
Abstract
Recent demands for the production of biofuels from lignocellulose led to an increased interest in engineered cellulases from Trichoderma reesei or other fungal sources. While the methods to generate such mutant cellulases on DNA level are straightforward, there is often a bottleneck in their production since a correct posttranslational processing of these enzymes is needed to obtain highly active enzymes. Their production and subsequent enzymatic analysis in the homologous host T. reesei is, however, often disturbed by the concomitant production of other endogenous cellulases. As a useful alternative, we tested the production of cellulases in T. reesei in a genetic background where cellulase formation has been impaired by deletion of the major cellulase transcriptional activator gene xyr1. Three cellulase genes (cel7a, cel7b, and cel12a) were expressed under the promoter regions of the two highly expressed genes tef1 (encoding translation elongation factor 1-alpha) or cdna1 (encoding the hypothetical protein Trire2:110879). When cultivated on d-glucose as carbon source, the Δxyr1 strain secreted all three cellulases into the medium. Related to the introduced gene copy number, the cdna1 promoter appeared to be superior to the tef1 promoter. No signs of proteolysis were detected, and the individual cellulases could be assayed over a background essentially free of other cellulases. Hence this system can be used as a vehicle for rapid and high-throughput testing of cellulase muteins in a homologous background.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lignocellulose, composed mainly of cellulose, hemicellulose, and lignin, is the most abundant renewable carbon source on earth and therefore an attractive resource to use it for the production of different chemical building blocks or biofuels. Due to the complex structure and the recalcitrance of lignocellulosic feedstocks, the conversion to their monomers is much more difficult to achieve than using, e.g., starch-based feedstocks (Himmel et al. 2007). As a result, the breakdown of the polymeric sugars, especially cellulose, requires large amounts of enzymes (Merino and Cherry 2007). Recent demands to reduce the production costs for biofuels from lignocellulosic plant matter led to a general renaissance in cellulase research (Carroll and Somerville 2009; Wilson 2009; Kumar et al. 2008). Fungal cellulases are attractive for enzymatic cellulose conversion as they are highly active and can be expressed at levels exceeding 100 g per liter in fungal hosts such as Trichoderma reesei (Hypocrea jecorina, Cherry and Fidantsef 2003; Kubicek et al. 2009).
To date, many important commercial enzyme preparations for biomass conversion of cellulose or hemicelluloses are from T. reesei. These T. reesei cellulase mixes include different types of cellulases including cellobiohydrolases (CEL7A and CEL6A), endoglucanases (e.g., CEL7B and CEL6B) and different β-glucosidases (e.g., CEL3A) which synergistically degrade cellulose (Henrissat et al. 1985).
While the economic production of cellulosic ethanol can be tackled by various approaches, protein engineering of cellulases to produce enzymes with improved catalytic activity (K cat) or enzyme stability is a major issue. In addition, different hosts and environments are screened for novel cellulase activities (Zhang et al. 2006). The engineering of enzymes by rational design or directed evolution uses different methodologies to optimize existing enzymes by construction of mutant libraries followed by their testing to identify mutant proteins with improved properties. This involves, e.g., the mutagenesis of potential active center residues and their subsequent kinetic analysis and is based upon the identification of the invariant residues identified in the sequence-based CAZy family classification (http://www.cazy.org; Cantarel et al. 2009). While the methods to generate such mutant libraries are straightforward, there is a bottleneck in the testing and production of the mutant enzymes. Today, such muteins can be routinely expressed in Escherichia coli, Saccharomyces cerevisiae, and Pichia pastoris, but the functional requirement for modifications such as disulfide bridges or delicate N- and O-glycosylation present at, e.g., the linker domains of the cellulase proteins render most of these expression hosts inappropriate. Examples are the production of the cellobiohydrolase CEL7A in P. pastoris and S. cerevisiae which resulted in hyper- or overglycosylated enzymes with compromised activities (Boer et al. 2000; Godbole et al. 1999; Jeoh et al. 2008; Penttilä et al. 1988; Van Arsdell et al. 1987). The alternative—to produce these mutant enzymes in T. reesei itself—is hampered by the fact that traces of native cellulases are even produced on cellulase-noninducing carbon sources or during sporulation. The resulting protein is therefore contaminated with other enzyme activities which render the kinetic analysis prone to errors (Carle-Urioste et al. 1997).
