
Abstract
The behaviour of arsenic (As) derived from tailings was investigated at the Yara Siilinjärvi apatite mine and industrial site in eastern Finland. The study assessed factors influencing the migration and fate of As and compared the anthropogenic As load to the natural geogenic background. Environmental risks related to As were assessed by examining the As concentrations in humus, glacial till, aquatic sediments, groundwater, and surface water. The occurrence and fractionation of As and the presence of secondary precipitates and geochemical transformations in the tailings and in the ambient soil and sediment were evaluated by selective extraction. The water-derived emissions were evaluated by field measurements, hydrogeochemical analysis, and modelling. Results indicate elevated environmental risks due to dust and seepage emissions from the tailings since the concentrations and mobility of As and other potentially harmful elements (PHEs) such as Co, Ni, and Zn were elevated relative to the geogenic background. These elements were mainly associated with Fe (oxy)hydroxides in the soil and their mobility was closely linked to Fe biogeochemistry. Additionally, although the concentrations of As and PHEs were high in the tailings pond and seepage water, they decreased in ambient groundwater and surface water, indicating Fe (oxy)hydroxide stability. This was supported by hydrogeochemical modelling, which indicated precipitation of Fe oxides and hydroxides. According to speciation modelling, As was present mainly as toxic trivalent arsenious acid (H3AsO3) in groundwater and as the less toxic pentavalent As acid (H2AsO4 − and HAsO4 2−) in surface water.
Zusammenfassung
Das Verhalten von Arsen (As) in/aus Tailingablagerungen wurde im Yara Siilinjärvi Apatitabbau- und verarbeitungsgebiet (Ostfinnland) untersucht. Die Studie zeigt die Faktoren, welche die Verlagerung und den chem. Abbau von As beeinflussen. Die anthropogene As-Fracht wird mit dem natürlichen, geogenen Hintergrund verglichen. Risiken für die Umwelt durch Arsen werden durch Untersuchungen der As-Konzentration in Humus, glazialem Till, aquatischen Sedimenten sowie im Grund- und Oberflächenwasser ausgewiesen. Das Vorkommen und die Fraktionierung von As und das Auftreten sekundärer Fällungsprodukte sowie geochemischer Transformation in den Tailingablagerungen und in umliegenden Böden und Sedimenten werden durch selektive Extraktionen bestimmt. Die wasserbürtige Emission ist durch Felduntersuchungen, hydrogeochemische Analysen sowie einer Modellierung berechnet worden. Die Ergebnisse zeigen erhöhte Umweltrisiken infolge Staub- und Sickerwasseremissionen aus den Tailingablagerungen, wenn die relativ Konzentration und Mobilität von As sowie weiteren potenziell schädlichen Elementen (PHEs) wie Co, Ni und Zn gegenüber dem geogenen Hintergrund erhöht ist. Diese Elemente sind hauptsächlich an Fe(oxy)hydroxiden der Böden gebunden, wobei ihre Mobilität maßgeblich von der Eisen-Biogeochemie beeinflusst wird. Obwohl die As und PHE-Konzentrationen sowohl in den Tailingablagerungen als auch im Sickerwasser hoch sind, verringern sie sich aufgrund der Fe(oxy)hydroxid-Stabilität im umgebenden Grund- und Oberflächenwasser. Dieses konnte durch hydrogeochemische Modelle nachgewiesen werden, bei denen Fällungen von Fe-oxiden und –hydroxiden berechnet wurden. Die Speziationsmodellierungen zeigten, dass As im Grundwasser hauptsächlich als toxische trivalente arsenige Säure (H3AsO3), im Oberflächenwasser als weniger toxische pentavalente Arsensäure (H2AsO4 − und HAsO4 2−) vorliegt.
Resumen
Se investigó el comportamiento del arsénico (As) proveniente de las colas de la mina de apatita y del sitio industrial Yara Siilinjärvi en el este de Finlandia. El estudio relevó los factores que influyen la migración de As y comparó el As antropogénico agregado al geogénico y que constituye el valor natural (fondo). Los riesgos ambientales relacionados al As se relevaron examinando las concentraciones de As en humus, sedimentos de origen glaciar, sedimentos acuáticos, aguas subterráneas y superficiales. Mediante extracción selectiva, se evaluó la aparición y el fraccionamiento de As, la presencia de precipitados secundarios y las transformaciones en las colas y en suelos y sedimentos. Las emisiones debidas al agua fueron evaluadas por mediciones de campo, análisis hidrogeoquímico y modelado. Los resultados indican elevados riesgos ambientales debido a las emisiones de polvo y las filtraciones desde las colas ya que las concentraciones y la movilidad de As y otros elementos potencialmente peligrosos (PHEs) tales como Co, Ni y Zn fueron relativamente elevados comparándolos con los valores de fondo. Estos elementos estaban principalmente asociados con (oxi)hidróxidos de Fe en el suelo y sus movilidades están fuertemente asociados a la biogeoquímica del Fe. Además, aunque las concentraciones de As y PHEs eran altas en los diques de cola y en las filtraciones de agua, decrecían en las aguas subterráneas y superficiales indicando estabilidad del (oxi)hidróxido de Fe. Esto fue justificado por modelado hidrogeoquímico que indicó la precipitación de óxidos e hidróxidos de Fe. De acuerdo al modelo de especiación, As estaba presente principalmente en estado trivalente y tóxico, como ácido arsenioso, (H3AsO3) en agua subterránea y en formas ácidas en estado pentavalente menos tóxico (H2AsO4- y HAsO42-) en el agua superficial.
