactate to pyruvate is sometimes raised, especially in children and in adults with encephalomyopathies, but is frequently normal in patients with progressive myopathies. Creatine kinase concentrations in serum are occasionally increased in people with mitochondrial diseases, mostly in the disorders that cause muscle damage [18].
The sensitivity of FGF-21 in identifying mitochondrial disorders with muscle involvement compared to those for lactate, pyruvate, lactate to pyruvate ratio, and creatine kinase suggest that the measurement of FGF-21 in serum by ELISA would be a useful first-line test in patients with suspected RCDs [18]. Recent evidence has indicated that there is a direct association between daily physical activity and serum FGF-21 levels [28]. There is also a positive association between mitochondrial myopathy and increased FGF-21 levels in the sera of mice [4]. Single RC-deficient muscle fibers induce the expression of the metabolic regulator FGF-21, with increased levels also seen in mouse plasma. These results strongly suggest that FGF-21 induction is a muscle-specific response, up-regulated especially in RC-deficient fibers, and that FGF-21 expression increases upon the disease progression, suggesting that their induction is related to the pathogenesis [4].
Suomalainen [18] presented a major step forward in the investigative options for patients with mitochondrial RCDs. They have focused their research on finding a biomarker to identify mitochondrial disease. In a mouse model with mitochondrial myopathy, they found that FGF-21 was induced in muscles and that concentrations in the serum were raised [4]. FGF-21 in serum seems to be a sensitive and specific quantitative biomarker for muscle pathology in a wide range of mitochondrial disorders in adults and children, including deficiencies in one or more of the respiratory chain enzyme complexes in the skeletal muscle. FGF-21 concentrations in serum seemed to increase with increasing clinical severity of the mitochondrial disease and muscle pathology [18]. Tyynismaa and colleagues [4] detected a threefold increase of FGF-21 levels in Deletor mice plasma, compared to controls. These results raised an interesting possibility that FGF-21 could be secreted from the diseased muscle fibers upon RC deficiency. RCD of single or multiple complexes resulted in high concentrations of FGF-21 in serum, including complex I deficiency, which is the most common form [29]. Respiratory chain defects induce FGF-21 expression in mouse skeletal muscle, which leads to raised concentrations in serum [30]. Systemic availability of FGF-21 could play a role in the metabolic alterations that are often are associated with mitochondrial diseases [3].
Conclusions
Our data showed that serum FGF-21 levels in patients with primary MIDs were significantly higher than in control subjects, but showed no significant difference between patients with FA (as a secondary MID) than control subjects. Among these primary MIDs, women had moderately increased FGF-21 concentrations in serum. These results support the role of FGF-21 as a key regulator of metabolism in humans, and suggest that serum FGF-21 levels are associated with mitochondrial diseases and can be potentially used as a biomarker for primary MIDs. Although FGF-21 concentrations in serum were useful to differentiate patients with primary MIDs from healthy controls, FGF-21 concentrations were unable to diagnose FA in patients. This study provides the first clinical demonstration of the associations of serum FGF-21 levels with both primary MIDs and FA in humans.
