
Abstract
Chain reversals are often nucleation sites in protein folding. The β-hairpins of FBP28 WW domain and IgG are stable and have been proved to initiate the folding and are, therefore, suitable for studying the influence of charged residues on β-hairpin conformation. In this paper, we carried out NMR examination of the conformations in solution of two fragments from the FPB28 protein (PDB code: 1E0L) (N-terminal part) namely KTADGKT-NH2 (1E0L 12–18, D7) and YKTADGKTY-NH2 (1E0L 11–19, D9), one from the B3 domain of the protein G (PDB code: 1IGD), namely DDATKT-NH2 (1IGD 51–56) (Dag1), and three variants of Dag1 peptide: DVATKT-NH2 (Dag2), OVATKT-NH2 (Dag3) and KVATKT-NH2 (Dag4), respectively, in which the original charged residue were replaced with non-polar residues or modified charged residues. It was found that both the D7 and D9 peptides form a large fraction bent conformations. However, no hydrophobic contacts between the terminal Tyr residues of D9 occur, which suggests that the presence of a pair of like-charged residues stabilizes chain reversal. Conversely, only the Dag1 and Dag2 peptides exhibit some chain reversal; replacing the second aspartic-acid residue with a valine and the first one with a basic residue results in a nearly extended conformation. These results suggest that basic residues farther away in sequence can result in stabilization of chain reversal owing to screening of the non-polar core. Conversely, smaller distance in sequence prohibits this screening, while the presence oppositely-charged residues can stabilize a turn because of salt-bridge formation.
Similar content being viewed by others


1 Introduction
Formation of chain reversals as centers of folding initiation by interactions between hydrophobic amino-acid residues in proteins was proposed for the first time by Matheson and Scheraga [1]. Muňoz and his coworkers [2] presented an idea for the mechanism of the creation of the central part of the β conformations (β-hairpins) by hydrophobic forces and hydrogen bonds. Further study by Karplus et al. [3] underlined that the hydrophobic forces are very important in the protein folding process. In the works that have been published recently [4, 5], it was determined that both local conformational preferences to form a bent structure, and long-range interactions (hydrophobic interactions and hydrogen bonds) are a crucial part in turn formation; however creations of salt bridges by the peptide groups of the main chain does not play the main role in this process.
One approach to investigate very first stages of the formation of the tertiary structure of a protein is based on conformational studies of the parts of proteins with well-defined secondary/supersecondary structure [6–10]. Investigation of small segments of proteins convey information of local interactions which are separated from the context of the whole protein, and therefore enable us to discern the short-range interactions that are important in formation of the secondary structure blocks in biomolecules. In aqueous solution, some fragments of small proteins can form similar structure to that that a given fragment has in full protein, although such structure is more flexible, has only a small fraction of native hydrogen bonds and residue–residue packing is not exactly as that in the full protein [11]. It has been already known that, for protein folding and stability, the local interaction play a crucial role. Therefore, it should be expected that selected fragments which have the secondary structure will populate native-like structures even with lack of the tertiary contacts.
The conformational analysis of the peptide with the following sequence: Ac-(X)2-(A)7-(O)2-NH2 (where X, A, and O denote diaminobutyric acid, alanine, and ornithine, respectively) by using the NMR method [12] led us to the conclusion that its structure is bent. Further potentiometric-titration studies [13] supported this conclusion because showed that pKa1 for this system is abnormally low (2.72), compared to the value of the model compound of the ornithine side chain. This observation suggests that the distance between the ionizable groups is small, implying a bent shape. Yet further studies [14], enabled us to formulate a hypothesis that the shape of alanine-based oligopeptides, flanked by charged amino-acid residues, depends probably more on the size of the amino-acid side chains of the non-neutral flanking groups of the sequence and less of the kind of their charge (like or opposite). The bent shape of the structure which those peptides assume because of the presence of charged groups (if the side chains are not too long) makes it easier to screen the uncharged alanine segment between them from the solvent. In the peptides which contained flanking residues with longer charged side chains, the effect of protection of alanine core was manifested to a smaller extent [14].