A useful alternative would be the production of mutant cellulases in a host whose cellulase formation has been impaired. Cellulase induction in T. reesei is dependent on the function of the Zn2Cys6 transcriptional regulator XYR1, and knockout mutants in xyr1 are cellulase negative (Stricker et al. 2006). In addition, these cellulases are even not produced during sporulation in such a Δxyr1 strain (Metz et al. 2011). However, it is not yet known whether XYR1 also influences other traits related to cellulase formation such as protein transport or secretion in T. reesei. For example a comparison of the transcriptomic response of an Aspergillus oryzae overproducer of the orthologous xlnR to an xlnR deletion strain did not only reveal changes in the expression pattern of various glycoside hydrolases and genes encoding for pentose catabolism but also changes in various transport processes (Noguchi et al. 2009).
In this paper, we therefore tested the hypothesis that a xyr1-knockout strain could be used as a production host for individual cellulase components under constitutive expression signals. Our data show that this strain is a useful vehicle for production of pure cellulases for kinetic analysis.
Materials and methods
Strains and culture conditions
T. reesei QM9414 (ATCC 26921) and the Δxyr1 strain (Stricker et al. 2006) derived from it were used throughout this study. They were maintained on potato dextrose agar (PDA) plates at 28°C. For cellulase expression strains were grown for the indicated time in 250 mL medium in 1 L flasks at 28°C on a rotary shaker at 250 rpm (Multitron 2, Infors AG). A modified medium (Vaheri et al. 1979) containing 10 g/L carbon source, 1.4 g/L (NH4)2SO4, 2.0 g/L KH2PO4, 0.3 g/L MgSO4·7H2O, 0.4 g/L CaCl2·2H2O, 1 g/L peptone, 5 mM urea, 0.02% (w/v) Tween80, and 1/50 (v/v) of the trace element solution (0.25 g/L FeSO4·7H2O, 0.08 g/L MnSO4·H2O, 0.07 g/L ZnSO4·7H2O, 0.1 g/L CoCl2·2H2O) was adjusted to pH 5.0 by citric acid and not further controlled.
Expression plasmid construction and fungal transformation
Vectors for expression of cel7a (cbh1), cel7b (egl1), and cel12a (egl3) under the tef1 (translation elongation factor 1 alpha) promoter were constructed by inserting the respective PCR amplified cellulase gene in pPtef1-hph (Akel et al. 2009) and named pPtef1-cel7a, -cel7b, or -cel12a. The final expression vector contains 740 bp of the promoter region the T. reesei tef1, followed by the coding and terminator region of the respective cellulase gene and the hygromycin B phosphotransferase (hph) expression cassette as selection marker. The three cellulase genes including their respective terminator region were amplified from genomic DNA of T. reesei QM9414 with the primer pairs Egl1_ClaI_F/Egl1_HindIII_R for cel7b, Egl3_ClaI_F/Egl3_HindIII_R for cel12a Cbh1_ClaI_F and Cbh1_SalI_R for cel7a, thereby introducing the denoted cloning sites (Table 1).
For expression under the cdna1 promoter region, the tef1 promoter region was removed by an XhoI/ClaI digest and the PCR amplified, XhoI/ClaI restricted cdna1 promoter region was inserted resulting in pPcdna1-cel12a. For pPcdna1-cel7b construction, the tef1 promoter region was removed from pPtef1-hph by an XhoI/HindIII digest followed by the insertion of the XhoI/ClaI restricted cdna1 promoter region and the ClaI/HindIII restricted cel7b gene via ligation.
Fungal transformation was performed as described (Gruber et al. 1990) using uncut plasmid DNA for transformation. Selection media contained 50 μg/mL hygromycin B (Roth). For sporulation, the transformants were transferred to small PDA plates and purified by plating conidiospores onto PDA plates with 0.1% Triton X-100 as colony restrictor. Single colonies which showed regular growth were transferred to selective media. To test stability, the transformants were passaged three to four times to nonselective media before they were retested on selective media for growth and expression of the individual cellulases.
