
Abstract
Summary
Periostin (POSTN) as a regulator of osteoblast differentiation and bone formation may affect susceptibility to osteoporosis. This study suggests POSTN as a candidate gene for bone mineral density (BMD) variation and vertebral fracture risk, which could better our understanding about the genetic pathogenesis of osteoporosis and will be useful in clinic in the future.
Introduction
The genetic determination of osteoporosis is complex and ill-defined. Periostin (POSTN), an extracellular matrix secreted by osteoblasts and a regulator of osteoblast differentiation and bone formation, may affect susceptibility to osteoporosis.
Methods
We adopted a tag-single nucleotide polymorphism (SNP) based association method followed by imputation-based verification and identification of a causal variant. The association was investigated in 1,572 subjects with extreme-BMD and replicated in an independent population of 2,509 subjects. BMD was measured by dual X-ray absorptiometry. Vertebral fractures were identified by assessing vertebral height from X-rays of the thoracolumbar spine. Association analyses were performed with PLINK toolset and imputation analyses with MACH software. The top imputation finding was subsequently validated by genotyping. Interactions between POSTN and another BMD-related candidate gene sclerostin (SOST) were analyzed using MDR program and validated by logistical regression analyses. The putative transcription factor binding with target sequence was confirmed by electrophoretic mobility shift assay (EMSA).
Results
Several SNPs of POSTN were associated with BMD or vertebral fractures. The most significant polymorphism was rs9547970, located at the −2,327 bp upstream (P = 6.8 × 10−4) of POSTN. Carriers of the minor allele G per copy of rs9547970 had 1.33 higher risk of vertebral fracture (P = 0.007). An interactive effect between POSTN and SOST upon BMD variation was suggested (P < 0.01). A specific binding of CDX1 to the sequence of POSTN with the major allele A of rs9547970 but not the variant G allele was confirmed by EMSA.
Conclusions
Our results suggest POSTN as a candidate gene for BMD variation and vertebral fracture risk.
Similar content being viewed by others

Introduction
Osteoporosis is a common skeletal disorder characterized by compromised bone strength leading to an increased risk of fracture. Bone mineral density (BMD) is a widely used proxy measure and accounts for ∼70% of bone strength [1]. Genetic studies have firmly established that BMD is under strong genetic control with a heritability estimate of 0.6–0.85 [2–4]. In the last few decades, many linkage and association studies have been conducted to identify genes that underlie low bone mass and reported some disease-related genes. Nevertheless, despite several genome-wide association studies (GWAS) that have attempted to unravel the genetic components of osteoporosis, the loci identified thus far combined account for <5% of the variance in BMD [5]. Some truly associated variants might be filtered out in current GWAS, due to the highly stringent method used for the correction of multiple testing, which could inflate the false-negative rate. While GWAS enables high-throughout evaluation of thousands of single nucleotide polymorphisms (SNPs), many of these markers have no known function. In an attempt to further understand the genetic pathogenesis that is responsible for the predisposition to or progression of osteoporosis, the association study based on candidate genes with prior functional knowledge of their influence on bone metabolism remains an attractive and cost-effective way to identify genes and variants for osteoporosis.
Bone is a highly dynamic structure that undergoes constant remodeling. Osteoporosis occurs when bone resorption by osteoclasts exceeds bone formation by osteoblasts. Periostin (POSTN) is an extracellular matrix secreted by osteoblasts. It regulates the recruitment and adhesion of osteoprogenitors from essential sources such as bone marrow and blood [6]. POSTN is a member of the vitamin-K-dependent gamma carboxylated protein family and is homologous to other family members such as fasciclin I, MPB70, and bIG-H3 proteins. All these secreted proteins regulate cell adhesion [7, 8]. The extracellular domain of POSTN is evolutionarily conserved from humans to bacteria [9]. POSTN was first identified in MC3T3-E1 osteoblast-like cells [8], and it was preferentially expressed in periosteum in vivo [10]. The overexpression of a basic helix–loop–helix transcription factor, Twist, is related to the increased expression of POSTN by binding to its promoter in preosteoblasts [11]. Twist plays a key regulatory role in early osteogenesis [12]. Inactivation of POSTN leads to a severe reduction of osteoblast-specific differentiation markers, such as type I collagen, osteocalcin, osteopontin, and alkaline phosphatase [13]. Recently, an animal study demonstrated that the Postn protein is essential for the down-regulation of sclerostin (Sost) and thereby plays an important role in the determination of bone mass and microstructural in response to loading [14]. SOST is important in bone and mineral metabolism, and its polymorphisms have previously been shown to associate with BMD [15].