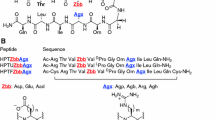
The β-hairpins from the FPB28 protein and from protein G are known to initiate folding of these proteins [4, 5]. They are, therefore, independently forming and not induced β-hairpins and, consequently, are excellent object with which to study the factors that influence β-hairpin formation. The β-hairpin from the FBP28 WW domain has two charged (lysine) residues on the opposite strand. According to our previous research [15], these residues might stabilize the structure. Conversely, the β-hairpin fragment of the IgG protein B3 domain has negatively charged (aspartic acid) residues on one side and a positively charged (lysine) residue on the opposite side. These residues probably stabilize the structure by forming a salt bridge [4, 5]. Consequently, these two systems and their variants are very suitable to investigate the influence of charged residues on β-hairpin formation (Fig. 1).

a NMR structure [44], of the FBP28 WW domain (PDB code: 1E0L); the boxed fragments of the FBP28 WW domain: FPB28 (12-18) D7, and FPB28 (11-19) D9 were synthesized and examined. b X-ray structure [45] of the protein G (PDB code: 1IGD) derived from Streptococcus—B3 domain of immunoglobulin binding protein; the boxed fragment of the 1IGD (51-56) Dag1 and its mutants were synthesized and examined
In our previous work [15] we investigated two peptides: FPB28 (11–19) (D9), with sequence YKTADGKTY-NH2, and FPB28 (12–18) (D7), with sequence KTADGKT-NH2, (Fig. 1a), by using potentiometric titration at different temperatures, circular dichroism spectroscopy (CD), and differential scanning calorimetry (DSC) measurements, respectively. Subsequently, by using the same experimental techniques, we studied [16] the DDATKT-NH2 peptide derived from protein IgG (PDB code: 1IGD 51–56) (Dag1), its variant in which D52 in the original sequence was replace with valine (DVATKT-NH2; Dag2), and two variants in which D52 was replaced with valine and D51 was replaced with ornithine or lysine, respectively (OVATKT-NH2; Dag3, and KVATKT-NH2; Dag4) (see Fig. 1b). These studies suggested that both D7 and D9 peptides undergo a folding-unfolding transition (as indicated by the presence of a maximum in the corresponding DSC curves); nevertheless, a trend of the calorimetric curve indicates that the equilibrium between conformation is established for both peptides. For D7 and D9, the Tm determined from the heat capacity curves is at t ~ 50 °C. Simultaneously, the potentiometric titration studies showed that the values of pKa1 and pKa2 are below 50 °C, which suggest that the two charged groups (lysines) are neighbours and the D7 and D9 have a tendency to be bent at low temperatures (especially D9). Above t = 50 °C (the melting point), pKa1 and pKa2 rise for D9, whereas for D7 the increase is less significant. This results suggests that the peptide turn bend to an shapeless structure and that the basic amino-acids (lysines) stabilizes the ordered state [15].
An analysis of temperature dependence of pKa values in the fragment of IgG protein and its derivatives enabled us to estimate the temperature of conformational transition [16]. In the works summarized above [12–16] we did not, however, determine explicitly the structure of the investigated peptides. Therefore, to complete our study, in this work, we investigated the conformation of D7, D9 and Dag1, Dag2, Dag3, Dag4 peptides by using the NMR spectroscopy and molecular dynamics (MD) simulations with restraints.
2 Materials and Methods
2.1 Peptide Synthesis
All peptides under study were synthesized by using the procedure describing in our earlier work [14]. The purities of the peptides were 99.98, 99.99, 99.99, 99.95, 99.98, 99.98 % for D7, D9, Dag1, Dag2, Dag3 and Dag4, respectively, as assessed by the analytical HPLC and MALDI-TOF analyses.
2.2 NMR Measurements
The NMR experiments of D7, D9 and Dag1, Dag2, Dag3 and Dag4 peptides were measured on the spectrometer VARIAN 500-MHz Unity-Plus instrument in the Intercollegiate NMR Laboratory at the Technical University of Gdansk. The following 2D 1H–1H were recorded: DQF-COSY [17], TOCSY [18] (80 ms) and ROESY [19] (200 ms) at 303 K. The samples were dissolved in H2O/2H2O (9:1 by vol). The pH value of D7 and D9 peptides was about 7 and of all Dag peptides about 5.5. The concentration of each of the samples was ~5 mM. The spectra were processed by using VARIAN 4.3 software (Varian Instruments, Palo Alto, CA) and analyzed with the SPARKY program [20]. The spectra were calibrated against DSS (sodium 4,4-dimethyl-4-silapentane-1-sulfonate) signal [21]. Proton signals were assigned based on the TOCSY spectra. The ROE inter-proton cross-peaks of all peptides under study were derived from 2D 1H–1H NMR ROESY spectra. In the first step, the ROESY peak volumes were converted to upper distance bounds by using CALIBA [22] of the DYANA package [23]. In the next step, torsion angles, based on the Bystrov–Karplus equation [24], were generated by using the HABAS algorithm of the DYANA package [25]. The upper distance limits and torsional angles were used as restraints in MD calculations.