Molecular analysis of transformants
Genomic DNA of the transformants was extracted using a phenol–chloroform method (Seiboth et al. 2002). Transformants were screened for the presence of the different cellulase expression cassettes by amplifying the expression cassette by PCR with one oligonucleotide specific for the promoter region of tef1 or cDNA1 and the second specific for the terminator region of the respective cellulase gene. Southern analysis was used to verify the integration of the construct and determine the approximate copy number of the integrated cellulase genes. For Southern blot analysis, genomic DNA (~15–20 μg) of QM9414, the Δxyr1 strain and the transformants were digested with BamHI or PstI for cel7b, and XhoI or PstI for cel12a copy number determination. Digested DNA was size fractionated by gel electrophoresis and transferred to a Biodyne B 0.45-μm membrane (Pall Corporation). DNA labeling, hybridization, and detection were performed according to the DIG High Primer DNA Labeling and Detection Starter Kit I protocols (Roche Applied Science). DIG-labeled probes were amplified with oligonucleotides Egl1_ClaI_F/Egl1_HindIII_R for cel7b and Egl3_ClaI_F/Egl3_HindIII_R for cel12a. The above-mentioned restriction enzymes cut the respective cellulase gene approximately in the middle of the coding region which results upon hybridization with the probe in two hybridizing bands of similar intensity. The copy number of integrated cellulase genes was hence determined by the number of additional fragments (n − 1) to the endogenous cellulase gene copy.
SDS-PAGE
Supernatants were loaded to SDS gel after EtOH precipitation as follows: 500 μL of culture supernatant was mixed with 1 mL 96% EtOH and stored o/n at −20°C. Following centrifugation at 13,000 rpm for 20 min at 4°C, the protein pellets were resuspended with 40 μL ddH2O. Samples were run with constant current (15 mA/gel) on 12% denaturing SDS gel.
Cellulase activity assays
Cel7b transformants were assayed using 4-methylumbelliferyl-β-d-cellobioside (MUC) and cel7a transformants using 4-methylumbelliferyl-β-d-lactoside (MULAC) as substrate. Substrates (MUC/MUCLAC) were dissolved in DMSO, and the activities were measured with 50 nmol substrate, 50 mM NaAc buffer (pH 5.0) and 50 μL supernatant in a reaction volume of 200 μL/well. Fluorescence was measured with a Gemini XS spectrofluorometer with 1 min intervals for 1 h at 40°C using ddH2O as blank, with the following parameters: excitation 330 nm, emission 456 nm, and cutoff 455 nm. Initial linear release rate of fluorescence was used to calculate activities.
The carboxymethylcellulase assays for CEL12A were done in quadruplets. To remove interfering d-glucose in the culture filtrate, the extracellular protein was precipitated by the addition of 2 vol. of ethanol and then resuspended in ddH2O. Reagents were preheated to 50°C and 96-well plates were covered with a plastic mat during incubations to prevent evaporation. Thirty-microliter supernatants were added to 96-well PCR plates and heated to 50°C. Thirty microliters 2% carboxymethylcellulose (CMC) in NaOAc Buffer (pH 4.8) was added to wells simultaneously, mixed quickly, and incubated for 30 min at 50°C. Sixty microliters DNS reagent was added to wells, mixed shortly, and incubated at 95°C for 5 min for color development. PCR plate was then placed on ice, and absorbances of 100 μL of mixtures were read at 550 nm after transferring them to flat bottomed 96-well microplate. CMCase activity was calculated as described previously (Ghose 1987). One unit catalyzes the liberation of 1 μmol glucose from sodium carboxymethyl cellulose per min. Specific activities are related to the total extracellular protein produced.
Total protein concentration and dry biomass determination
Total protein concentrations in the culture supernatants were measured by the Bradford assay (Bradford 1976). For dry biomass determination, 20 mL of culture were centrifuged at 5,000×g for 10 min, the pellet was resuspended with ddH2O to get rid of salts and centrifuged again. The pellets were dried in an oven (70°C) to constant weight for 1 week.