摘要
研究了芬兰Yara Siilinjärvi磷灰矿尾矿中砷(As)的行为特征,评价了影响砷迁移与归宿的因素,对比了人为砷荷载与砷天然背景值。通过检测腐殖质、冰碛物、水生沉淀、地下水和地表水中的砷浓度,评价了砷的污染风险。通过选择性提取分析了尾矿及周围土壤和沉积物中砷的存在、分馏、次生沉淀和化学转化。采用野外监测、水文地球化学分析和水文地球化学模拟等方法评价了尾矿废水特征。结果表明,源自尾矿的粉尘和废水使砷和其它潜在有害元素(PHEs如钴、镍、锌等)的浓度及活性高于背景值,砷的污染风险提高。这些有害元素主要与土壤中铁(氧)氢氧化物有关,而它们的活性与铁的生物地球化学过程密切相关。虽然尾矿库和溶滤液中砷和其它潜在有害元素浓度较高,但它们在周围地下水和地表水中的浓度已经降低,表明铁(氧)氢氧化物已经稳定。水文地球化学模拟能够证明铁氧化物和氢氧化物沉淀、稳定的过程。化学形态分析结果表明,砷在地下水和地表水中的形态分别为有毒的三价砷酸(H3AsO4)和毒性稍弱的五价砷酸(H2AsO4 −和HAsO4 2−)。
Similar content being viewed by others

Introduction
Arsenic (As) is a ubiquitous metalloid that is found in low concentrations in the atmosphere, in aquatic environments, in soils and sediments, and in organisms. It occurs as a major constituent in more than 200 minerals as elemental As, arsenides, sulfides, oxides, arsenates, and arsenites (Smedley and Kinniburgh 2002). Its presence in the environment can be due to natural processes, such as weathering, biological, and volcanic activity, or anthropogenic activities (Cullen and Reimer 1989; Huang et al. 2011; Pongratz 1998). As is problematic because of its relatively high mobility over a wide range of redox conditions and its toxicity to humans, animals, and plants (Turpeinen et al. 1999). In natural conditions, As speciation is controlled by chemical properties such as pH and redox conditions, along with absorption, desorption, and ion exchange reactions (Ferguson and Gavis 1972; Sadiq 1997). As adsorption is high at low pH and decreases with increasing pH (Xu et al. 1991). At all pH values, arsenate bonds more easily than arsenite and therefore, arsenite is also more mobile than arsenate (Kim et al. 2012). Moreover, once released, As is highly bioavailable (Naidu 2012). However, in acidic aquatic environments, even a slight increase in pH may lead to adsorption of As onto Fe (oxy)hydroxide particles that settle, decreasing the amount of dissolved As. Because of this, dissolved As does not usually migrate far from its source in surface water (Straskraba and Moran 2006).
Human activities, such as mining and industrial activities, can greatly affect the stability, mobility, and toxicity of As by changing the chemistry and redox conditions of water and soil (Hakala and Hallikainen 2004; Henke 2009; Lahermo et al. 1996). Therefore, even in areas naturally poor in As, anthropogenic activities may concentrate As in solid wastes or drainage and create a serious threat to human health and the environment (Henke 2009; Lahermo et al. 1996).
Since different As species exhibit wide-ranging levels of toxicity to various organisms, it is important to analyse As speciation and fractionation, not just total concentration. This can also be assessed using hydrogeochemical modelling software, such as PHREEQC (Parkhurst and Appelo 1999). Chemical fractionation by selective extraction has also been widely used to assess solid-phase distribution, retention, and mobility of As and other harmful substances in soils and sediments (e.g. Filguieras et al. 2002; Kahelin and Kallio 2004; Räisänen and Hämäläinen 1991; Räisänen et al. 1992; Tessier et al. 1979). It can also be used to identify the adsorbents that are controlling the mobility of As in the environment (e.g. Filguieras et al. 2002; Räisänen and Hämäläinen 1991; Räisänen et al. 1992; Tessier et al. 1979). Chemical fractionation reveals the proportion of easily leachable and available As in soil and water and can thus be used in ecological risk assessment (ERA) (c.f. Filguieras et al. 2002; Kahelin and Kallio 2004). A combination of geochemical and hydrogeochemical methods has been helpful in assessing the migration of industrial-derived emissions and alterations in the environment (e.g. Heikkinen et al. 2009a,b; Parviainen 2012). Moreover, ERA can be used to identify a certain hazard and evaluate the probability that an adverse effect will occur as a result of an exposure to a stressor (Wang et al. 2012).
Every year 250,000 t of tailings are recovered as a byproduct of sulphuric acid production in Siilinjärvi. The sulphuric acid is required to process the apatite ore into phosphoric acid which is used in the fertilizer and animal feed production. The sulphuric acid is produced by reprocessing the byproduct pyrite from the Pyhäsalmi copper and zinc mine located in Pyhäjärvi, in western Finland, about 130 km northwest of the Yara industrial site. We aimed to elucidate the factors contributing to the fate and risks of As migrating from the tailings pond to the surrounding environment at the Yara Siilinjärvi industrial site and apatite mine, in eastern Finland. Also, the anthropogenic As load from the tailings was compared to the natural, geogenic background to estimate the ecological risks associate with the As emissions. We used a combination of geochemical and hydrogeochemical analysis, modelling, and risk assessment.
Methods
Study Area
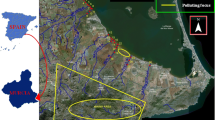
The study area, located at 63°6′N 27°45′E (Fig. 1), consists of a tailings pond and its ambient environment close to the Yara Suomi Oyj’s industrial site, fertilizer plant, and apatite mine. The plant started operating in 1969 and the mine has operated since 1979. The plant mainly produces fertilizers and phosphoric acid but also other industrial chemicals. The mine produces apatite, biotite, and calcite (ELY 2006). The Fe tailings are a byproduct of the sulphuric acid factory and have been deposited at the site since 1969. The study site is located 3 km from the city of Siilinjärvi and the nearest residential houses are ca. 500 m from the tailings. Lake Kuuslahti is located 200 m from the tailings. The land use in the vicinity of the study site mainly consists of forest and agricultural land. In 2012 a few million tons of tailings was deposited at the site, which is 21 ha in area (AVI 2011).

Location and overview of the study site showing the sampling sites in the vicinity of calcinate tailings at Yara industrial site
The pyrite (FeS2) is calcinated at a high temperature. In calcination, sulphur is oxidized to gaseous sulphur dioxide; the decomposing agent is Fe oxide. Before 1983, pyrrhotite (Fe1−XS) or pyrrhotite mixed with cobalt-concentrate or pyrite were the main materials used to produce sulphuric acid. The third calcination facility started in 1983, which increased the production of the tailings as well as the amount of seepage from the tailings (Huttunen 2012). The tailings consist mainly of hematite (99.4 %), with an Fe content of 66 %. According to earlier studies (AVI 2011; Vestola et al. 2012), the Fe tailings at Yara Suomi Oyj has a high As content (500–654 mg kg−1), which exceeds the upper guideline for soil in Finland (100 mg kg−1). The tailings also contain high levels of Ba, Cu, Co, and Zn. Arsenic occurs as arsenic trifluoride (AsF3) and with other metal impurities (Co, Zn, Cu) in an oxidized form. The tailings have been analysed by the certified leaching tests of the European Committee for Standardization (CEN). In the leaching tests, the solubility of As, Cu, Zn, and SO4 were high, exceeding the limits for hazardous waste (AVI 2011; ELY 2006; Vestola et al. 2012). The water content of the tailings was approximately 19 % and since the particle size is very fine (80 % <90 μm), the tailings are a potential source of dust emission, especially during extreme weather events. Therefore, Yara Suomi Oy is performing preventive actions (irrigation, covering with soil, revegetation) to control dust emissions (AVI 2011; ELY 2006).