These functional reports propose a role for POSTN in human osteoblast differentiation and bone formation. This prompted us to perform a genetic association study between SNPs along the POSTN gene and osteoporosis phenotypes. We first selected the tag SNPs (tSNPs) of the POSTN gene and studied their relationship with BMD variation in a Hong Kong Southern Chinese (HKSC) population that included 1,572 subjects with extreme BMD. We then used the imputation approach to study the phenotypic associations with a more extensive fine map of polymorphisms around the gene region using the Asian population data of HapMap phase II as the reference. The significant association was further confirmed in another independent Hong Kong Osteoporosis Study (HKOS) prospective cohort with BMD (n = 2,509) and vertebral fracture (n = 1,746) data. In addition, the finding from animal study may suggest the interactive effect between POSTN and SOST genes on regulating of BMD; thus, the interaction analysis was also conducted between these two genes in this study. Finally, the potentially biological function of the identified variant of POSTN gene was studied.
Methods
Subjects
HKSC cohort with extreme BMD
A total of 1,572 unrelated subjects (81.3% women) with either high or low BMD were selected from a growing database at the Osteoporosis Centre of the University of Hong Kong (>9,000 HKSC volunteers). Subjects that were reported to have diseases or environmental factors that may affect BMD and bone metabolism were excluded. The recruitment procedure and exclusion criteria have been detailed elsewhere [16]. BMD was measured at the lumbar spine (LS) and femoral neck (FN) by dual X-ray absorptiometry (Hologic QDR4500, Waltham, MA, USA). The in vivo precision of the machine was 1.2% and 1.5% for LS and FN BMD, respectively. Low BMD subjects were individuals with a BMD z-score ≤ −1.28 (the lowest 10% of the population) at either LS or FN; high BMD subjects had BMD z-score ≥ +1.0 (the highest 15% of the population) at one or both skeletal sites [17, 18]. Height was measured using a wall-mount stadiometer and weight with an electronic scale.
HKOS prospective cohort (for replication)
This random population is also a part of the on-going HKSC database with BMD (n = 2,509) and vertebral fracture (n = 1,746) data. A total of 1,794 unrelated postmenopausal women (≥45 years) and 715 men (≥50 years), without receiving osteoporosis treatment or any drug known to influence bone metabolism, were included as described previously [19]. Vertebral fractures were assessed by digital measurements of morphologic changes on a lateral radiograph of the thoracolumbar spine. A vertebral body was considered fractured if there was a reduction of at least 3 SD in anterior, mid or posterior ratios compared with normative means [20]. The information on vertebral fracture was available for a total of 1,746 subjects.
All subjects gave informed consent. The study was approved by the institutional review board of the Hong Kong West Cluster Hospitals of the Hospital Authority and the University of Hong Kong and was conducted according to the Declaration of Helsinki.
SNP genotyping
A total of 10 SNPs in the POSTN gene were selected for genotyping: seven tSNPs with reported minor allele frequency (MAF) ≥0.05 in Chinese and three potentially functional SNPs located in exons. The tSNPs were identified using data from the phase II HapMap CHB (r 2 ≥ 0.8). SNPs for HKSC extreme cohort were genotyped using the high-throughput Sequenom MassARRAY platform (Sequenom, San Diego, CA, USA). DNA from high and low BMD subjects were randomly assigned to the 96-well plates and genotyping performed with sample status blinded. Genotyping was repeated in 5% of the samples for verification: Data were confirmed to have an error rate <0.1%. The TaqMan system (Applied Biosystems, Foster City, CA, USA) was used for SNP genotyping in the verification and replication steps.
Statistical methods
Both single marker and haplotype association analyses were performed using the PLINK software [21]. Any SNP with call rate <90%, MAF <0.01 or Hardy–Weinberg equilibrium (HWE) P < 0.001 was excluded. The binary logistic regression was used to test the association between each SNP and BMD variation of the HKSC extreme cohort and vertebral fractures under the additive model. The association of SNP with BMD variation in the replication cohort was detected by the linear regression analysis. In the block-based haplotype association analysis, the haplotype global test is an omnibus test (if there are H haplotypes, a single test with H-1 degrees of freedom is conducted). The haplotype-specific test evaluates each specific haplotype versus all other haplotypes (i.e., tests with 1 degrees of freedom). The conditional haplotype testing model was used to assess the correlation structure and identify which SNP was most likely to be the causal variant. The hypothesis is that if the global haplotype association disappears in the omnibus test when conditioned on SNP “A” but remains significant under the control of other SNPs, then SNP “A” accounts for the observed association. The age, height, weight, and gender were included as covariates in all of the association analyses. Statistical tests were performed for both LS and FN BMD. The false discovery rate (FDR) method, which is an effective way to address the problems of multiple comparisons, was used in this study to correct for multiple testing.
The imputation of genotypes for untyped SNPs from HapMap in the POSTN gene and its flanking regions, approximately 5 kb upstream and downstream, was conducted by a hidden Markov model programmed in MACH v1.0 [22]. We used the phase II HapMap Asian data (CHB and JPT) as the reference panel. In brief, this method combines genotypic data of studied samples with the reference genotype data and then infers genotypes of untyped SNPs based on probability. The most frequently sampled genotype will be the final imputed one. We used the most likely genotype for the association analysis. The estimated squared correlation (r 2) between imputed and true genotypes was used to assess the imputation quality in MACH. SNPs with r 2 < 0.3 were defined as low imputation quality and were excluded. The most significant untyped SNP was validated by direct genotyping in the HKSC extreme cohort and was replicated in the HKOS prospective cohort. The weighted z-transform test was used in the meta-analysis of SNP with BMD variation in this study.