Results
Cellulase expression vector construction
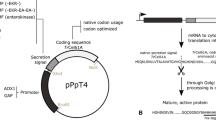
For the expression of cellulases in a Δxyr1 strain, we had to switch their endogenous promoter regions. We chose the promoters of two genes that were isolated during the screening for highly expressed genes during growth on d-glucose as carbon source—i.e., tef1 (translation elongation factor 1-α-encoding; abbreviated as “t”) and the uncharacterized “cdna1” (abbreviated as “c,” Nakari et al. 1993; Nakari-Setälä and Penttilä 1995). With the help of the previously released T. reesei genome sequence, it was now possible to localize the complete cDNA1 gene on scaffold 23:43726–44652 (http://genome.jgi-psf.org/Trire2/Trire2.home.html). In agreement with earlier results, a high number of ESTs are assigned to the cDNA1 gene. BLASTP search of the translated nucleotide sequence showed that the protein is only moderately conserved showing a 39 to maximal 46% aa identity to other fungal hypothetical proteins from Nectria haematococca, Gibberella zeae, Verticillium alboatrum, Magnaporthe grisea, Sclerotinia sclerotiorum, and Botryotinia fuckeliana. Three cellulase genes were chosen for testing cellulase expression in the Δxyr1 background in T. reesei: one cellobiohydrolase encoded by cel7a (cbh1) and two endoglucanases encoded by cel7b (egl1) and cel12a (egl3). cel7b and cel12a were expressed under both promoters, whereas cel7a was tested only with the tef1 promoter region. All three cellulases together with their terminator region were amplified by PCR and ligated downstream of the respective promoter region (Fig. 1)

Expression plasmids for cellulase production. Schematic presentation of the two cellulase expression plasmids used in this study. Both plasmids contain the hygromycin B expression cassette as fungal selection marker followed by either the tef1 or the cDNA1 promoter region and the coding and terminator region of the respective cellulase gene (e.g., the endoglucanase encoding gene cel7b)
Recombinant expression of three cellulases in the Δxyr1 strain
After transformation of the Δxyr1 strain with different expression plasmids and purification of putative transformants, the integration of the expression vector was tested by PCR and Southern analysis (data not shown). Strains with one additional copy of the expression cassette were chosen for further analysis and named according to the promoter region and cellulase expressed (e.g., t-cel7b for a transformant expressing cel7b under the tef1 promoter region). Their growth rate on glucose was similar to the parental strain. Strains produced 2.5 g/L dry biomass after 20 h and reached a maximum of 5 g/L after 30 h of cultivation. To test for cellulase production in these strains, we followed the total extracellular protein production over time: all strains secreted protein into the medium until around 36 h of cultivation. Best transformants reached extracellular protein concentrations of more than 40 mg/L, which exceeds the Δxyr1 recipient strain which formed up to 18 mg/L protein as background (Fig. 2). To visualize cellulase expression, the proteins in the culture filtrate were separated by SDS-PAGE: Consistent with previous findings (e.g., Jeoh et al. 2008; Shoemaker et al. 1983; Sprey and Uelker 1992), CEL7B is detected as a broad band between 60 and 70 kDa (Fig. 2a), CEL12A is found around 25 kDa (Fig. 2b), and CEL7A, which is found around 65 kDa, was evident from a band around 70 kDa in the gels (Fig. 2c). No degradation products of the recombinant produced cellulases which would have been evident from a decrease of the cellulase band or an increase in bands with lower molecular mass with time were observed. In the Δxyr1 control background, only two faint protein bands appeared after 28 h of cultivation which were only visible when a higher volume of culture supernatant was used (Fig. 2a, c).