The bedrock and sediments of the study area are naturally poor in As. The bedrock is mostly a Meso- and Neoarchean-basement gneiss complex, with rocks of various heterogenic migmatites, homogenic tonalitic, granodioritic granitoids, and granites. Migmatite also includes trondhjemitic to granodioritic veins and more homogenous gneiss, with varying composition of granodiorite to quartz diorite. On the west side of the study area is a north-trending, 16 km long and 1.5 km wide alkaline rock complex of carbonatite/glimmerite-syenite. It contains apatite core that is mined as a raw material for phosphoric acid and phosphate fertilizers. The tailings site is located above the basement gneiss complex (Lukkarinen 2008). The quaternary deposits of the area consist mainly of glacial till. In places there are some more permeable deposits of glaciofluvial gravel and sand but no continuous groundwater reserve is present in the area. In valleys and closer to lakes, the surface sediments are lacustrine clays, fine silts, and fine sands (Lukkarinen 2008). The As concentration of local bedrock is below 20 mg kg−1 and due to the chemical characters of the local bedrock the As concentrations of the till (<10 mg kg−1), stream sediments (<5 mg kg−1), stream water (<1 μg L−1) and ground water (<10 μg L−1) are naturally low (e.g. Koljonen et al. 1992; Lahermo et al. 1996; Tarvainen 2004; Tarvainen and Mannio 2004).
The area’s runoff flows towards Kuuslahti Lake, located approx. 200 m from the tailings. The tailings are surrounded by double ditches to collect the seepage water. The runoff and seepage water from the tailings (360 m3 d−1) are collected by the inner ditch and flows to seepage ponds located between the lake and the tailings (Fig. 1). This water is then reused as process water in the sulphuric acid factory. The outer ditch collects runoff water from the surrounding environment and leads them through two periodically monitored ditches to Kuuslahti Lake. The water in the two ditches is prevented from mixing by a compressed layer between the ditches. This layer continues below the bottom of the outer “clean water” ditch. Polluted groundwater from the tailings migrates to the fractured bedrock below the tailings site, from which it is pumped and directed back to the tailings ponds to prevent the polluted groundwater from flowing towards Kuuslahti Lake. The pumping started in 1999–2000 (AVI 2011). The tailings pond consists of old and new parts. The bottom and sides of the newer pond are a manually compressed mixture of till and calcite with a low permeability. The top part of the structure is under-drained to lower the water pressure in the tailings and till. This structure prevents seepage water flow into the surrounding environment. The bottom of the oldest part of the tailings site is not manually compressed, but the old site is no longer used and is mostly landscaped (AVI 2011).
Sampling and Chemical Analyses
Water and sediment sampling points were selected based on earlier studies provided by Yara and environmental authorities as well as hydrogeological and sedimentological characteristics of the site. A total of 49 samples were taken from 16 spots near the tailings in May and September, 2012 (Fig. 1). In addition, humus and subsoil samples were taken 5.5 km from the north of the study site to evaluate the geogenic background concentration. In addition to these background samples, values obtained from the National benchmark database Tapir (GTKs national benchmark database TAPIR) and from earlier studies (AVI 2011; ELY 2006; Koljonen et al. 1992; Lahermo et al. 1996; Tarvainen 2004; Tarvainen and Mannio 2004) were used to evaluate the natural geogenic background.
The till samples were taken from topsoil (5 samples) and subsoil (24 samples), above and below the groundwater level, using a handheld percussion drill. The humus and stream sediments were taken using a plastic shovel, sediment net, and soil corer. A fresh tailings sample was collected in a bucket from the conveyor belt before it was deposited. The 23 cm long lake bottom sediment core was taken from the Kuuslahti Lake using a Sediment Limnos sampler in September 2012. The physical properties (pH, Eh, EC) of soil and sediment samples were measured in situ using Mettler Toledo® instrument.
The soil samples were analyzed and pre-treated in the accredited (SFS-EN ISO/IEC 17025:2005) Labtium laboratory in Kuopio. The mobile and solid-phase fractionation of trace metals were determined using a three-step non-sequential (parallel) selective extraction procedure, in which the separate samples were leached with reagents of increasing dissolution strength (e.g. Filguieras et al. 2002; Kahelin and Kallio 2004; Räisänen and Hämäläinen 1991; Tessier et al. 1979). For quality control, blank samples were analysed in parallel with the research samples. Prior to selective extraction, samples were either freeze-dried (organic samples), or warm dried at 40 °C (minerogenic samples) and sieved further to <2 mm grain size. For total concentration measurements, the samples were ground in a hardened steel bowl (Kahelin and Kallio 2004; Niskavaara 1996; Räisänen and Hämäläinen 1991). The total concentration of metals was made by dissolution with hot aqua regia (12 M HCl + 4 M HNO3, 3:1) for minerogenic samples and with hot nitric acid (7 M HNO3) for samples that contained more than 10 % organic material, i.e. humus and sediments samples. The readily exchangeable fraction consisting of chemically adsorbed elements and phases was determined by ammonium acetate extraction (1 M NH4Ac, 1:60) with continuous shaking at pH 4.5 for 2 h. The fraction adsorbed on poorly crystalline (oxy)hydroxides was determined by ammonium oxalate extraction (0.2 M NH4-oxalate, 1:100) with continuous shaking at pH 3.0 for 3 h. Since the stronger extractions are also expected to dissolve phases leached in weaker solutions, the amount of each fraction is calculated by subtracting the concentration obtained in weaker extractions from the concentration obtained in the next stronger extraction step (e.g. Filguieras et al. 2002; Kahelin and Kallio 2004; Niskavaara 1996; Räisänen et al. 1992; Tessier et al. 1979). The concentrations were measured using inductively coupled plasma optical emission spectroscopy (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS) (e.g. Kahelin and Kallio 2004; Räisänen and Hämäläinen 1991). Mineralogical analysis was made for the tailings sample using X-ray fluorescence (XRF). The lake bottom sediment samples were dated using the radiocaesium (137Cs) method based on locating the current depth of the distinct 1986 Chernobyl nuclear and nuclear weapon testing (1963 fallout maximum) in the sediment profile by detecting the caesium activity with an Ortec gamma spectrometer. Records of the vertical distribution of 137Cs in sediments can be used to provide nearly accurate dates; hence, concentrations from the bottom of the core were used as background values to evaluate the contamination of the Lake Kuuslahti sediments caused by industrial and mining activities (Jaakkola et al. 1981; Klaminder et al. 2012). The final dating was based on linear extrapolation without correcting for sediment compaction. The lake bottom sediments were analysed only for total concentrations with hot nitric acid (7 M HNO3), since high organic content may accelerate the precipitation of Fe, resulting in altered concentrations.