2.3 Molecular Dynamics Calculations with NMR-Derived Restraints
Molecular dynamics calculations (MD) were carried out by using the AMBER 11 program [26, 27] with the AMBER ff99SB force field at constant volume and temperature (the NVT scheme). All simulations were performed in a periodic box of TIP3P water [28]. The linear dimensions of the box were 40.94 Å × 59.4 Å × 41.1 Å with volume 99,836.397 Å3 for D7, 59.71 Å × 59.38 Å × 41.07 Å with volume 145,620.203 Å3 for D9, 59.714 Å × 40.602 Å × 41.07 Å with volume 99,576.006 Å3 for Dag1, Dag2, Dag3, and 59.71 Å × 59.34 Å × 41.07 Å with volume 145,620.203 Å3 for Dag4, respectively. The particle-mesh Ewald procedure [29, 30] was used for the calculations of long-range electrostatic interactions at T = 303 K. All simulations were performed in a periodic box of TIP3P water (The pH of D7, D9, Dag1, Dag2, Dag3 and Dag4 peptides solutions was 3.8, 4.2, 3.3, 4.3, 5.6, 5.9, respectively. This means that all Lys side-chain amino groups were protonated and that the Asp side-chain carboxyl group was deprotonated. The Berendsen thermostat was implemented to maintain constant temperature. The peptides studied were terminally-blocked and counter ions were added for both systems studied to neutralize the peptide charge. The total simulation time was 10 ns for each trajectory, and the integration time step was 2 fs. The time-averaged restraint method (TAV) [29, 31, 32] was used to include experimental values for the calculations, with interproton-distance restraints (calculated from the intensities of the ROE signals Fig. 2). In our earlier work [12–14, 33, 34], we noticed that all-α-helical conformations are often formed if anti-ROE restraints are not imposed. Therefore, anti-ROEs between j ≥ i + 2 residue pairs (where i and j denote residue numbers) were included for those pairs of protons for which no ROE signals were observed. This means that the protons involved were assumed to be separated by more than 6 Å, which is justified because ROE signals would be observed if the distances were smaller than 6 Å.

The ROE effects and the values of the vicinal 3JHN/Hα coupling constants for D7 (a), D9 (b), Dag1 (c), Dag2 (d), Dag3 (e) and Dag4 (f) peptide
The numbers of interproton-distance restraints were: 69 for D7, 74 for D9, 72 for Dag1, 85 for Dag2, 59 for Dag3, 88 for Dag4 (including anti-ROE). The interproton distances for all three peptides were restrained with the force constant k d = 20 kcal/(mol × Ǻ2), and the angles with k θ = 2 kcal/(mol × rad2), respectively. The force constants corresponding to anti-ROE restraints were the same as those corresponding to ROE restraints.
5,000 conformations (the conformations were collected every 1,000 steps) were obtained for each peptide and the last 1,200 of those for D7 and D9 and last 800 for Dag1, Dag2, Dag3 and Dag4 were analyzed. The number 5,000 refers to the number of snapshots and not to the number of conformations. In production calculations, a single trajectory per peptide was run. Initially, for selected peptides we ran four independent trajectories and compared the resulting structures. They did not depend on trajectory. Moreover, to address the ergodicity issue, additional simulations at T = 400 K and T = 500 K, respectively, were run for D9 and Dag1. These simulations resulted in the same conformational families as those obtained at T = 300 K.
The set of the final conformations was clustered by using the MOLMOL program [35]. MOLMOL uses a hierarchical minimal spanning tree method [36–39]. The rms-deviation cut-off values of 2.5 Å for D7 and 3.0 Å for D9, respectively, over the Thr1-Lys7, Tyr1-Tyr9 residues and of 2.5 Å for Dag1, Dag2, Dag3 and Dag4 over Asp1-Thr6 (Dag1, Dag2), Orn1-Thr6 (Dag3), Lys1-Thr6 (Dag4) residues was used for the clustering. These rmsd cut-off values are relatively large in view the small size of the molecules under study. However, because of high flexibility of the molecules under study, these relatively large cut-off values were required to dissect the sets of conformations into a reasonably small number of clusters.