Endoglucanase CEL7B, endoglucanase CEL12A, and cellobiohydrolase CEL7A production in the cellulase negative background of T. reesei Δxyr1 transformants. Expression profile of typical Δxyr1 transformants expressing CEL7B (a), CEL12A (b), or CEL7A (c) under the tef1 (t) or cdna1 (c) promoter regions as indicated. Left SDS-PAGE gel of the supernatant of selected transformants after Coomassie blue staining. The amount of supernatant loaded to each gel is indicated in parentheses. Right total extracellular protein concentration (in milligrams per liter) of selected transformants expressing the respective cellulase under the cDNA1 (filled diamond) or tef1 promoter region (filled square). Δxyr1 (empty triangle) is included as a negative control
Quantification of cellulase expression in the Δxyr1 strain
To test whether the secreted cellulases were active, CEL7A and CEL7B enzymes in the culture filtrates were quantified using the specific fluorogenic substrates MULAC (van Tilbeurgh et al. 1982) or MUC (Claeyssens and Aerts 1992), respectively. The results, shown in Fig. 3a, indicate that only the CEL7B producing transformants show activity towards MUC, strain c-cel7b-6 thereby giving highest values (dotted lines). Solid lines in Fig. 3a show the specific activities in micromoles/minute/milligram extracellular protein, illustrating that the specific activity was highest during the early time points (till around 28 h of growth). Thereafter, the specific activities decreased, consistent with the delayed secretion of the two faint proteins and other proteins expressed in the Δxyr1 background. Extra sugar addition (10 g/L) after 20 h of growth increased enzyme purity and yield (c-cel7b-6+).

Cellulase activities in the supernatants of the cel7b, cel7a, and cel12a transformants hydrolysis rates of MUC, MULAC, and CMC by culture supernatants of CEL7B (a), CEL12A (b), and CEL7A (c) producing strains respectively were assayed at each sampling point. Volumetric activities (dashed lines) are reflective of total active enzyme amount in the culture supernatant and specific activities (solid lines) are reflective of enzyme purity. Compared to the transformants the parental strain T. reesei Δxyr1 (empty triangle) has negligible activities towards MUC, MULAC, and CMC. The strains selected are identical to Fig. 2 and express the respective cellulase either under the cDNA1 (filled diamond) or tef1 promoter region (filled square). In the case of strain c-cel7b-6, extra d-glucose (10 g/L) was added after 20 h of incubation (empty diamond)
CEL7A producing transformants were active on MULAC, and their volumetric and specific activities displayed the same time course as that of CEL7B (Fig. 3c). Activity of CEL12A was quantified by following the release of reducing sugars from CMC (Ghose 1987). The volumetric activities obtained confirmed the production seen from SDS-PAGE, and the specific activities were—as with CEL7A and CEL7B—also highest during the early cultivation periods (Fig. 3b). It is also evident from a comparison of the activities produced by the different strains that the cdna1 promoter enabled higher protein production when using single-copy transformants and is thus to be preferred over the tef1 promoter under these conditions.
Discussion
T. reesei is a model system for the degradation of plant biomass to monomeric sugars applied in biofuel production. Its genome was recently sequenced (Martinez et al. 2008) and several sophisticated tools such as strains with improved gene targeting, strain crossing, HTP gene manipulation, and genome sequencing have been developed (reviewed by Seiboth et al. 2011). One step which asks for improvement is the testing of properties of its gene products involved in plant cell wall degradation. Since most of the cellulases and hemicellulases are glycosylated proteins, the use of commonly available hosts may produce misleading results since prokaryotic hosts are not able to perform the necessary posttranslational modifications at all and other eukaryotic hosts produce usually overglycosylated proteins with altered activities (Boer et al. 2000; Godbole et al. 1999; Nakazawa