Groundwater samples were collected using a tube bailer after water volumes in the wells were replaced by pumping. Surface water samples were collected in bottles from the ambient ditches, seepage ponds, and the ditches discharging to the Lake Kuuslahti. Two lake water samples were collected at depths of 2 and 17 m from Kuuslahti Lake using a Water Limnos sampler on September 2012. The alkalinity of the water samples was measured in situ using a HACH® titrator (sulphuric acid titration). The physical properties (pH, Eh, EC, DO, T) of water samples were measured in situ using a YSI® Professional Plus instrument. The water samples were analyzed for total and soluble metal and metalloid concentrations, anions, pH, redox, and alkalinity. The acid-soluble metals and metalloids in non-filtered water were measured by decomposing the sample with nitric acid. The concentrations of metals and metalloids dissolved in water were analysed using a filtered water sample. Both types of samples for metal and metalloid analyses were preserved using 0.5 mL of NHO3 immediately after sampling at the site and analysed in laboratory using ICP-MS and ICP-OES. Anion determination was done from untreated sample using an ion chromatograph (IC).
The metal speciation in water was modelled using the geochemical equilibrium model PHREEQC (Parkhurst and Appelo 1999) using the MinteqA2 database. Spearman’s rho correlation coefficients were calculated for soil and water analysis data using SPSS software (SPSS Statistics 21, IBM Corporation).
Ecological Risk Assessment
Ecological risks can be assessed by calculating the risk characterization ratio (RCR) for different elements in soil, water, or sediment. RCR was calculated as the ratio of the total concentrations or PEC (predicted environmental concentration) and predicated no-effect concentration (PNEC). PNEC is a risk assessment parameter that represents the concentration of a pollutant that is unlikely to harm the species involved, with respect to the effect being studied. Should the PEC exceed the PNEC, the concentration may be harmful for the environment in proportion to the ratio of the PEC to PNEC. If the PEC is less than the PNEC, there is little risk. The calculated risk assessment provides an estimate of the level of exposure, on the basis of which the risk estimate is conducted (Traas 2001; Wang et al. 2012). ERA studies are generally performed in compliance with international (e.g. OECD, ECHA, EPA) and/or national guidelines. Finnish benchmark values for selected elements (Hg, Cd, Co, Cr, Cu, Pb, Zn, V, Tl, B, Ba, Mo, Se, Sn, As) are provided in the TAPIR national benchmark database of the Geological Survey of Finland (Tapir database).
The quantitative ERA carried out in this research was based on a conceptual site model (CSM), soil and water sampling, and geochemical analyses. The ERA did not include ecotoxicological tests or bioassays. The values for the assessment are derived from literature, national and international benchmark studies, and guideline values, as well as the field monitoring data (natural background). The first assessment of the soil samples was made by comparing the soluble fraction (NH4-acetate extracted) and chemically adsorbed fraction (NH4-oxalate extracted) towards the total fractions (aqua regia or nitric acid extracted) of As. To quantitatively assess the potential ecological risk of As and other PHEs at the site, the environmental values were compared with the PNEC values for soil and fresh water sediments by the European Chemicals Agency (ECHA), the PNEC values for soil by Finnish Environmental Institute (Reinikainen 2007), local background concentrations obtained in this study, and benchmark values from a nationwide survey in Finland by the GTK (Koljonen et al. 1992; Lahermo et al. 1996; Tapir database; Table 1). Additionally, soluble concentrations of As and other PHEs in water samples were compared to total concentrations and to natural background concentrations obtained from the literature (Koljonen et al. 1992; Lahermo et al. 1996; Tarvainen 2004; Tarvainen and Mannio 2004). The PNEC values of ECHA, Finnish concentration limits and quality standards for household water, and the quality standards for groundwater were compared to our measured concentrations (Table 1).
Results and Discussion
Contaminant Migration from Tailings
Stability of Fe-oxides has an effect on the mobility of PHEs and especially As from tailings. According to the XRF mineralogical analyses, the Fe content and especially Fe-oxide content of the tailings was high (Table 2). Based on the geochemical analysis (Table 2), the As content of the tailings exceeded the upper reference values for contaminated soils and relatively high proportion of As was mobile or chemically adsorbed (i.e. potentially mobile) forms (Fig. 3).
Chemical analysis of soil, sediment, and humus revealed elevated concentrations of As and other PHEs in the humus and topsoil (due to dust emission) close to the tailings as well as in the subsoil further from the tailings pond due to migration in the saturated zone (Table 3). The effect of dust emission can be visually observed in sampling points approximately 50–100 m from the tailings (sampling sites 1, 8, and 17, Fig. 1). The concentrations of As in humus ranged from 8 to 164 mg kg−1, being highest 50 m from the tailings at sampling point H8 (Table 3) and exceeded the upper reference value for soil contamination. In humus, the total concentrations of As and other PHEs were elevated compared to the background values, but in tills, the total concentrations were mainly below the background levels (Table 3). However, the concentrations of As and other PHEs with increasing distance from the tailings were low in soils in unsaturated zone and higher in groundwater zone, which can be explained by smaller influence of dust emissions compared to contaminant transport in groundwater. The emissions from tailings e.g. sulphur have neither caused soil acidification as the pH in tills and in humus is typical for Finnish soils (Table 3; Kontio and Kähkönen 1991; Kähkönen 1993; Räisänen and Pulkkinen 1991).