3 Results
3.1 NMR
The assignments of the proton NMR spectra of the D7, D9 and Dag1, Dag2, Dag3 and Dag4 peptides were done according to the standard Wüthrich procedure [40] by using the DQF-COSY, TOCSY and ROESY spectra. The chemical shifts of the proton resonances for all six peptides are listed in Table 1a–f. The fingerprint region of the TOCSY spectra, with peak assignments of D7 (A), D9 (B), Dag1 (C), Dag2 (D), Dag3 (E) and Dag4 (F) are shown in Figure 1S (supplementary materials).
The rotating-frame Overhauser enhancement (ROE) effects corresponding to the interproton distances and the 3JHN/Hα coupling constants of D7, D9, Dag1, Dag2, Dag3 and Dag4 are shown in Fig. 2 and |i − j| > 1 interactions (where i and j are the residue number) are summarized in Table 2.
3.2 D7 and D9 Peptides
For D7 peptide the Hα(i)–HN (i + 1), Hβ(i)–HN(i + 1) and HN(i)–HN(i + 1) sequential ROE signals were observed (Fig. 2a). We also observed few short-range (i)–(i + 2) or (i)–(i + 3) interactions between residues located mainly in the middle part of the D7 sequence (see Fig. 2a).
The D9 peptide differs from the D7 peptide by two Tyr residues at both ends of the peptide chain, which certainly influence the structure of D9. We observed strong sequential Hα(i)–HN(i + 1) signals along the whole amino acid sequence (see Fig. 2b) in the ROESY spectrum. The HN(i)–HN(i + 1) were observed between residues Thr3-Ala4-Asp5-Gly6-Lys7 and Thr8 (Fig. 2b). We also observed long-range (i)–(i + 6) and (i)–(i + 7) interactions between Tyr1-Thr8 and Lys2-Thr8 residues, respectively.
3.3 Dag1, Dag2, Dag3 and Dag4 Peptides
We identified 56, 64, 60 and 67 ROE signals for Dag1, Dag2, Dag3 and Dag4, respectively. For each of the analyzed Dag peptides Hα(i)–HN(i + 1) and Hβ(i)–HN(i + 1) sequential ROE connectivities were always observed (see Fig. 2c–f). We also observed HN(i)–HN(i + 1) sequential ROE interactions for all Dag peptides, especially in the central part of the peptide chain.
4 Discussion
We found in the TOCSY spectra (Figure 1S) only one set of chemical resonances for all amino acid residues, what indicates that all peptides exist as a mixture of conformations with the tendency of the inter-conversion.
The HN(i)–HN(i + 1) sequential ROE interactions can provide either an α-helix or β-turn-like structure [40]. In the β-turn like structure, long-range interactions, which fasten together the two lines, should also be observed. Such signals are observed between Thr2-Asp4, Ala3-Gly5 and Thr2-Gly5 residues for D7 peptide (Fig. 2a; Table 2). This results suggests that the shape of the conformation of D7 is bent in the middle part of the sequence and stabilized by this particular interactions. In NMR spectra we did not observe any interactions, that could clip the two ends together. However extension of D7 sequence of one amino acid residue on the N and C-terminus clearly indicated the bent-conformation preferences of these sequence.
The observed long-range interactions for D9 indicates that the D9 peptide adopts a bent structure and has a U-shape, which is in good agreement with pKa values measured for these peptides in our earlier potentiometric study [15].
All observed sequential HN/HN interactions showed that the distance between the HN(i)–HN(i + 1) atoms of Ala3/Thr4/Lys5 in all Dag peptides never exceeds 3.8 Å. For the fully extended conformation, the distance between HN(i)–HN(i + 1) atoms is about 4.3 Å. This observation suggests that the main chains of Dag1, Dag2, Dag3 and Dag4 peptide have some tendency to bend in the middle part of the sequence. On the other hand, long-range interactions are not observed for theses peptides, which suggests that their ends are, on average, not close to each other. This fact can be explained in terms of increased flexibility of the ends of the peptides which might, in turn, be caused by the presence of structures with a sequence of turns directing the chain in opposite directions (such as, e.g., two consecutive turns of a helix). Only the second-neighbor interactions between Val2 and Thr4 residues (see Table 2) are observed for Dag2, Dag3, and Dag4. This observation is in clear contrast to the D7 and D9 peptides, for which long-range interactions were observed.