The interactive effect between POSTN and SOST genes was evaluated using our GWAS data with about 500K SNPs in 800 female subjects with extreme BMD that has been described in detail previously [18]. These 800 GWAS extreme subjects belong to the HKSC extreme cohort, which was used as the discovery cohort in this study (n = 1,572). Several polymorphisms in these two genes showed nominally significant association with BMD in our GWAS (P < 0.05), although they failed to reach the genome-wide significant level (Table S3, ESM 1). The most significant SNP of POSTN from this candidate gene study and four SNPs (rs9899889, rs865429, rs1234612, and rs2301682) in the SOST and ∼20 kb flanking regions from the GWAS data were used for the interaction analysis. The interactions were assessed by the MDR program [23]. MDR is a nonparametric data mining approach, which pools multi-locus genotypes with high dimensions into one dimension model. It evaluated the predictor using cross-validation method and permutation testing. The combinatorial examination by these two approaches would minimize false positive rates. Cross-validation consistency and testing accuracy were calculated for each combination of tested SNPs. The final best model was the one with maximal cross-validation consistency and minimal prediction error. The statistical significance was assessed by comparing the average prediction accuracy from the observed data to the distribution of average prediction accuracy under the null hypothesis of no associations derived empirically from 1,000 permutations. All possible two-, three-, and four-way SNP interactions were tested using 20-fold cross-validation in an exhaustive search (considering all possible SNP combinations). The conditional logistic regression analysis was performed using SPSS (v16.0) to confirm the reported interactive effects in MDR, which may be caused by the main effects from the component loci instead of the epistatic interactions. A logistic regression analysis with P < 0.05 could support the corresponding significant MDR interaction model.
Electrophoretic mobility shift assay
The human complementary DNA clone of CDX1 (pCMV6-CDX1) was produced by OriGene (OriGene Technologies, Rockville, MD, USA). CDX1 protein preparation was made by transfecting pCMV6-CDX1 construct into HEK293 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were harvested 48-h post-transfection, and nuclear extractions were performed using a nuclear extraction kit (Panomics, Fremont, CA, USA). Protein concentration was measured using the DC protein assay kit (Bio-Rad, Hercules, CA, USA), with bovine serum albumin as a standard. The following double-stranded oligonucleotides were synthesized (Sigma-Aldrich Corp., St. Louis, MO, USA) and used in electrophoretic mobility shift assay (EMSA): (1) the labeled major allele A probe, corresponding to POSTN sequences centering rs9547970 (underlined and bolded in the following sequences), prepared by annealing of the biotin-labeled oligonucleotide 5′-AAAAGAGAGGTCTTAAATCTTTCTTTTCACACT-3′ with the complementary sequence 5′-AGTGTGAAAAGAAAGATTTAAGACCTCTCTTTT-3′; (2) the minor allele G probe, prepared by annealing the biotin-labeled oligonucleotide 5′-AAAAGAGAGGTCTTGAATCTTTCTTTTCACACT-3′ with the complementary sequences 5′-AGTGTGAAAAGAAAGATTCAAGACCTCTCTTTT-3′; and (3, 4) the corresponding unlabeled major allele A and minor allele G probes.
The EMSA was performed using the EMSA kit (Panomics, Fremont, CA, USA). We incubated 10 ng of biotin-labeled probe with 15.64 μg of nuclear extract of HEK293 cells transfected with pCMV6-CDX1 for 30 min at 15°C in a 10-μl reaction volume containing 2 μl 5× binding buffer (aqueous buffered solution for TF binding) and 1 μg poly d(I-C). Nuclear extract of untreated HEK293 served as negative control. For competitive reactions, we used the above unlabeled probe for competition at 660-fold molar excess of the labeled probe. After incubation, samples were separated by electrophoresis on a 6% non-denaturing polyacrylamide gel with 0.5× Tris–borate–EDTA buffer. DNA–protein complexes were electroblotted to Pall Biodyne B nylon membrane (Pall Corp., Pensacola, FL, USA) and visualized by exposure to Chemiluminescent Detection Film (Agfa, Shanghai, China).
Results
The characteristics of the subjects are summarized in Table 1 for both the extreme and prospective cohorts and Table S1 (ESM 1) for the LS and FN sub-groups of the extreme cohort. The two populations differed in age, height, and weight, and these three factors were included as covariates in the association analyses to eliminate their potential confounding effects. Two SNPs rs1133973 and rs3205088 were monomorphic in our population. The remaining eight SNPs passed the genotyping quality control criteria with the overall genotyping call rate >98.7%, duplicate error rate <1%, MAF >1%, and HWE P > 0.001 (Table 2).
