
Abstract
Aims/hypothesis
The incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotrophic peptide (GIP) are released from intestinal endocrine cells in response to luminal glucose. Glucokinase is present in these cells and has been proposed as a glucose sensor. The physiological role of glucokinase can be tested using individuals with heterozygous glucokinase gene (GCK) mutations. If glucokinase is the gut glucose sensor, GLP-1 and GIP secretion during a 75 g OGTT would be lower in GCK mutation carriers compared with controls.
Methods
We compared GLP-1 and GIP concentrations measured at five time-points during a 75 g OGTT in 49 participants having GCK mutations with those of 28 familial controls. Mathematical modelling of glucose, insulin and C-peptide was used to estimate basal insulin secretion rate (BSR), total insulin secretion (TIS), beta cell glucose sensitivity, potentiation factor and insulin secretion rate (ISR).
Results
GIP and GLP-1 profiles during the OGTT were similar in GCK mutation carriers and controls (p = 0.52 and p = 0.44, respectively). Modelled variables of beta cell function showed a reduction in beta cell glucose sensitivity (87 pmol min−1 m−2 [mmol/l]−1 [95% CI 66–108] vs 183 pmol min−1 m−2 [mmol/l]−1 [95% CI 155–211], p < 0.001) and potentiation factor (1.5 min [95% CI 1.2–1.8] vs 2.2 min [95% CI 1.8–2.7], p = 0.007) but no change in BSR or TIS. The glucose/ISR curve was right-shifted in GCK mutation carriers.
Conclusions/interpretation
Glucokinase, the major pancreatic glucose sensor, is not the main gut glucose sensor. By modelling OGTT data in GCK mutation carriers we were able to distinguish a specific beta cell glucose-sensing defect. Our data suggest a reduction in potentiation of insulin secretion by glucose that is independent of differences in incretin hormone release.
Similar content being viewed by others


Introduction
The incretin effect explains the augmentation of insulin secretion by oral glucose compared with similar glycaemia achieved by i.v. glucose and is mediated through gut-derived hormones. The most important incretin hormones are glucose-dependent insulinotrophic peptide (GIP) and glucagon-like peptide 1 (GLP-1), which both potentiate glucose-stimulated insulin release by pancreatic beta cells. There is controversy in the literature as to how these specialised cells sense the gut glucose load. Glucokinase [1], sodium–glucose cotransporter (SGLT) [2] and G protein-coupled receptors [3] have all been proposed as functioning chemosensors in the gut enteroendocrine cells.
The enzyme glucokinase has been proposed as the chief glucose sensor in the gut, analogous to its role in pancreatic beta cells [1, 4–6]. When GIP-producing K cells in the duodenum were engineered to produce insulin under the control of the GIP gene promoter, they functioned in place of beta cells to maintain whole-body glucose homeostasis [5, 7]. In vitro data from immortalised L cell lines suggest that GLP-1 release from L cells and insulin release from beta cells share some common glucose-sensing machinery including glucokinase [8, 9]. In addition to glucokinase-regulated enteroendocrine cells releasing GIP and GLP-1, glucose-responsive neurons in the myenteric plexus may also be regulated by glucokinase [1, 10, 11].
We aimed to study patients with naturally occurring glucokinase gene (GCK) mutations to assess the role of glucokinase in GIP and GLP-1 secretion. Heterozygous GCK mutation carriers have a rightward shift in their glucose/insulin secretion rate (ISR) dose–response curves compared with normal controls, consistent with their reduced glucose-sensing function of pancreatic beta cell glucokinase [12]. Since luminal, but not systemic, glucose is a potent trigger of incretin hormone release, if glucokinase was also the chief glucose sensor in the gut K and L cells then, we hypothesised, the secretion of incretin hormones (GIP and GLP-1) in response to an identical oral glucose load would be lower in heterozygous GCK mutation carriers compared with controls (despite a higher plasma glucose level in the mutation carriers). We also used these OGTT data to employ a mathematical model of beta cell function to quantify abnormalities in pancreatic glucose sensitivity and potentiation in GCK mutation carriers compared with controls.
Methods
Participants for clinical studies
Seventy-seven participants from GCK mutation families were recruited in the UK. Forty-nine carried the GCK mutation and 28 familial controls did not. All participants were between 16 and 70 years and had no history of type 2 diabetes in first-degree relatives. Pregnant women and those taking steroids were excluded. The study protocol was approved by local research ethics committees and the studies were performed in accordance with the Declaration of Helsinki. All participants gave written informed consent.
Methods for clinical studies
Measurements were taken according to a common protocol. Participants fasted from 22:00 hours the night before assessment, and avoided excessive exercise and alcohol for the previous 24 h. Three plasma samples for glucose, insulin, C-peptide, GLP-1 and GIP were collected from the fasting participants before an OGTT. Four further plasma samples were taken at 30 min intervals following a 75 g oral glucose load. A single blood sample was taken for DNA extraction. Body composition was assessed by bioelectrical impedance (Bodystat 1500; Douglas, Isle of Man, British Isles). Waist and hip measurements were taken.