It should be also emphasized that both 1H-alpha chemical shifts and vicinal 3JHN/Hα coupling constants (with values ranging 5.9–8.3 Hz, see Fig. 2c–f) of all analyzed Dag peptides indeed indicate the random coil structure, however the conservative (i)–(i + 2) interaction observed in the Dag2, Dag3 and Dag4 peptides denote a strong inclination to have a turn-like structure. Except for the interaction mentioned above we also observed many (i)–(i + 1) connectivities between residues located in the middle part of the sequence in all analyzed Dag peptides. It has been demonstrated [41] that the Dag1 peptide tends to adopt a bent conformation due to its amino acid composition, especially at lower temperatures. Moreover, the HN(i)–HN(i + 1) ROE inter links appear in the Dag1 peptide along the entire amino acid sequence while, in Dag2, Dag3 and Dag4 peptides, such signals are observed only between residues 3–4, 4–5 and 5–6. This observation suggests that the main chain of Dag1 peptide is slightly more stable than that of Dag2, Dag3 and Dag4.
In summary, analysis of the NMR spectra clearly indicates the existence of bent shape conformation of the D9 peptide with a turn located between residues {Ala-Asp-Gly}. The presence of long-range connectivities in D9 suggests that this peptide has a quite well defined structure despite its relatively small size. For D7, only a short-range interaction between Ala3(αH) and Gly5(NH) is observed, which suggests the presence of a turn in this region. The presence of a turn in D7 (there are i − i + 2 and i − i + 3 interactions seen in Fig. 2a) is supported by NMR data. It should be stressed that both 1Hα chemical shifts and vicinal 3JHN/Hα coupling constants estimated for all six analyzed peptides indicate the statistical coil structure; however, the conservative (i)–(i + 2), (i)–(i + 3) interactions observed in the D7, Dag2, Dag3 and Dag4 peptides and (i)–(i + 6), (i)–(i + 7) interactions observed in the D9 peptide might denote a tendency to form a turn-like structures.
4.1 Molecular Dynamics
The main families of conformations of two peptides (two for each of them), obtained by MD simulations with TAV derived from NMR measurements and clustered by using the MOLMOL [35] program, are shown in Fig. 3. The two dominant families constitute 40.5 and 37 % of the conformational ensembles of D7 and 39 and 28 % of D9, respectively. The two families of D7 consist of slightly bent conformations and differ by the position of the turn, which occurs at {Ala3-Asp4-Gly5} for the first and at {Thr2-Ala3-Asp4} for the second family (Fig. 3a). The conformations of (Fig. 3b) D9 have more bent shape compared to those of D7 and only the ends are flexible while the central part seems to have a well-defined bent structure. It should be noted that D9 in contradiction to D7 has, in the sequence, two flanking hydrophobic residues which play important role in hairpin formation. As in D7, the conformations of D9 of the two dominant families differ by the position of the turn (Fig. 3b). To check if the presence of some of the observed structural features could result from applying the force field, for D9 we carried out a test run without using any restraints. It was found that the dominant conformations were completely different from those obtained with restraints; in particular, no hairpin-like structures appeared (data not shown). It can, therefore, be stated that the experimental information and not the force field used determined the obtained conformations.

The average conformations of families 1 of D7 and D9 superpose well on the corresponding section of the native FBP28 WW domain (PDB: 1E0L) (Fig. 4a, b), with Cα root mean square deviation (rmsd) of 1.477 and 2.822 Å for D7 and D9, respectively. This observation suggest that the loop corresponding to the D7 and D9 sequence serves as the nucleation site in the folding of the FBP28 WW domain.