Association between POSTN and BMD variation in tSNP-based analysis
Table 2 lists the single-marker association results with BMD variation in the extreme cohort. Two tSNPs (rs7322993 and rs7338244) in the POSTN gene showed significant associations with BMD variation after the correction of multiple testing (P FDR < 0.05, OR > 1). Both of their minor alleles were related to the higher risk of low BMD. SNP rs7322993 had the strongest association (P = 0.001), and the P values were 0.006 and 0.029 for BMD at LS and FN, respectively, in site-specific analyses.
We examined the association between common haplotypes of POSTN and BMD variation. The LD structure of POSTN is illustrated in Figure S1 (ESM 1). The haplotype in POSTN was associated with BMD variation in the global test (P = 0.038) (Table S2, ESM 1). Table S2 (ESM 1) also shows the specific effects of each haplotype. Seven haplotypes were identified, each with a frequency >3%. The haplotype CGTTGAAG, including both the low BMD-related alleles of rs7322993 and rs7338244, was most strongly associated with BMD variation (P = 0.025) and a higher risk of low BMD was expected. The haplotype data corresponded to the single marker analysis, but did not add further information to outcome.
Validation of rs9547970 with imputation-based association testing
We used the imputation method to study SNPs that were not genotyped in our study, but available from the HapMap database. This enabled a more comprehensive fine map of polymorphisms along POSTN and a better understanding of this association. We estimated the strength of evidence for a phenotypic association with typed and untyped SNPs located between ∼5 kb upstream and ∼5 kb downstream of POSTN. Sixty-two SNPs from the Asian population data of HapMap phase II were extracted for imputation. In addition to the genotyped SNPs, 54 imputed SNPs (MAF ≥ 0.01 and the imputation quality r 2 ≥ 0.3) were tested for associations with BMD variation.
The strongest evidence for an association with BMD variation in the imputed data is undoubtedly for the untyped SNP rs9547970 (P FDR < 0.05), which is located at −2,327 bp upstream of POSTN (Fig. 1). An additional 27 SNPs displayed convincing evidence of association (P < 0.005) and were in high LD (r 2 > 0.5) with rs9547970 based on the HapMap Asian population data. The most significant untyped-SNP rs9547970 had a high imputation quality (r 2 = 0.9983). Subsequently, it was also directly genotyped in the 1,572 extreme samples to verify its association with BMD variation. The frequency of minor allele G (0.265) of rs9547970 was similar to the MAF of HapMap CHB (0.233). The results demonstrated the association between rs9547970 and BMD variation, with P (OR, 95%CI) values of 6.8 × 10−4 (1.41, 1.16–1.73) in all subjects, 0.007 (1.38, 1.09–1.76) and 0.019 (1.42, 1.06–1.91) for LS and FN subgroups, respectively. Its G allele was related to the higher risk of low BMD (Table 3).

Association results of BMD variation with single SNPs from the imputed genotyping data after the adjustment of age, height, weight, and gender in all of the 1,572 extreme subjects. X-axis the genomic position (B36); Y-axis the −log10 (P value) of association results (left scale) and the fine scale recombination rate (B36, right scale); circle dots genotyped SNPs; square dots untyped SNPs. The colors of dots are coded according to the degree of linkage disequilibrium (r 2) with rs9547970 identified as the most significant SNP in this study (P FDR < 0.05), and this imputed top SNP was then directly genotyped in the 1,572 extreme subjects for validation; rs1977278, the SNP had strongest association with BMD variation in the Framingham Study; rs7322993 and rs7338244, the selected tSNPs showed significant associations with BMD variation after the correction of multiple testing in the tSNP-based analyses (P FDR < 0.05)
To further explore the relationship between the significant SNPs rs7322993, rs7338244, and rs9547970 with BMD variation, we performed the conditional haplotype analysis using these three SNPs. The global association was significant (P < 0.01) and remained significant when conditioned on rs7322993 or rs7338244 (P < 0.05) but disappeared when conditioned on rs9547970 (P > 0.1). This provides further evidence that the significant associations of rs7322993 and rs7338244 derive from the LD correlation with rs9547970 (D′ = 1, r 2 ≥ 0.5) also that rs9547970 is the most promising candidate to explain the identified association.
Replication in an independent population-based cohort
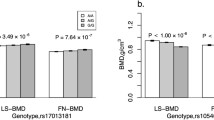
The association between rs9547970 and BMD variation was replicated in the HKOS prospective cohort. This is a pre-hypothesis test; thus, one-sided P < 0.05 can be taken as a successful replication. The one-sided P value (beta) was 0.023 (−0.078) for LS BMD and 0.039 (−0.061) for FN BMD (Table 3). The effect direction of G allele was consistent with the initial analysis in the HKSC extreme subjects, which was related to low BMD. The effect size of rs9547970 estimated in the HKSC extreme cohort could be biased because of selection for extreme subjects. Thus, we conducted the estimation in our HKOS prospective cohort, and the allelic variance of rs9547970 of POSTN explained ∼0.25% and ∼0.15% of BMD variance at LS and FN, respectively. The raw BMD value was 0.030 and 0.011 (g/cm2) less in minor allele GG carriers compared with AA carriers for LS and FN, respectively (Fig. S2, ESM 1). Using weighted z-transform test, the meta-analyzed P values of rs9547970 were 0.003 and 0.01 for LS BMD and FN BMD, respectively.