As and PHEs in surface waters tend to accumulate in ditch sediments. The highest concentration of As and PHEs in solid phase was observed in the surrounding ditch sediment sample (S2, Table 3). The concentrations of As and other PHEs exceeded upper reference values also in the tailings pond sample S11. However, although the As concentrations in the beginning of the southern discharge (S13) and northern discharge ditch (S6) sediment were somewhat high, the concentrations decrease towards the end of the ditches (S7 and S14), and it seems that due to the increase in pH between the tailings pond and the ditches of the discharged water, As adsorbs onto Fe (oxy)hydroxide particles and settles to the bottom already in the beginning of the ditches (Tables 4, 6). The concentrations of the discharge ditch sediments were higher than the average concentration of stream sediments in Siilinjärvi area (3 mg kg−1) (Tarvainen and Mannio 2004). Low total and soluble As and PHE concentrations detected in ditch water samples (SW6, SW7, SW13 and SW14) support the contaminant adsorption to ditch sediment. Comparison of total and soluble As and other PHE concentrations shows that the amount of contaminants in particulate form are retained through settlement and adsorption, especially in the northern ditch.
Lake sediments are known to act as a sink for contaminants derived from their catchment. A sediment core taken from Lake Kuuslahti provides insight into the pre-mining background values as well as elemental conditions during and after mining and industrial activity. Based on Cs-dating of the lake bottom sediment (Fig. 2), the core represents information from late 1940s to present and thus reveals the loading before the factory and the open pit mine opened. The vertical distribution of the total concentrations of As and other PHEs shows clearly how the concentrations have increased since industrial and mining activities began in the 1970s. The As content of the lake bottom sediment tripled since the third calcination facility began operating in 1983, while the Cd, Co, Zn, Cu, and S concentrations increased four to tenfold at about the same time. The changes in the content of the sediment began with sharp peaks in the Cd, Co, Cu, Ni, Zn, and S profiles in the early 1980s, accompanied by less prominent, but clear increases in the As profile (Fig. 2). After peaking in the early 1980s, the concentrations of Cu, Ni, and S decreased relatively rapidly, whereas As, Cd, Co, and Zn decreased more slowly. These decreases were most likely due to growing environmental awareness and stricter environmental permit terms. Based on the chemical composition of the sediments, the concentrations of several contaminants exceed background concentrations (S15, Table 3). However, the sediments provide a record of the net loading from the ambient environment with all sources and mechanisms combined, not a single source.

The vertical distribution of the total concentrations of As, Co, Cd, Cu, Ni, Pb, S and Zn in the lake bottom sediments of Lake Kuuslahti
Statistically significant positive correlation was detected not only between As, Co, Ni, and Zn, but also with the S and Fe in the soil and sediment samples (Table 4; Fig. 4a, b). This indicates that the concentrations of As and other PHEs were elevated by emissions from tailings, but also possibly by sulphidic precipitation or adsorbtion by secondary Fe minerals such as Fe-oxides and hydroxides. The selective extractions showed that the emissions from the tailings not only increased the total concentration of As, but also increased the potentially mobile and available fractions (Fig. 3). Remarkable proportion of the total As in the soil, humus, and sediment was associated with secondary precipitates rather than sulphide phases (Fig. 3). In addition relatively high proportion of the As fraction was ammonium acetate extractable and thus easily mobilized and bioavailable. As expected from the high solubility of As in water, the As tended to be more mobile in areas with higher groundwater levels. The proportion of the ammonium acetate fraction was fairly high, especially in humus and sediment samples. The highest proportion of available As in humus was detected about 50 m from the tailings, at sampling point H8. The majority of As in tailings pond sediment (S11) was in ammonium acetate extractable. In addition, mobile and chemically adsorbed (oxalate extractable) fractions of As in the surrounding sediments and discharge ditches (S2, S6, S7, S13, S14) were high (Fig. 4).

The map of the study site showing the proportional distribution of chemical fractionation of arsenic in soil samples. The chemical fractionation of arsenic expressed as percentage, is defined by the ratio of the ammonium acetate and oxalate fractions to the total concentrations (aqua regia or HNO3 extraction)

As correlation with a Fe and b S. Amm.Ox. = 0,2 M NH4-oxalate extracted fraction and Amm.ac. = 1 M NH4-acetate extracted fraction. Aqua Regia extraction for till samples, nitric acid extraction for humus and sediment samples
The high proportion of oxalate-extracted Fe in the soil samples alongside statistically significant positive correlations between oxalate-extracted Fe and PHEs show the adsorption of As and other PHEs by Fe (oxy)hydroxides. This was also supported by the saturation indices (SI) from PHREEQC. SI is zero when water is saturated with a mineral, negative when the water is undersaturated, and positive when water is supersaturated (Parkhurst and Appelo 1999; Parkhurst et al. 1985). Geochemical modelling indicated that As minerals should be dissolving and Fe oxides and hydroxides (e.g. goethite) precipitating, further showing how As mobility is closely linked to Fe biogeochemistry (cf. Dixit and Hering 2003).
The As concentrations in soil and water were elevated near the tailings, but the As is retained by chemical adsorption to secondary minerals such as Fe-, Al-, and Mg-oxides and hydroxides in ditch sediment and in soil (e.g. Straskraba and Moran 2006). Also, the As–Fe correlation increases with decreasing pH, due to the fact that when ferrous Fe oxidizes and hydrolyzes, the pH is decreased below 6 and As is less mobile and easily adsorbed by Fe (oxy)hydroxides (e.g. Favas and Pratas 2012). This prevents high concentrations of dissolved As from migrating far from the tailings by dust or surface water. However, if pH increases above 6, the surface charge of Fe adsorbents becomes dominantly negative, or the redox conditions change to reducing, adsorbed As may be released again into water due to reductive dissolution of Fe(III) (oxy)hydroxides that have sorbed As (e.g. Belzile and Tessier 1990; Bose and Sharma 2002; Dixit and Hering 2003). Therefore, it is important to understand the physico-chemical conditions in the environment when planning site remediation or predicting migration from tailings. It is essential to minimize the dust emissions from the tailings area and to continue pumping seepage water from the bedrock fracture zone.