The structural alignment between the most representative conformation of a D7, b D9 peptides with the corresponding fragment of FBP28 WW structure and c Dag1, d Dag2, e Dag3, f Dag4 peptides with the corresponding fragment of 1IGD protein structure
The representatives of the main families of Dag1, Dag2, Dag3 and Dag4 peptides are shown in Fig. 4c–f. It can be seen (Fig. 4c, d) that Dag1 and Dag2 have clearly bent shapes, the bend being more pronounced for Dag1. It can be seen (Fig. 4c, d) that the conformations of these peptides superpose well on the corresponding section of the native IgG protein (PDB: 1IGD). In earlier NMR studies at low temperature [41], the interactions between Asp2 and Lys5 were observed for the Dag1 peptide. These studies have also suggested that the Dag1 fragment can be nucleation site of the IgG protein. Our study suggests that Dag2 also retains the nucleation propensity, which is, therefore not lost upon replacement of Asp2 with Val in Dag1.
Some tendency to form a chain reversal still persists for Dag3 (Fig. 4e) and, to a lesser extent, for Dag4 (Fig. 4f); however, the overall shape of Dag4 is already quite extended. Differences between shapes of the four peptides (Dag1–4) under studies are also reflected by the averaged value of the radius of gyration: 5.5, 6.0, 6.4, 7.0 Å for Dag1, Dag2, Dag3 and Dag4, respectively. This results are in harmony with our previous potentiometric studies [16], which showed that the pKa values of their basic side-chain groups are not much different from those of the reference compounds representing the isolated side chains. These peptides do not superpose that well on the corresponding section of IgG as Dag1 and Dag2 (Fig. 4e, f), which suggests that nucleation propensity is lost upon substituting a basic residue for Asp.
5 Conclusions
We carried out conformational studies by using the experimental (NMR) and theoretical methods of two peptides derived from the N-terminal β-hairpin of the form in binding protein (FPB28): KTADGKT-NH2 and YKTADGKTY-NH2, of the peptide with sequence of the immunoglobulin binding protein G from Streptococcus (DDATKT-NH2; Dag1) (the C-terminal β-hairpin of the B3 domain), of its variant, in which one aspartic acid (negatively charged) residue was replaced with a non-polar valine residue (DVATKT-NH2; Dag2), and of its two variants in which, in addition, the second aspartic acid residue was replaced with a basic (positively charged) residue (OVATKT-NH2; Dag3 and KVATKT-NH2; Dag4). It was found that both fragments of the FBP28 form bent conformations. However, the structure is more definite for the longer (D9) sequence. The D7 peptide is bent in the middle of the sequence but more flexible at the ends, in comparison with D9, because of the lack of two Tyr residues at both ends of the peptide chain, which certainly influence the structure of D9. It should be emphasized that no hydrophobic contacts between the terminal Tyr residues of D9 occur. This observation suggests that the presence of a pair of like-charged lysine residues could stabilizes chain reversal by forming a charged “coat” around the molecule.
The six-residue fragment of IgG (Dag1), and its derivatives, have some tendency to bend in the middle part of the sequence. However, the Dag3 and Dag4 peptides have clearly diminished propensity to forming chain reversals, compared with the Dag1 and Dag2 peptides. Thus, the side chains, which are oppositely charged, in Dag1 and in Dag2 have an influence on conformation to be more compact because of a higher probability of forming salt bridges in comparison with Dag3 and Dag4, while introduction of a positively charged amino-acid residue into the sequence leads to more extended conformations. It should be noted that the two positively charged residues are separated by three residues in Dag3 and Dag4 and by four residues in D7 and D9, which can be the reason why the presence of two positively charged residues disrupts chain reversal for Dag3 and Dag4, while it seems to stabilize it for D7 and D9. For shorter separation, the presence of a salt-bridge, which was observed in a previous study of Dag1 [41] seems to stabilize chain reversal. The mechanism of hairpin formation was investigated in several other studies [4, 5, 41–43, 46–48]. It was found that both local propensities that lead to the formation of a turn in a certain segment of a polypeptide chain and the formation of short-range hydrophobic contacts play a comparable role in hairpin formation. Hairpin formation thus starts from turn formation followed by zippering [46–48]. In the present work we suggest that, apart from hydrophobic interactions, the screening by charged residues can also contribute to zippering.