Furthermore, results supported the association of rs9547970 with vertebral fractures even after the adjustment of LS BMD and the covariates of age, height, weight, and gender (P = 0.007, OR 1.33, 95%CI 1.08–1.62, Table 3). Carriers of the minor allele G per copy of rs9547970 had 1.33 higher risk of vertebral fracture, consistent with the association of G allele with low BMD. To detect the effect of age difference between two groups on vertebral fractures, besides age, the age2 was also added to the model as a covariate, and the result was similar to the model without age2. This suggested that the association of rs9547970 with vertebral fractures was derived from the genetic effect independent of the effect of age.
Interactions between POSTN and SOST genes
A previously functional study on bone metabolism suggested the molecular interaction between POSTN and SOST [14]. Results from MDR also suggested an interactive effect of POSTN and SOST genes upon BMD variation (P < 0.001). The best models for each trait were listed in Table 4, of which two-way SNPs model were associated with BMD variation in all subjects and four-way model for LS BMD and three-way model for FN BMD. We validated these three potential interaction models using the conditional logistic regression method. Results showed that these three models were highly supported by logistic regression (P < 0.01). The results from both MDR and conditional logistical regression analyses indicated the existence of gene–gene interactions between POSTN and SOST genes influencing BMD variation.
EMSA showed the disappearance of CDX1 binding site in the variant allele of rs9547970
To detect the potential function of the identified variant, we used the FASTSNP program to predict the function of rs9547970 [24]. Bioinformatics analysis suggests that the allele change (A/G) at rs9547970 should demolish one binding site of CDX1 (caudal type homeobox 1) (MIM 600746). We therefore conducted an EMSA to confirm the potential changes of CDX1 binding to POSTN caused by rs9547970.
In the gel shift assay (Fig. 2), the 33-bp oligonucleotides that contained both allelic variants of rs9547970, representing native and mutated CDX1 binding sites, were assayed with nuclear extract of HEK293 cells transfected with pCMV-CDX1. We found a specific binding of CDX1 from nuclear extract of HEK293 cells transfected with pCMV6-CDX1 to the wild-type site centering the rs9547970 major allele A of POSTN. No binding was observed with oligonucleotide containing the minor allele G. Binding to the major A allele resulted in a complex that was specifically competed by 660-fold excess of unlabeled probe containing the major A allele. The results indicate that the A/G change at rs9547970 demolishes a CDX1 binding site in the POSTN gene.

Electrophoretic mobility shift and competition assays with nuclear extract of HEK293 cells transfected with pCMV-CDX1 and allelic variants of SNP rs9547970 in POSTN. 1 Labeled G probe + nuclear extract(Cdx1−); 2 labeled A probe + nuclear extract (Cdx1−); 3 labeled G probe; 4 labeled G probe + nuclear extract (Cdx1+); 5 labeled G probe + nuclear extract (Cdx1+) + unlabeled G probe; 6 labeled A probe; 7 labeled A probe + nuclear extract (Cdx1+); 8 labeled A probe + nuclear extract (Cdx1+) + unlabeled A probe. Cdx1−, from untreated HEK293; Cdx1+, from HEK293 transfected with pCMV-CDX1
Discussion
We reported the association of common polymorphisms along the POSTN gene (13q13.3) with osteoporosis phenotypes. SNP rs9547970 was determined to be the variant that could best explain the identified association. The EMSA experiment further demonstrated the specific binding of CDX1 to sequence around rs9547970 with major allele A.
Previous functional studies have demonstrated the important role of POSTN in the regulation of osteoblast differentiation and bone formation [6, 8, 11, 13]. In this study, there was also evidence to strongly suggest an association of polymorphisms in the POSTN gene with osteoporosis phenotypes. We first used a tSNP-based method to facilitate this association study. This is sufficient to capture most of the variation in the studied region and can reduce the genotyping effort for association mapping. The results supported the association of the POSTN gene with BMD variation from both the single marker (P FDR < 0.05) and haplotype analysis (P < 0.05). These tSNP-based results presumably suggest that the POSTN gene may influence BMD variation.