et al. 2008; Jeoh et al. 2008). The aim of this study was therefore to test whether T. reesei could be used as the native host for overexpression of selected cellulases with high purity and free of other cellulase contaminants. Such cellulase contaminants are inevitable even if the overexpressed protein would be tagged for purification, because cellulases are known to tightly adsorb to each other, particularly in diluted solutions (Sprey and Lamberta 1984). The hypothesis that was tested was whether a strain in which the XYR1 transcriptional regulator of cellulase and hemicellulase gene transcription is nonfunctional would produce cellulases at a reasonable level and in high purity when fused to a strong expression signal. Using three model cellulase (CEL7A, CEL7B, and CEL12A), we show that strong expression signals such as those from the tef1 and especially the cdna1 expression signals result in a reasonable high production of these enzymes on d-glucose as carbon source. In contrast to earlier reports where the cloning of an expression vector based on the cDNA1 promoter region failed (Nakari-Setälä and Penttilä 1995), we encountered no problem to construct such an expression vector and provide a readily usable expression vector based on the cDNA1 promoter region. We should note that the present studies were performed in shake flasks, and the secreted protein concentrations reached under these conditions can easily be increased tenfold and more by using controlled fermentation conditions (unpublished data). Interestingly, the Δxyr1 strain was not completely devoid of extracellular protein production and secreted a low amount of extracellular proteins during cultivation on d-glucose. However, our data rule out that these proteins would affect cellulase activity assays under these conditions: all three enzyme activity assays specifically demonstrated activity only in the overexpressing strains and were absent from the Δxyr1 strain. Zymograms for cellulase activity confirmed that the major band detected in the SDS-PAGE nicely overlapped with the cellulase activity (our unpublished results). In addition, the culture filtrates did not reveal any signs of proteolytic degradation which is frequent during cultivation on inducing carbon sources (Haab et al. 1990; Hagspiel et al. 1989). The nature of the two proteins secreted by Δxyr1 has not been examined in this study. However, the lower Mr band exhibits a mass resembling that of the elicitor of plant response protein EPL1. The orthologue of this gene has been shown to be constitutively expressed in another Trichoderma sp., Trichoderma atroviride, during growth on d-glucose (Seidl et al. 2006b), and it is possible that the respective protein produced by Δxyr1 strain is thus T. reesei EPL1.
In this study, we used d-glucose as a low-cost carbon source. Due to the use of the Δxyr1 strain as host, carbon sources such as cellulose, xylan, lactose, or d-xylose cannot be applied as substrates since the host strain would fail to assimilate them (Stricker et al. 2006; Seiboth et al. 2007; our unpublished data). However, glucose may not be always the best carbon source for protein production because it is known to nonenzymatically form Schiff’s bases with amino group side chains in proteins and eventually lead to their activation (Sullivan 1996). Therefore alternative carbon sources such as glycerol or protein hydrolysates, whose utilization is not XYR1 dependent could be used with probably equal results. Tef1 is known to be well expressed under all these conditions. The cdna1 promoter is currently tested by us for its response on nonglucose carbon sources. The system configured here for shake flask experiments can further be refined since conditions for highly reproducible growth of T. reesei in microtiter plates have been established which allow also the testing for the formation of extracellular enzyme activities (Seidl et al. 2006a). Taken together, our expression system based on the cDNA1- and tef1-driven expression vectors and the cellulase negative host strain Δxyr1 is suitable for a high-throughput system for testing of cellulase muteins.