References
Itoh N, Ornitz DM (2004) Evolution of the Fgf and Fgfr gene families. Tren Gen 20(11):563–569
Ornitz DM, Itoh N (2001) Fibroblast growth factors. Gen Biol 2(3):3005.3001–3005.3012
Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA (2005) FGF-21 as a novel metabolic regulator. J Clin Inv 115(6):1627–1635
Tyynismaa H, Raivio T, Hakkarainen A, Ortega-Alonso A, Lundbom N, Kaprio J, Rissanen A, Suomalainen A, Pietiläinen KH (2011) Liver fat but not other adiposity measures influence circulating FGF21 levels in healthy young adult twins. J Clin Endocrinol Metab 96(2):E351–E355
Nishimura T, Nakatake Y, Konishi M, Itoh N (2000) Identification of a novel FGF FGF-21 preferentially expressed in the liver. Biochimica et Biophysica Acta (BBA)-Gen Struct Exp 1492(1):203–206
Li H, Bao Y, Xu A, Pan X, Lu J, Wu H, Lu H, Xiang K, Jia W (2009) Serum fibroblast growth factor 21 is associated with adverse lipid profiles and γ-glutamyltransferase but not insulin sensitivity in Chinese subjects. J Clin Endocrinol Metab 94(6):2151–2156
Chen W, Li L, Yang G, Li K, Qi X, Zhu W, Tang Y, Liu H, Boden G (2008) Circulating FGF-21 levels in normal subjects and in newly diagnose patients with type 2 diabetes mellitus. Exp Clin Endocrinol Diabet 116(1):65–68
Moore DD (2007) Sister act. Science 316(5830):1436–1438
Reitman ML (2007) FGF21: a missing link in the biology of fasting. Cell Metab 5(6):405–407
Zhang X, Yeung DCY, Karpisek M, Stejskal D, Zhou ZG, Liu F, Wong RLC, Chow WS, Tso AWK, Lam KSL (2008) Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57(5):1246–1253
Wang H, Qiang L, Farmer SR (2008) Identification of a domain within peroxisome proliferator-activated receptor γ regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol 28(1):188–200
Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, Walsh K (2008) FGF21 is an Akt-regulated myokine. FEBS Lett 582(27):3805–3810
Wente W, Efanov AM, Brenner M, Kharitonenkov A, Köster A, Sandusky GE, Sewing S, Treinies I, Zitzer H, Gromada J (2006) Fibroblast growth factor-21 improves pancreatic β-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes 55(9):2470–2478
Johnson CL, Weston JY, Chadi SA, Fazio EN, Huff MW, Kharitonenkov A, Köester A, Pin CL (2009) Fibroblast growth factor 21 reduces the severity of cerulein-induced pancreatitis in mice. Gastroenterology 137(5):1795–1804
Tacer KF, Bookout AL, Ding X, Kurosu H, John GB, Wang L, Goetz R, Mohammadi M, Kuro-o M, Mangelsdorf DJ (2010) Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 24(10):2050–2064
Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E (2007) Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5(6):426–437
Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab 5(6):415–425
Suomalainen A (2011) Biomarkers for mitochondrial respiratory chain disorders. J Inherit Metab Dis 34(2):277–282
Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Gen 6(5):389–402
Skladal D, Halliday J, Thorburn DR (2003) Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 126(8):1905–1912
Chinnery PF (2000) Mitochondrial disorders overview. Gene reviews funded by the NIH developed at the University of Washington, Seattle initial posting
Schapira A (2002) Primary and secondary defects of the mitochondrial respiratory chain. J Inherit Metab Dis 25(3):207–214
Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM (2007) Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63(1):35–39
DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348(26):2656–2668
Di Donato S (2009) Multisystem manifestations of mitochondrial disorders. J Neurol 256(5):693–710
Smeitink JAM (2003) Mitochondrial disorders: clinical presentation and diagnostic dilemmas. J Inherit Metab Dis 26(2):199–207
Shaham O, Slate NG, Goldberger O, Xu Q, Ramanathan A, Souza AL, Clish CB, Sims KB, Mootha VK (2010) A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proc Natl Acad Sci 107(4):1571–1575
Cuevas-Ramos D, Almeda-Valdes P, Gómez-Pérez FJ, Meza-Arana CE, Cruz-Bautista I, Arellano-Campos O, Navarrete-López M, Aguilar-Salinas CA (2010) Daily physical activity, fasting glucose, uric acid, and body mass index are independent factors associated with serum fibroblast growth factor 21 levels. Eur J Endocrinol 163(3):469–477
Janssen RJRJ, Nijtmans LG, Heuvel LP, Smeitink JAM (2006) Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis 29(4):499–515
Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkilä S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola-Mjøsund KE, Tulkki V (2010) Mitochondrial myopathy induces a starvation-like response. Hum Mol Gen 19(20):3948–3958
Acknowledgments
We would like to thank for all the Iranian patients with mitochondrial diseases from the Medical Genetics Department at the Special Medical Center, Tehran, Iran. This work was supported by a grant from Dr. M. Houshmand, to whom we give our grateful appreciation.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Salehi, M.H., Kamalidehghan, B., Houshmand, M. et al. Association of fibroblast growth factor (FGF-21) as a biomarker with primary mitochondrial disorders, but not with secondary mitochondrial disorders (Friedreich Ataxia). Mol Biol Rep 40, 6495–6499 (2013). https://doi.org/10.1007/s11033-013-2767-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-013-2767-0