Arsenic Hydrogeochemistry
Hydrogeochemical studies revealed a strong hydrochemical anomaly (high concentrations of As in ground and surface water) near the tailings. The highest As concentration was detected in the seepage water (sampling SW2 in Fig. 1 and Table 5), but the As concentrations decrease rapidly away from it, apparently due to the pH increase between sampling points SW2 and SW4 and (Fig. 1; Table 5). As quickly adsorbs onto Fe (oxy)hydroxide and settles to the bottom of the surrounding ditch, decreasing the dissolved As (cf. Straskraba and Moran 2006). The difference between total and soluble As concentrations was negligible; the As occurs mainly in soluble form in ground and surface water.
Concentrations of As exceeded the limits for household water (10 µg L−1) and the environmental standard for groundwater (5 µg L−1) at sampling point GW5, <200 m from Lake Kuuslahti (Fig. 1). Additionally, the concentrations of other PHEs (Co, Cd, Zn, Cu, and Ni) and SO4 were elevated in groundwater at sampling points 5 and 9 (Fig. 1; Table 5), so it seems that despite the preventive pumping of the seepage water, the water from the tailings and seepage ponds is able to migrate towards Lake Kuuslahti. However, since the concentrations of As and other PHEs are low in the lake water (SW15), these elements seem to be adsorbed before discharging to the lake.
In most of the water samples, As concentrations showed strong positive correlations with Co, Li, Ni, Fe, Mg, S, Zn, Mo, Mn, and SO4 (Table 6), but the concentrations decreased with increasing pH. The correlation with Fe, Mn, and Mg indicates that As mobilization is mainly controlled by adsorption by metal (oxy)hydroxides at the site. This is supported by the high concentrations of metal (oxy)hydroxides, especially Fe oxides, in the soil and sediment samples and the statistically significant positive correlation between Fe oxides and PHE concentrations in these samples (Table 4). In addition, geochemical modelling supports the precipitation of Fe (oxy)hydroxides, such as goethite (SI > 1.3…7.0).
The pH of the water samples ranged from 2.5 to 7.7, being highest in Lake Kuuslahti (SW15) and lowest in the seepage water (SW2). The oxidation reduction potential (ORP) ranged from 19 to 466 mV, being highest at SW2 and lowest in GW10 next to the lake. The water samples were mainly oxic, except for the groundwater samples close to the tailings pond and the lake water sample taken at a depth of 19 m. Various studies (e.g. Cullen and Reimer 1989; Ferguson and Gavis 1972; Francesconi and Kuehnelt 2002; Sadiq 1997) have shown that As speciation is highly affected by pH and redox conditions. In oxic, less acidic, neutral, and alkaline water, arsenate is usually the dominant form of As and it commonly hydrates to the dissociated As acid anions, H2AsO4 − and HAsO4 2− (Henke 2009). In comparison, under even slightly reducing conditions, arsenite becomes dominant (Kim et al. 2012) and inorganic As primarily exists as dissolved arsenious acid (H3AsO3) at a pH below 9.2. According to the modelling, As occurred in groundwater as the less toxic trivalent arsenious acid (H3AsO3) at pH < 6 and mainly as pentavalent As acid (H2AsO4 − and HAsO4 2−) at pH > 6. In ditch and lake water, As occurred mainly as pentavalent arsenate-compounds, H2AsO4 − and HAsO4 2−, regardless of the pH (Table 5). Based on water analysis and hydrogeochemical modelling, the As concentrations and variations in As speciation are closely linked to SO4 and Fe concentration and especially Fe speciation (Fig. 5). This can be explained by the Fe speciation and higher mobility of As(III) at low pH and in the reduced environment, due to weaker adsorption of As(III) to Fe(III) hydroxides (Bose and Sharma 2002). The higher the concentration of Fe(II) and lower the pH, the more As(III) was present in the water.

Correlation between As(III) and As(V) with Fe(II) and Fe(III) species based on hydrogeochemical speciation modeling by PHREEQC
Effect of Anthropogenic Emissions to Ecological Risks
Industrial activity increased the ecological risk by releasing As from the tailings and increasing the amount of chemically-adsorbed, easily mobilized, and available As in the soil. Since the ecosystem developed in the presence of naturally low background concentrations of As, elevated concentrations may pose a risk to the ambient environment. Various studies (e.g. Vaajasaari et al. 2004) have shown that As contamination of soil has a direct impact on microbial community and structure by reducing the population in soil and water.
In humus, the total concentrations of As, Co, Cd, Cu, and Ni, and in till, the total concentration of As and Co, exceeded the PNEC values of ECHA for soil. The total concentrations of As, Cd, Co, Cu, and Zn in lake bottom sediment (S15) exceeded the ECHA PNEC value for fresh water sediment (Tables 2, 4). The levels of As, Co, Cu, and Cd reached the ECHA PNEC value in the surrounding ditch (S2) and northern discharging ditch (S6, S7) samples. In the southern discharging ditch (S13, S14), only Co exceeded the PNEC. The concentrations of As and other PHEs in soil were below the Finnish maximum acceptable concentration in soil of industrial site (250 mg kg−1), but exceeded the maximum ineffective concentrations in soil.
The As concentration in 64 % of the water samples exceeded the ECHA PNEC value for fresh water (0.5 µg L−1). In addition, several of the samples contained Co, Cd, Cu, and Zn at levels exceeding PNEC. Based on hydrogeochemical modelling, most of the As in water was the less toxic arsenate, and the concentrations in discharging ditches (SW6, SW7, SW13, SW14) and Lake Kuuslahti (SW15) were low. Yet, the As in ground and surface water at the site is mainly dissolved and is thus highly bioavailable, so the concentrations may still pose a risk to aquatic organisms (c.f Naidu 2012). In addition, although the elements seem to be stable in Lake Kuuslahti, the lake bottom is dredged for cargo ships transporting fertilizers from the factory, and the harmful substances may be released into the lake water, elevating the possibility of adverse effects on aquatic ecosystem.
Conclusions
This study of the Yara industrial site indicated that dust and seepage emissions from tailings has increased the total concentrations of As and other PHEs as well as the amount of potentially mobile and available contaminants, compared to the natural geogenic background. Elements in the soils were mainly associated with Fe(oxy)hydroxides, and so their mobility is closely linked to Fe biogeochemistry. Although concentrations of As and PHEs were high in tailings pond and seepage water, the concentrations were low in ambient ground and surface water, due to the stability of Fe(oxy)hydroxides in current chemical conditions. This was supported by hydrogeochemical modelling, which showed precipitation of Fe oxides and hydroxides. Concentrations of As and other PHEs were elevated many times in nearby lake sediment after industrial activities began in the 1970s, but have decreased during the last few decades. According to speciation modelling, As was mainly present in groundwater as the toxic trivalent arsenious acid (H3AsO3) at pH < 6, and mainly as the less toxic pentavalent As acids (H2AsO4 − and HAsO4 2−) at pH > 6 and in surface water. If the physico-chemical conditions change, As adsorbed to Fe(oxy)hydroxides may be mobilized, and As speciation may change into the more toxic form. Our study showed that a combination of hydrogeochemical and geochemical approaches can be helpful in assessing tailings-derived contamination and metal mobility.