The tSNP-based method is a cost-effective approach but has limitations in the determination of real causal variants. We therefore used the available data of HapMap Asian population to study the variation in the POSTN gene with a much higher coverage. The association analyses of imputed data revealed the most significant SNP rs9547970 located at −2,327 bp upstream of POSTN. This was validated by direct genotyping in the HKSC extreme cohort (P = 6.8 × 10−4) and by replication in the HKOS prospective cohort (one-sided P < 0.05) with effect direction of rs9547970 consistently relating to low BMD. Our further analysis revealed that rs9547970 was the most promising candidate causal variant, and the significance of other SNPs arose from the high LD with rs9547970. Similar to other susceptible allele, the allelic variance of rs9547970 explained only a small portion (<0.5%) of the variance in BMD, but the information from this study adds to the understanding of the genetic control of BMD and fracture risk. In addition, we also evaluated the gender-specific effect of POSTN gene on BMD variation using the HKSC extreme subjects. Associations were found in female (P < 0.001) while not in male (P > 0.05) in gender-specific analyses, while effects on BMD were not significantly different between the two genders (P > 0.1) in the gender-interaction analysis using the regression model. Failure to detect the association in male subjects might be due to the small number of males in our population. Furthermore, evidence from the replication step was also observed for an association of rs9547970 with high risk of vertebral fractures, consistent with its association with low BMD. Compared with the modest effect of POSTN on BMD variation, the very high odds ratio on vertebral fracture risk is likely due to an overestimation, as there are several other confounders related to fracture risk that could not be adjusted in the analysis. Future study is required to assess the actual effect of POSTN on fracture determination. However, the findings from this study suggest that the POSTN gene is likely to play a contributory role to BMD and fracture risk prediction. In addition, the association of POSTN with vertebral fracture remained significance even after the adjustment of LS BMD. This confirms that BMD alone is inadequate to comprehensive measure of bone strength and structure and predict the risk of fracture [25, 26]. These association results were limited to Chinese population in this study, and further replications in other ethnic groups are necessary.
The association between POSTN gene and BMD was supported by previously published genome-wide linkage and genome-wide association studies. The NEMO Family Study suggested that the 13q12-14 region may contain quantitative trait loci linked to BMD variation [27]. According to the available results from dbGaP, in the 100K association data of bone mass in the Framingham Heart Study [28], SNPs in the POSTN gene showed associations with BMD variation, although they were not prominent in this GWAS. A polymorphism rs1977278 was associated with LS BMD (P = 0.008, n = 1,141) using the additive generalized estimating equation model. Another SNP, rs7336380, showed a modest association with LS BMD (P = 0.018). Both SNP rs1977278 and rs7336380 are in relative high LD with rs9547970 (r 2 > 0.5) based on the CHB HapMap data. These two SNPs in our population was also associated with low LS BMD under the same direction of effect (P = 0.012 for rs1977278 and P = 0.013 for rs7336380). The association significance of rs1977278 and rs7336380 were further supported and strengthened in the meta-analysis of HKSC extreme cohort and Framingham Heart Study with P values being 4.82 × 10−4 and 1.14 × 10−3 for LS BMD, respectively. Publically available Caucasian databases from populations with GWAS in BMD such as the Framingham Heart Study and deCODE GWAS Study do not have information on rs9547970, and it would be very interesting to genotype this SNP for replication studies in Caucasian populations.
Cross-species comparison indicated that the proximal 5 kb upstream of the translational start site of POSTN comprised evolutionarily conserved domains [29]. SNPs of the 5′ flanking region may be involved in the regulation of gene expression. Thus, we searched for possible transcription factor binding sites in this region using the FASTSNP program. Results were confirmed by EMSA experiment and suggested a putative binding site for CDX1 in the presence of major allele A, but not the risk allele G. SNP rs9547970 may alter the transcriptional activity of the POSTN gene, thereby affecting bone formation. The CDX1 gene is a member of the caudal-related homeobox transcription factor family. The encoded DNA-binding protein regulates intestine-specific gene expression and enterocyte differentiation. It induces expression of the intestinal alkaline phosphatase gene and inhibits beta-catenin/T-cell factor transcriptional activity [30]. The functional significance of Homeobox (Hox) genes in embryonic skeletogenesis has been well documented by knockout and deficiency studies; Hox gene expression is reactivated during bone regeneration. The presence of putative Cdx1-binding sites within the regulatory sequences of Hox genes and in vitro transactivation of Hoxa-7 by Cdx1 indicates a direct interaction [31, 32]. Further replication and functional analyses are required to confirm the hypothesis that there is a direct regulation between CDX1-binding and the expression level of POSTN.
Our comprehensive imputation-based analysis identified rs9547970 as the variant that best explains the observed association in this study. Although most promising as the causal variant, it is also possible that rs9547970 is in LD with other unobserved and independent functional variants. According to the imputation based analysis, there was no strong evidence to support this, although the possibility of other independent rare variants of MAF <0.01 in the POSTN gene cannot be ruled out. Resequencing of the entire gene in a large number of individuals would provide more information to clarify the association of the POSTN gene with osteoporosis risk. Our findings were not observed in recently reported GWAS in Caucasian populations [33–35]. This may be due to ethnic differences and sampling and statistical methods. Nevertheless, our study sample was selected from a large population with relatively high homogeneity. The selected sampling strategy can substantially increase power over random sampling for detection of allelic association [36]. According to the Genetic Power Calculator [37], our HKSC extreme cohort has more than 95% power to detect an association for a functional locus accounting for 1% phenotypic variation (P = 0.002, MAF = 0.3, D′ = 0.8). Moreover, the identified association was replicated in another independent population using different genotyping technique and sampling method.