Abbreviations
- FBP28 protein:
-
Formin binding protein
- CD:
-
Circular dichroism
- DSC:
-
Differential scanning calorimetry
- NMR:
-
Nuclear magnetic resonance
- TOCSY:
-
Two-dimensional nuclear magnetic resonance spectroscopy
- ROESY:
-
Rotating frame nuclear Overhauser effect spectroscopy
- DQF-COSY:
-
Double quantum filtered correlation spectroscopy
- Tm :
-
Melting temperature
References
Matheson RR, Scheraga HA (1978) A method for predicting nucleation sites for protein folding based on hydrophobic contacts. Macromolecules 11:819–829
Muňoz V, Henry ER, Hofrichter J, Eaton WA (1998) A statistical mechanical model for β-hairpin kinetics. Proc Natl Acad Sci USA 11:5872–5879
Dinner AR, Lazaridis T, Karplus M (1999) Understanding beta-hairpin formation. Proc Natl Acad Sci USA 16:9068–9073
Skwierawska A, Makowska J, Ołdziej S, Liwo A, Scheraga HA (2009) Mechanism of formation of the C-terminal β-hairpin of the B3 domain of the immunoglobulin binding protein G from Streptococcus. I. Importance of hydrophobic interactions in stabilization of β-hairpin structure. Proteins Struct Funct Bioinform 75:931–953
Lewandowska A, Oldziej S, Liwo A, Scheraga HA (2010) Mechanism of Formation of the C-Terminal beta-Hairpin of the B3 Domain of the Immunoglobulin-Binding Protein G from Streptococcus. IV. Implic Mech Fold Parent Protein Biopolym 93:469–480
Bystroff C, Baker D (1998) Prediction of local structure in proteins using a library of sequence-structure motifs. J Mol Biol 281:565–577
Wetlaufer DB (1998) Folding of protein fragments. Adv Prot Chem 34:61–92
Wright PE, Dyson HJ, Lerner RA (1998) Conformation of peptide fragments of proteins in aqueous-solution—implications for initiation of protein folding. Biochemistry 27:7167–7175
Pecorari F, Minard P, Desmadril M, Yon JM (1993) Structure and functional complementation of engineered fragments from yeast phosphoglycerate kinase. Prot Engng 6:313–325
ChenWJ Huang PT, Liu J, Liao TH (2004) Involvement of the N- and C-terminal fragments of bovine pancreatic deoxyribonuclease in active protein folding. Biochemistry 44:10653–10663
Searle MS, Williams DH, Packman LC (1995) A short linear peptide derived from the N-terminal sequence of ubiquitin folds into a water-stable non-native β-hairpin. Nat Struct Biol 2:999–1006
Makowska J, Rodziewicz-Motowidło S, Bagińska K, Vila JA, Liwo A, Chmurzyński L, Scheraga HA (2006) Polyproline II conformation is one of many local conformational states and is not an overall conformation of unfolded peptides and proteins. Proc Natl Acad Sci USA 103:1744–1749
Makowska J, Baginska K, Makowski M, Jagielska A, Liwo A, Kasprzykowski F, Chmurzynski L, Scheraga HA (2006) Assessment of two theoretical methods to estimate potentiometric titration curves of peptides: comparison with experiment. J Phys Chem B 110:4451–4458
Makowska J, Baginska K, Skwierawska A, Liwo A, Chmurzynski L, Scheraga HA (2008) Influence of charge and size of terminal amino-acid residues on local conformational states and shape of alanine-based peptides. Biopolymers 90:772–782
Makowska J, Uber D, Chmurzyński L (2012) Thermodynamics of protonation equilibria of two fragments of N-terminal β-hairpin of FPB28 WW domain. J Phys Chem B116:653–659
Makowska J, Uber D, Lubowiecka D, Chmurzyński L (2014) Studies of conformational preferences of derivatives fragments of protein G (1IGD) using temperature dependent potentiometric titration methodology. J Chem Thermodyn 70:88–94
Piantini U, Sørensen OW, Ernst RR (1982) Multiple quantum filters for elucidating NMR coupling networks. J Am Chem Soc 104:6800–6801
Bax A, Freeman R (1985) Enhanced NMR resolution by restricting the effective sample volume. J Magn Reson 65:355–360
Bax A, Davis DG (1985) 2D ROESY with cw spinlock for mixing phase sensitive using States-TPPI method. J Magn Reson 63:207–213
Goddard TD, Kneller DG, SPARKY 3, University of California, San Francisco
Tiers GVD, Coon RI (1961) Preparation of sodium 2,2-dimethyl-2-silapentane-5-sulfonate, a useful internal reference for NSR spectroscopy in aqueous and ionic solutions. J Org Chem 26:2097–2098