References
Akel E, Metz B, Seiboth B, Kubicek CP (2009) Molecular regulation of arabinan and l-arabinose metabolism in Hypocrea jecorina (Trichoderma reesei). Eukaryot Cell 8(12):1837–1844. doi:10.1128/EC.00162-09
Boer H, Teeri TT, Koivula A (2000) Characterization of Trichoderma reesei cellobiohydrolase Cel7A secreted from Pichia pastoris using two different promoters. Biotechnol Bioeng 69(5):486–494
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37(Database issue):D233–D238
Carle-Urioste JC, Escobar-Vera J, El-Gogary S, Henrique-Silva F, Torigoi E, Crivellaro O, Herrera-Estrella A, El-Dorry H (1997) Cellulase induction in Trichoderma reesei by cellulose requires its own basal expression. J Biol Chem 272(15):10169–10174
Carroll A, Somerville C (2009) Cellulosic biofuels. Annu Rev Plant Biol 60:165–182
Cherry JR, Fidantsef AL (2003) Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol 14(4):438–443
Claeyssens M, Aerts G (1992) Characterisation of cellulolytic activities in commercial Trichoderma reesei preparations: an approach using small, chromogenic substrates. Bioresour Technol 39:143–146
Ghose TK (1987) Measurement of cellulase activities. Pure Appl Chem 59(2):257–268
Godbole S, Decker SR, Nieves RA, Adney WS, Vinzant TB, Baker JO, Thomas SR, Himmel ME (1999) Cloning and expression of Trichoderma reesei cellobiohydrolase I in Pichia pastoris. Biotechnol Prog 15(5):828–833. doi:10.1021/bp9901116
Gruber F, Visser J, Kubicek CP, de Graaff LH (1990) The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr Genet 18(1):71–76
Haab D, Hagspiel K, Szakmary K, Kubicek CP (1990) Formation of the extracellular proteases from Trichoderma reesei QM 9414 involved in cellulase degradation. J Biotechnol 16(3–4):187–198. doi:10.1016/0168-1656(90)90035-A
Hagspiel K, Haab D, Kubicek CP (1989) Protease activity and proteolytic modification of cellulases from a Trichoderma reesei QM 9414 selectant. Appl Microbiol Biotechnol 32(1):61–67. doi:10.1007/BF00164824
Henrissat B, Driguez H, Viet C, Schülein M (1985) Synergism of cellulases from Trichoderma reesei in the degradation of cellulose. Nat Biotechnol 3:722–726. doi:10.1038/nbt0885-722
Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315(5813):804–807
Jeoh T, Michener W, Himmel ME, Decker SR, Adney WS (2008) Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnol Biofuels 1(1):10. doi:10.1186/1754-6834-1-10
Kubicek CP, Mikus M, Schuster A, Schmoll M, Seiboth B (2009) Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol Biofuels 2:19
Kumar R, Singh S, Singh OV (2008) Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives. J Ind Microbiol Biotechnol 35(5):377–391
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EG, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, Schoch CL, Yao J, Barabote R, Nelson MA, Detter C, Bruce D, Kuske CR, Xie G, Richardson P, Rokhsar DS, Lucas SM, Rubin EM, Dunn-Coleman N, Ward M, Brettin TS (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26(5):553–560
Merino ST, Cherry J (2007) Progress and challenges in enzyme development for biomass utilization. Adv Biochem Eng Biotechnol 108:95–120
Metz B, Seidl-Seiboth V, Haarmann T, Kopchinskiy A, Lorenz P, Seiboth B, Kubicek CP (2011) Expression of biomass degrading enzymes is a major event during conidium development in Trichoderma reesei. Eukaryot Cell 10(11):1527–1535. doi:10.1128/EC.05014-11
Nakari T, Alatalo E, Penttilä ME (1993) Isolation of Trichoderma reesei genes highly expressed on glucose-containing media: characterization of the tef1 gene encoding translation elongation factor 1 alpha. Gene 136(1–2):313–318
Nakari-Setälä T, Penttilä M (1995) Production of Trichoderma reesei cellulases on glucose-containing media. Appl Environ Microbiol 61(10):3650–3655
Nakazawa H, Okada K, Kobayashi R, Kubota T, Onodera T, Ochiai N, Omata N, Ogasawara W, Okada H, Morikawa Y (2008) Characterization of the catalytic domains of Trichoderma reesei endoglucanase I, II, and III, expressed in Escherichia coli. Appl Microbiol Biotechnol 81(4):681–689. doi:10.1007/s00253-008-1667-z