Sample analysis
All blood samples were stored at −80°C. All biochemistry was performed in the same UK laboratories. Plasma glucose was measured by standard automated laboratory methods. Insulin was measured by an immunoenzymometric assay (Insulin EASIA; Biosource-Invitrogen, Merelbeke, Belgium). C-peptide was measured by a immunochemiluminometric assay (Siemens Medical Solutions Diagnostics, Newbury, UK). GIP and GLP-1 concentrations were measured after extraction from plasma with 70% ethanol. GIP was measured using RIA with C-terminally-directed antiserum R65 [13], which cross-reacts fully with human GIP and the primary metabolite GIP3–42, but not GIP 8000. Human GIP and 125I-labelled human GIP (70 MBq/nmol) were used for standards and tracer. Plasma GLP-1 was measured against standards of synthetic GLP-1 7–36 amide using antiserum code no. 89390 [14] (specific for the amidated C-terminus of GLP-1 and therefore not reactive with GLP-1-containing peptides from the pancreas). The results of the assay accurately reflect the rate of secretion of GLP-1 because the assay measures the sum of intact GLP-1 and the primary metabolite, GLP-1 9–36 amide, into which GLP-1 is rapidly converted [15]. For both incretin hormone assays sensitivity was <1 pmol/l, intra-assay CV <6% at 20 pmol/l, and recovery of standard added to plasma before extraction about 100% when corrected for losses inherent in the plasma extraction procedure.
DNA from all study participants was sequenced for GCK mutations. PCR products were purified and sequenced using BigDye Terminator chemistry (Applied Biosystems, Warrington, UK). Reactions were analysed on an ABI Prism 3100 DNA Sequencer (Applied Biosystems) and sequences compared using Mutation Surveyor v2.51 software (SoftGenetics, State College, PA, USA).
Data analysis and mathematical modelling
Incretin hormone secretion at five time-points during the OGTT, modelled components of beta cell function and any decline in beta cell function with age were each compared between heterozygous GCK mutation carriers and controls.
Empirical indices of beta cell function included calculation of the homeostasis model assessment of beta cell function index (HOMA(B)), from fasting insulin and glucose values through the computer model v2.2 [16, 17]. Other empirical indices of beta cell function were derived from OGTT values (glucose in mmol/l and insulin in pmol/l): insulinogenic index (IGI), calculated as (30 min insulin−basal insulin)/(30 min glucose−basal glucose), and the early insulin response (EIR) calculated using the formula (30 min insulin−basal insulin)/(30 min glucose) [18].
Mathematical modelling was used to obtain more sophisticated variables describing the beta cell insulin secretory process. The model [19, 20] describes the relationship between insulin secretion (pmol min−1 m−2) and glucose concentration coupled with a model of C-peptide kinetics. Importantly it provides three main indices of beta cell function: beta cell sensitivity to glucose (glucose sensitivity), beta cell sensitivity to the rate of change of glucose (rate sensitivity) and a potentiation factor (expressing relative potentiation of insulin secretion from the beginning to the end of the OGTT). Potentiation is related to factors such as prolonged exposure to hyperglycaemia, non-glucose substrates, incretin hormones and neurotransmitters. The model also computed basal insulin secretion rate (BSR) and the total insulin secretion (TIS) during the OGTT. The model variables were estimated from the glucose and C-peptide concentrations by regularised least-squares and implemented using Matlab (The MathWorks, Natick, MA, USA) [21].
We also examined a number of surrogate measures of insulin sensitivity using fasting measures or formulas incorporating fasting and OGTT data (the homeostasis model assessment of insulin sensitivity [HOMA(S)], the quantitative insulin sensitivity check index [QUICKI] and the index of composite whole-body insulin sensitivity [ISIcomp]).
Statistical analysis
Baseline means from participants with GCK mutations were compared with those without mutations using Student’s t test. Distribution of sex amongst the two groups was compared using χ2 tests. Mean hormone concentrations during five time-points of the OGTT were compared using repeated-measures analysis using SPSS v11.5 and are shown with 95% CIs. All other means were compared by ANCOVA using age and BMI as covariates and are presented with SEs. Data showing a skewed distribution were log transformed (log10). All tests were two-tailed and the significance level was 0.05.
Results
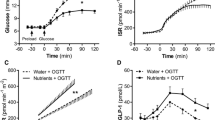
Of the 77 participants studied, 49 (64%) carried the GCK mutation. The baseline characteristics (Table 1) showed no significant difference other than glucose values, which were greater in GCK mutation carriers than non-carriers (Fig. 1a).