Although GWAS are clearly a major advance for gene discovery, the results from those studies also suggest that more osteoporosis-related variants and genes are yet to be discovered. To date, confirmed loci account for <5% of the BMD variation in the general population, leaving heritability largely unexplained. Many more common variants with increasingly smaller effects and rare variants with possible large effects could contribute to the undiscovered genetic component. In addition, the gene–gene interactions are acknowledged as important contributors to genetic variation in human complex traits. The functional study in animal mode demonstrated that the matricelluar Postn protein is required for Sost inhibition and thereby plays an important role in the determination of bone mass and microstructural [14]. Being consistent with the functional finding, our interaction analyses using MDR suggested the existence of interactive effect between POSTN and SOST genes on BMD variation (P < 0.001), and the results were validated by logistic regression analyses (P < 0.01). This finding supports that BMD variation may be determined by interactive effects between candidate genes other than their individual influence and gene–gene interactive effects could be a significant cause for BMD variation.
In summary, this study reported the associations of variations along the POSTN gene with low BMD and vertebral fracture risk.
References
NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy (2001) Osteoporosis prevention, diagnosis, and therapy. JAMA 285(6):785–795
Dequeker J, Nijs J, Verstraeten A, Geusens P, Gevers G (1987) Genetic determinants of bone mineral content at the spine and radius: a twin study. Bone 8:207–209
Arden NK, Baker J, Hogg C, Baan K, Spector TD (1996) The heritability of bone mineral density, ultrasound of the calcaneus and hip axis length: a study of postmenopausal twins. J Bone Miner Res 11:530–534
Ng MY, Sham PC, Paterson AD, Chan V, Kung AW (2006) Effect of environmental factors and gender on the heritability of bone mineral density and bone size. Ann Hum Genet 70:428–438
Cheung CL, Xiao SM, Kung AWC (2010) Genetic epidemiology of age-related osteoporosis and its clinical application. Nat Rev Rheumatol 6(9):507–517
Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A (1999) Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res 14:1239–1249
Coutu DL, Wu JH, Monette A, Rivard GE, Blostein MD, Galipeau J (2008) Periostin, a member of a novel family of vitamin K-dependent proteins, is expressed by mesenchymal stromal cells. J Biol Chem 283:17991–18001
Takeshita S, Kikuno R, Tezuka K, Amann E (1993) Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J 294(Pt 1):271–278
Kawamoto T, Noshiro M, Shen M, Nakamasu K, Hashimoto K, Kawashima-Ohya Y, Gotoh O, Kato Y (1998) Structural and phylogenetic analyses of RGD-CAP/beta ig-h3, a fasciclin-like adhesion protein expressed in chick chondrocytes. Biochim Biophys Acta 1395:288–292
Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ (2001) Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev 103:183–188
Oshima A, Tanabe H, Yan T, Lowe GN, Glackin CA, Kudo A (2002) A novel mechanism for the regulation of osteoblast differentiation: transcription of periostin, a member of the fasciclin I family, is regulated by the bHLH transcription factor, twist. J Cell Biochem 86:792–804
Lee MS, Lowe GN, Strong DD, Wergedal JE, Glackin CA (1999) TWIST, a basic helix–loop–helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem 75:566–577
Litvin J, Selim AH, Montgomery MO, Lehmann K, Rico MC, Devlin H, Bednarik DP, Safadi FF (2004) Expression and function of periostin-isoforms in bone. J Cell Biochem 92:1044–1061
Bonnet N, Standley KN, Bianchi EN, Stadelmann V, Foti M, Conway SJ, Ferrari SL (2009) The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J Bio Chem 284(51):35939–35950
Huang QY, Li GH, Kung AW (2009) The −9247 T/C polymorphism in the SOST upstream regulatory region that potentially affects C/EBPalpha and FOXA1 binding is associated with osteoporosis. Bone 45(2):289–294
Kung AW, Lai BM, Ng MY, Chan V, Sham PC (2006) T-1213 C polymorphism of estrogen receptor beta is associated with low bone mineral density and osteoporotic fractures. Bone 39:1097–1106
Cheung CL, Chan BY, Chan V, Ikegawa S, Kou I, Ngai H, Smith D, Luk KD, Huang QY, Mori S, Sham PC, Kung AW (2009) Pre-B-cell leukemia homeobox 1 (PBX1) shows functional and possible genetic association with bone mineral density variation. Hum Mol Genet 18(4):679–687
Kung AW, Xiao SM, Cherny S, Li GH, Gao Y, Tso G, Lau KS, Luk KD, Liu JM, Cui B, Zhang MJ, Zhang ZL, He JW, Yue H, Xia WB, Luo LM, He SL, Kiel DP, Karasik D, Hsu YH, Cupples LA, Demissie S, Styrkarsdottir U, Halldorsson BV, Sigurdsson G, Thorsteinsdottir U, Stefansson K, Richards JB, Zhai G, Soranzo N, Valdes A, Spector TD, Sham PC (2010) Association of JAG1 with bone mineral density and osteoporotic fractures: a genome-wide association study and follow-up replication studies. Am J Hum Genet 86(2):229–239