Güntert P, Brawn W, Wüthrich K (1991) Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J Mol Biol 217:517–530
Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273:283–298
Bystrov VF, Progr NMR (1976) Spectrosc 10:41–81
Güntert P, Wüthrich K (1991) Improved efficiency of protein structure calculations from NMR data using the program DIANA with redundant dihedral angle constraints. J Biomol NMR 1:447–456
Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA (2010) AMBER 11. University of California, San Francisco
Weiner SJ, Kollman PA, Nguyen DT, Case DA (1986) An all atom force field for simulations of proteins and nucleic acid. J Comput Chem 7:230–252
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Ewald PP (1921) Die Berchnung optischer und elektrostatischer Gitterpotentiale. Ann Phys 64:253–287
Darden T, York D, Pedersen L (1993) ParticleMesh Ewald—an n.log(n) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Torda AE, Scheek RM, van Gunsteren WF (1989) Time-dependent distance restraints in molecular-dynamics simulations. Chem Phys Lett 57:289–294
Pearlman DA, Kollman PA (1991) Are time-averaged restraints necessary for nuclear-magnetic-resonance refinement—a model study for DNA. J Mol Biol 220:457–479
Makowska J, Baginska K, Liwo A, Chmurzynski L, Scheraga HA (2008) Acidic-basic properties of three alanine-based peptides containing acidic and basic side chains: comparison between theory and experiment. Biopolymers 90:724–732
Makowska J, Liwo A, Zmudzinska W, Lewandowska A, Chmurzynski L, Scheraga HA (2012) Like-charged residues at the ends of oligoalanine sequences might induce a chain reversal. Biopolymers 97:240–249
Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics 14:51–55
Shamosand MI, Hoey D (1995) Closest-point problems of the 16th IEEE symposium on the foundations of computer science. AQ 6:151–162
Rohlf FJ (1978) A probabilistic minimum spanning tree algorithm. Inf Process Lett 7:44–48
Bentley JL, Friedman JH (1978) IEEE Trans Comput C-27: 97–105
Bentley JL, Weide BW, Yao AC (1980) ACM Trans Math Software 6:563–580
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York 292
Skwierawska A, Żmudzińska W, Ołdziej S, Liwo A, Scheraga HA (2009) Mechanism of formation of the C-terminal β-hairpin of the B3 domain of the immunoglobulin binding protein G from Streptococcus. II. Interplay of local backbone conformational dynamics and long-range hydrophobic interactions in hairpin formation. Proteins: Struct, Funct, Bioinf 76:637–654
Mu Y, Nordenskiold L, Tam JP (2006) Folding, misfolding, and amyloid protofibril formation of WW domain FBP28. Biophys J 90:3983–3992
Sharpe T, Jonsson AL, Rutherford TJ, Daggett V, Fersht AR (2007) The role of the turn in β-hairpin formation during WW domain folding. Prot Sci 16:2233–2239
Macias MJ, Gervais V, Civera C, Oschkinat H (2000) Structural analysis of WW domains and design of a WW prototype. Nat Struct Biol 7:375–379
Derrick JP, Wigley DB (1994) The 3rd IgG-binding domain from Streptococcal protein-G—an analysis by X-ray crystallography of the structure alone and in a complex with Fab. J Mol Biol 243:906–918
Lindorff-Larsen K, Piana S, Dror RO, Shaw DE (2011) How fast-folding proteins fold? Science 334:517–520
Kurova P, Simone AD, Otyepka M, Best RB (2012) Force-field dependence of chignolin folding and misfolding: comparison with experiment and redesign. Biophys J 102:1897–1906
Enemark S, Rajagopalan R (2012) Turn-directed folding dynamics of b-hairpin-forming de novo decapeptide chignolin. Phys Chem Chem Phys 14:12442–12450
Acknowledgments
This work was supported by a grant from the Polish National Science Centre (NCN 2011/01/D/ST4/04497).Calculations were carried out using the resources of the Informatics Center of the Metropolitan Academic Network (IC MAN) in Gdansk.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Makowska, J., Żmudzińska, W., Uber, D. et al. A Study of the Influence of Charged Residues on β-Hairpin Formation by Nuclear Magnetic Resonance and Molecular Dynamics. Protein J 33, 525–535 (2014). https://doi.org/10.1007/s10930-014-9585-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-014-9585-7