Noguchi Y, Sano M, Kanamaru K, Ko T, Takeuchi M, Kato M, Kobayashi T (2009) Genes regulated by AoXlnR, the xylanolytic and cellulolytic transcriptional regulator, in Aspergillus oryzae. Appl Microbiol Biotechnol 85(1):141–154. doi:10.1007/s00253-009-2236-9
Penttilä ME, Andre L, Lehtovaara P, Bailey M, Teeri TT, Knowles JK (1988) Efficient secretion of two fungal cellobiohydrolases by Saccharomyces cerevisiae. Gene 63(1):103–112
Seiboth B, Hofmann G, Kubicek CP (2002) Lactose metabolism and cellulase production in Hypocrea jecorina: the gal7 gene, encoding galactose-1-phosphate uridylyltransferase, is essential for growth on galactose but not for cellulase induction. Mol Genet Genomics 267(1):124–132
Seiboth B, Gamauf C, Pail M, Hartl L, Kubicek CP (2007) The d-xylose reductase of Hypocrea jecorina is the major aldose reductase in pentose and d-galactose catabolism and necessary for beta-galactosidase and cellulase induction by lactose. Mol Microbiol 66(4):890–900
Seiboth B, Ivanova C, Seidl-Seiboth V (2011) Trichoderma reesei: a fungal enzyme producer for cellulosic biofuels. In: dos Santos Bernardes MA (ed) Biofuel production—recent developments and prospects, 1st edn. InTech, Rijeka, pp 309–340
Seidl V, Druzhinina IS, Kubicek CP (2006a) A screening system for carbon sources enhancing β-N-acetylglucosaminidase formation in Hypocrea atroviridis (Trichoderma atroviride). Microbiology 152(Pt 7):2003–2012. doi:10.1099/mic.0.28897-0
Seidl V, Marchetti M, Schandl R, Allmaier G, Kubicek CP (2006b) Epl1, the major secreted protein of Hypocrea atroviridis on glucose, is a member of a strongly conserved protein family comprising plant defense response elicitors. FEBS J 273(18):4346–4359. doi:10.1111/j.1742-4658.2006.05435.x
Shoemaker S, Watt K, Tsitovsky G, Cox R (1983) Characterization and properties of cellulases purified from Trichoderma reesei strain L27. Nat Biotechnol 1:687–690. doi:10.1038/nbt1083-687
Sprey B, Lamberta C (1984) Heterogeneity of cellulase complexes from Trichoderma reesei: a preparative isoelectric focusing study of some extracellular hydrolases. FEMS Microbiol Lett 23(2–3):227–232
Sprey B, Uelker A (1992) Isolation and properties of a low molecular mass endoglucanase from Trichoderma reesei. FEMS Microbiol Lett 92(3):253–257. doi:10.1111/j.1574-6968.1992.tb05270.x
Stricker AR, Grosstessner-Hain K, Würleitner E, Mach RL (2006) Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and d-xylose metabolism in Hypocrea jecorina. Eukaryot Cell 5(12):2128–2137
Sullivan R (1996) Contributions to senescence: non-enzymatic glycosylation of proteins. Arch Physiol Biochem 104(7):797–806. doi:10.1076/apab.104.7.797.13107
Vaheri M, Leisola M, Kaupinnen V (1979) Transglycosylation products of the cellulase system of Trichoderma reesei. Biotechnol Letts 1:41–46
Van Arsdell JA, Kwok S, Schweickart VL, Ladner MB, Gelfand DH, Innis MA (1987) Cloning, characterization, and expression in Saccharomyces cerevisiae of endoglucanase I from Trichoderma reesei. Nat Biotechnol 5:60–64. doi:10.1038/nbt0187-60
Van Tilbeurgh H, Claeyssens M, de Bruyne CK (1982) The use of 4-methylumbelliferyl and other chromophoric glycosides in the study of cellulolytic enzymes. FEBS Lett 149:152–156
Wilson DB (2009) Cellulases and biofuels. Curr Opin Biotechnol 20(3):295–299
Zhang YHP, Himmel ME, Mielenz JR (2006) Outlook for cellulase improvement: screening and selection strategies. Biotech Adv 24(5):452–481
Acknowledgments
FU and US were supported by TUBITAK (BIDEB 2210- National Scholarship Programme for MSc Students) and by the Turkish State Planning Agencies (DPT) Viskosel grant. CPK and BS were supported by the Austrian Science Foundation FWF (P19690, P19421, and P24219).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Uzbas, F., Sezerman, U., Hartl, L. et al. A homologous production system for Trichoderma reesei secreted proteins in a cellulase-free background. Appl Microbiol Biotechnol 93, 1601–1608 (2012). https://doi.org/10.1007/s00253-011-3674-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3674-8