Kung AW, Lee KK, Ho AY, Tang G, Luk KD (2007) Ten-year risk of osteoporotic fractures in postmenopausal Chinese women according to clinical risk factors and BMD T-scores: a prospective study. J Bone Miner Res 22:1080–1087
Black DM, Cummings SR, Stone K, Hudes E, Palermo L, Steiger P (1991) A new approach to defining normal vertebral dimensions. J Bone Miner Res 6:883–892
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Li Y, Abecasis GR (2006) Mach 1.0: Rapid Haplotype Reconstruction and Missing Genotype Inference. Am J Hum Genet S79:2290
Hahn LW, Ritchie MD, Moore JH (2003) Multifactor dimensionality reduction software for detecting gene–gene and gene–environment interactions. Bioinformatics 19(3):376–382
Yuan HY, Chiou JJ, Tseng WH, Liu CH, Liu CK, Lin YJ, Wang HH, Yao A, Chen YT, Hsu CN (2006) FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res 34:W635–W641
Cordey J, Schneider M, Belendez C, Ziegler WJ, Rahn BA, Perren SM (1992) Effect of bone size, not density, on the stiffness of the proximal part of normal and osteoporotic human femora. J Bone Miner Res 2:S437–S444
Tabensky AD, Williams J, DeLuca V, Briganti E, Seeman E (1996) Bone mass, areal, and volumetric bone density are equally accurate, sensitive, and specific surrogates of the breaking strength of the vertebral body: an in vitro study. J Bone Miner Res 11:1981–1988
Kaufman JM, Ostertag A, Saint-Pierre A, Cohen-Solal M, Boland A, Van Pottelbergh I, Toye K, de Vernejoul MC, Martinez M (2008) Genome-wide linkage screen of bone mineral density (BMD) in European pedigrees ascertained through a male relative with low BMD values: evidence for quantitative trait loci on 17q21-23, 11q12-13, 13q12-14, and 22q11. J Clin Endocrinol Metab 93:3755–3762
Kiel DP, Demissie S, Dupuis J, Lunetta KL, Murabito JM, Karasik D (2007) Genome-wide association with bone mass and geometry in the Framingham Heart Study. BMC Med Genet 1:S14
Lindsley A, Snider P, Zhou H, Rogers R, Wang J, Olaopa M, Kruzynska-Frejtag A, Koushik SV, Lilly B, Burch JB, Firulli AB, Conway SJ (2007) Identification and characterization of a novel Schwann and outflow tract endocardial cushion lineage-restricted periostin enhancer. Dev Biol 307:340–355
Guo RJ, Huang E, Ezaki T, Patel N, Sinclair K, Wu J, Klein P, Suh ER, Lynch JP (2004) Cdx1 inhibits human colon cancer cell proliferation by reducing beta-catenin/T-cell factor transcriptional activity. J Biol Chem 279:36865–36875
Subramanian V, Meyer BI, Gruss P (1995) Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell 83:641–653
Gersch RP, Lombardo F, McGovern SC, Hadjiargyrou M (2005) Reactivation of Hox gene expression during bone regeneration. J Orthop Res 23:882–890
Richards JB, Rivadeneira F, Inouye M, Pastinen TM, Soranzo N, Wilson SG, Andrew T, Falchi M, Gwilliam R, Ahmadi KR, Valdes AM, Arp P, Whittaker P, Verlaan DJ, Jhamai M, Kumanduri V, Moorhouse M, van Meurs JB, Hofman A, Pols HA, Hart D, Zhai G, Kato BS, Mullin BH, Zhang F, Deloukas P, Uitterlinden AG, Spector TD (2008) Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet 371:1505–1512
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Center JR, Nguyen TV, Bagger Y, Gulcher JR, Eisman JA, Christiansen C, Sigurdsson G, Kong A, Thorsteinsdottir U, Stefansson K (2008) Multiple genetic loci for bone mineral density and fractures. N Engl J Med 358:2355–2365
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Snorradottir S, Center JR, Nguyen TV, Alexandersen P, Gulcher JR, Eisman JA, Christiansen C, Sigurdsson G, Kong A, Thorsteinsdottir U, Stefansson K (2009) New sequence variants associated with bone mineral density. Nat Genet 41:15–17
Abecasis GR, Cookson WO, Cardon LR (2001) The power to detect linkage disequilibrium with quantitative traits in selected samples. Am J Hum Genet 68:1463–1474
Purcell S, Cherny SS, Sham PC (2003) Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19:149–150
Acknowledgments
This project is supported by Hong Kong Research Grant Council (HKU 768610M), NSFC/GRC Joint Research Scheme N-HKU-715/07, The KC Wong Education Foundation, and The Bone Health Fund, Seed Funding for Basic Research, Small Project Funding (201007176237), Osteoporosis and Endocrine Research Fund, The University of Hong Kong.
Conflicts of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 267 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Xiao, SM., Gao, Y., Cheung, CL. et al. Association of CDX1 binding site of periostin gene with bone mineral density and vertebral fracture risk. Osteoporos Int 23, 1877–1887 (2012). https://doi.org/10.1007/s00198-011-1861-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-011-1861-1