References
Belzile N, Tessier A (1990) Interactions between arsenic and iron oxyhydroxides in lacustrine sediments. Geochim Cosmochim Acta 54(1):103–109. doi:10.1016/0016-7037(90)90198-T
Bose P, Sharma A (2002) Role of iron in controlling speciation and mobilization of arsenic in subsurface environment. Water Res 36(19):4916–4926. doi:10.1016/S0043-1354(02)00203-8
Cullen W, Reimer K (1989) Arsenic speciation in the environment. Chem Rev 89:713–746. doi:10.1021/cr00094a002
Dixit S, Hering JG (2003) Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environ Sci Technol 37(18):4182–4189. doi:10.1021/es030309t
Favas PJC, Pratas JMS (2012) Arsenic bioavailability and mobility in contaminated soils and mine tailings. In: Ng JC, Noller BN, Naidu R, Bundschuh J, Bhattacharya P (eds) Understanding the geological and medical interface of arsenic, 1st edn. Taylor & Francis Group, London, pp 128–129
Ferguson JF, Gavis J (1972) A review of the arsenic cycle in natural water. Water Res 6(11):1259–1274. doi:10.1016/0043-1354(72)90052-8
Filguieras AV, Lavilla I, Bendicho C (2002) Chemical sequential extraction for metal partitioning in environmental solid samples. J Environ Monit 4(6):823–857. doi:10.1039/B207574C
Francesconi KA, Kuehnelt D (2002) Arsenic compounds in the environment. In: Frankenberg WT (ed) Environmental chemistry of arsenic, 1st edn. Dekker, New York, pp 51–94
Hakala E, Hallikainen A (2004) Exposure of the finnish population to arsenic, effects and health risks. In: Loukola-Ruskeeniemi K, Lahermo P (eds) Arsenic in Finland: distribution, environmental impacts and risks, 1st edn. Geological Survey of Finland, Espoo, pp 153–166 (In Finnish)
Heikkinen PM, Räisänen ML (2009a) Trace metal and As solid-phase speciation in sulphide mine tailings: indicators of spatial distribution of sulphide oxidation in active tailings impoundments. Appl Geochem 24(7):1224–1237
Heikkinen PM, Räisänen ML, Johnson RH (2009b) Geochemical characterisation of seepage and drainage water quality from two sulphide mine tailings impoundments: acid mine drainage versus neutral mine drainage. Mine Water Environ 28(1):30–49. doi:10.1007/s10230-008-0056-2
Henke K (2009) Arsenic in natural environments. In: Henke KR (ed) Arsenic: environmental chemistry, health threaths and waste treatment, 1st edn. Wiley, West Sussex, pp 69–235
Huang J-H, Hu K-N, Decker B (2011) Organic arsenic in the soil environment: speciation, occurrence, transformation, and adsorption behavior. Water Air Soil Pollut 219(1–4):401–415. doi:10.1007/s11270-010-0716-2
Huttunen L (2012) Unreported written information via email communication. The environmental manager of the yara finland siilinjärvi site
Jaakkola T, Tolonen K, Huttunen P, Leskinen S (1981) The use of fallout 137Cs and 239,240Pu for dating of lake sediments from Finland. Hydrobiologia 103(1):15–19. doi:10.1007/BF00028422
Kahelin H, Kallio E (2004) Determination of arsenic concentrations in geological and environmental samples. In: Loukola-Ruskeeniemi K, Lahermo P (eds) Arsenic in Finland: distribution, environmental impacts and risks, 1st edn. Geological Survey of Finland, Espoo, pp 153–166 (In Finnish)
Kähkönen AM (1993) Acid-sensitivity of lakes and the geochemistry of till fines in catchments of northern Finland. Unpublished master’s thesis. University of Helsinki Department of Geology. p 72
Kim K, Kim S-H, Jeong G-Y, Kim RH (2012) Relations of As concentrations among groundwater, soil, and bedrock in Chungnam, Korea: implications for As mobilization in groundwater according to the As-hosting mineral change. J Hazard Mater 199–200:25–35. doi:10.1016/j.jhazmat.2011.10.037
Klaminder J, Appleby P, Crook P, Renberg I (2012) Post-deposition diffusion of 137Cs in lake sediment: implications for radiocaesium dating. Sedimentology 59(7):2259–2267. doi:10.1111/j.1365-3091.2012.01343.x
Koljonen T, Elo S, Gustavsson N, Huhma H, Kauranne LK, Noras P, Pesonen LJ, Ruotoisenmäki T, Saltikoff B, Sillanpää M, Tanskanen H, Vaasjoki M, Vuorela P (1992) The Geochemical Atlas of Finland, part II: Till. Geological Survey of Finland, Espoo, pp 146–147
Kontio M, Kähkönen A-M (1991) Geochemistry and variations of the sensitivity of soils to acidification in northern Finland. In: Pulkkinen E (ed) Environmental geochemistry in Northern Europe, proceedings of the first symposium on environmental geochemistry in Northern Europe held in Rovaniemi, Finland 17–19 Oct 1989. Geological Survey of Finland, Special Paper 9:53–60
Lahermo P, Väänänen P, Tarvainen T, Salminen R (1996) Geochemical atlas of Finland, part III: environmental geochemistry–stream water and sediments. Geological Survey of Finland, Espoo (In Finnish)
Lukkarinen H (2008) Bedrock map of Finland 1:100000, mapsheets and the explanations to the mapsheets 3331 and 3242, The bedrock maps of Siilinjärvi and Kuopio. Geological Survey of Finland, Digital form map (In Finnish)
Naidu R (2012) Bioavailability and bio-accessibility of arsenic for ecological and human health risk assessment: the geological and health interface. In: Ng JC, Noller BN, Naidu R, Bundschuh J, Bhattacharya P (eds) Understanding the geological and medical interface of arsenic. Taylor & Francis Group, London, pp 3–10
Niskavaara H (1996) Comprehensive scheme of analyses for soil, sediment, humus and plant samples using inductively coupled plasma atomic emission spectrometry. Geol Surv Finland Spec Pap 20:167–176
Parkhurst DL, Appelo CAJ (1999) User’s guide to PHREEQC (Version 2): a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. In: Water resources investigations report. US Department of the Interior, US Geological Survey, pp 99–4259
Parkhurst D, Thorstenson D, Plummer L (1985) PHREEQE: a computer program for geochemical calculations. NEA publication, Paris, p 308
Parviainen A (2012) Evolution of sulfide oxidation and attenuation mechanisms controlling acid mine drainage in decommissioned low-sulfide tailings. Doctoral Disserations Aalto University, Department of Civil and Environmental Engineering. Aalto University publication series 107/2012
Pongratz R (1998) Arsenic speciation in environmental samples of contaminated soil. Sci Total Environ 224(1):133–141. doi:10.1016/S0048-9697(98)00321-0
Räisänen ML, Hämäläinen L (1991) Selective sequential dissolution of polluted and non-polluted sediments in environmental geochemistry in northern Europe. In: Pulkkinen E (ed) Environmental geochemistry in Northern Europe, proceedings of the first symposium on environmental geochemistry in Northern Europe held in Rovaniemi, Finland 17–19 Oct 1989. Geological Survey of Finland, Special Paper 9:157–162
Räisänen ML, Pulkkinen E (1991) Soil acidification in the Naruska region of eastern Finnish Lapland. In: Pulkkinen E (ed) Environmental geochemistry in Northern Europe, proceedings of the first symposium on environmental geochemistry in Northern Europe held in Rovaniemi, Finland 17–19 Oct 1989. Geological Survey of Finland, Special Paper 9:87–98
Räisänen ML, Hämäläinen L, Westerberg LM (1992) Selective extraction and determination of metals in organic stream sediments. Analyst 117(3):623–627. doi:10.1039/AN9921700623
Reinikainen J (2007) Derivation basis of threshold and guideline values for soil. The Finnish Environment 23/2007, p 164. http://hdl.handle.net/10138/38431. (In Finnish)
Sadiq M (1997) Arsenic chemistry in soils: an overview of thermodynamic predictions and field observations. Water Air Soil Pollut 93(1):117–136. doi:10.1023/A:1022135909197
Smedley PL, Kinniburgh DG (2002) A review of the source, behavior and distribution of As in natural water. Appl Geochem 17(5):517–568. doi:10.1016/S0883-2927(02)00018-5
Straskraba V, Moran RE (2006) Environmental occurrence and impacts of arsenic at the gold mining sites in the Western United States. Mine Water Environ 9(1–4):181–191
Tarvainen T (2004) Arsenic in soils. In: Loukola-Ruskeeniemi K, Lahermo P (eds) Arsenic in Finland: distribution, environmental impacts and risks, 1st edn. Geological Survey of Finland, Espoo, pp 45–49 (In Finnish)
Tarvainen T, Mannio J (2004) Arsenic in surface water and stream sediments. In: Loukola-Ruskeeniemi K, Lahermo P (eds) Arsenic in Finland: distribution, environmental impacts and risks, 1st edn. Geological Survey of Finland, Espoo, pp 97–101 (In Finnish)
Tessier A, Campbell PGC, Bisson M (1979) Sequential extraction procedure for speciation of particulate trace metals. Anal Chem 51(7):844–850. doi:10.1021/ac50043a017
The Regional Environmental Permit Agency of Eastern Finland (ELY) (2006) Kemphos Ltds, Kemira Ltds and Minelco Ltds environmental permit application concerning to companies production plants in Siilinjärvi and Kemphos Ltds water permit, Siilinjärvi. Permit decision No 79/06/02. Dno ISY-2004-Y-272. (In Finnish)
The Regional State Administrative Agency of Eastern Finland (AVI) (2011) Yara Finland Ltd. An application for updating the environmental permit concerning to the classification of tailings as a waste, Siilinjärvi. Permit decision No 99/2011/1. Dno ISAVI/69/04.08/2011. (In Finnish)
Traas TP (2001) Guidance document on deriving environmental risk limits. Rijksinstituut voor Volksgezondheid en Milieu (RIVM) report 601501012. p 117
Turpeinen R, Pantsar-Kallio M, Häggblom M, Kairesalo T (1999) Influence of microbes on the mobilization, toxicity and biomethylation of arsenic in soil. Sci Total Environ 236(1–3):173–180. doi:10.1016/S0048-9697(99)00269-7
Vaajasaari K, Schultz E, Sorvari J (2004) Arsenic in contaminated soils, assesment of environmental risks and ecotoxicology. In: Loukola-Ruskeeniemi K, Lahermo P (eds) Arsenic in Finland: distribution, environmental impacts and risks, 1st edn. Geological Survey of Finland, Espoo, pp 135–152 (In Finnish)
Vestola E, Mäkinen J, Laine-Ylijoki J, Bolmberg P, Neitola R, Korhonen T, Knauf O, Ruokojärvi A (2012) Cost efficient and intergrated biological treatment of metal containing wastes. Unpublished research report of PROBIO-project (Cost-efficient and integrated biological treatment for metal-containing wastes). Geological survey of finland, Technical research center of finland and savonia university of applied sciences
Wang S, Wang P, Men B, Lin C, He M (2012) Chemical forms and ecological risk of arsenic in the sediment of the Daliao River System in China. Environ Monit Assess 184(4):2237–2245. doi:10.1007/s10661-011-2113-8
Xu H, Allard B, Grimvall A (1991) Effects of acidification natural organic materials on the mobility of arsenic in the environment. Water Air Soil Pollut 57(1):269–278. doi:10.1007/BF00282890
Tapir database. National benchmark database, Geological Survey of Finland. http://www.geo.fi/tapir/
Acknowledgments
This study is part of project “Solutions for Arsenic Control in Mining Processes and Extractive Industry”, which was funded by the Finnish Funding Agency for Technology and Innovation (TEKES) during 2011–2013.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Turunen, K., Backnäs, S., Neitola, R. et al. Factors Controlling the Migration of Tailings-Derived Arsenic: A Case Study at the Yara Siilinjärvi Site. Mine Water Environ 35, 407–420 (2016). https://doi.org/10.1007/s10230-016-0393-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10230-016-0393-5