OGTT results showing glucose (a), GLP-1 (b), GIP (c) and insulin (d) concentrations with time in GCK mutation carriers (dashed line) and non-mutation carrying controls (solid line); p < 0.001, p = 0.32, p = 0.70, p = 0.14, respectively. Vertical bars are SEs
Mean GLP-1 (Fig. 1b) and GIP (Fig. 1c) concentrations during the 75 g OGTT were not different between GCK mutation carriers and non-carriers (p = 0.70 and p = 0.32, respectively). Peak GIP or GLP-1 (at 30 min following 75 g oral glucose) did not alter with age, sex, BMI or peak circulating plasma glucose.
Insulin (Fig. 1d) and C-peptide concentrations were similar between carriers and non-carriers (p = 0.14 and p = 0.20, respectively), but the modelled glucose/ISR dose–response curve was right-shifted (Fig. 2). A reduction in beta cell function in carriers vs non-carriers was found using all the empirical measures except IGI (Table 1). Of the modelled beta cell function variables, glucose sensitivity and potentiation factor were reduced in GCK mutation carriers, while BSR, TIS and rate sensitivity were unaltered (Table 1).

Relationship between plasma glucose concentrations and ISRs determined by modelling from the OGTT in participants with GCK mutations (dashed line) and non-mutation carrying controls (solid line). Vertical bars are SEs
The glucose sensitivity was not significantly correlated with age in either group (carriers: r 2 = 0.05, p = 0.13; controls r 2 = 0.03, p = 0.36). There were no significant differences detected in any of the measures of insulin sensitivity.
Discussion
Our results show clear effects of a heterozygous mutation in GCK on the beta cell in altering insulin secretion, but not in the gut in altering incretin hormone secretion. There was no difference in incretin hormone secretion among GCK mutation carriers and controls following identical gut exposure to a 75 g oral glucose load. All participants will have had a similar concentration of glucose in their gut lumen even though their plasma glucose levels differed according to GCK mutation status. Since gut glucose sensing is thought to be luminal and not systemic [22], this suggests that glucokinase is not acting as the main glucose sensor in the enteroendocrine cells that secrete GIP and GLP-1. This is an important result, as glucokinase has been shown to be the sensor of circulating glucose not only in the pancreatic beta cell [23], but also for the synthesis of glycogen in the liver [24] and in glucagon responses to hypoglycaemia [25].
Electrophysiological studies using immortalised L cell lines show these cells are electrically excitable and depolarise in response to a range of nutrients including glucose [8]. ATP generated from glucose metabolism in the enteroendocrine cells closes KATP channels and leads to membrane depolarisation and opening of the voltage-gated calcium channels, in a similar process to that in pancreatic beta cells and is necessary for stimulation of GLP-1 release. In addition, SGLT action (whereby luminal glucose is transported into the gut cell coupled to sodium entry, causing depolarisation from sodium influx at higher sugar concentrations) has also been demonstrated to precede GLP-1 secretion in GLUTag cells [2].
A recently proposed mechanism of incretin secretion is by sensing gut glucose through the taste cell pathways as used by the tongue [3]. The sweet taste receptor T1R2+T1R3 is expressed on the luminal membrane of gut enteroendocrine cells along with α-gustducin and other components known to function in taste transduction. Rodent and human enteroendocrine cell lines expressing these taste signalling elements have shown that enhanced GLP-1 and GIP secretion occurs with non-metabolisable artificial sweetener, and incretin hormone secretion is blocked by specific inhibitors of the sweet taste receptor or small interfering RNA for α-gustducin [26, 27]. Isolated small bowel and intestinal villi from α-gustducin-null mice showed markedly defective GLP-1 secretion in response to glucose [27].
The mathematical model we used to assess beta cell function based on five-sample OGTT data (with insulin and C-peptide measurements) enabled us to discern the specific glucose-sensing abnormality in GCK mutation carriers, while all the empirical indices, except IGI, suggested a non-specific reduction in beta cell function. Since IGI reflects the increment in insulin relative to the increment in glucose, this was similar in both GCK mutation carriers and control participants, reflecting a purely glucose-sensing abnormality in the former. The rightward shift in the glucose/ISR dose–response curve from simple five-point OGTT-derived data is very similar to that obtained previously with much more complex and prolonged protocols involving graded i.v. glucose infusion in GCK mutation participants [12].
OGTT modelling suggested a reduction in potentiation factor in this group. This is in agreement with previous studies [28] that have shown that hyperglycaemic states are accompanied by a decrease in the potentiation factor, which is progressively reduced as glucose tolerance worsens. The mechanisms underlying this phenomenon are unknown; however, studies on the incretin effects in glucose intolerance and type 2 diabetes suggest that this defect is intrinsic to the beta cell rather than dependent on the secretion of incretin hormones [29]. Indeed, besides incretin hormone-mediated potentiation in insulin release, there is also glucose potentiation and neural modulation.
The glucose toxicity theory proposes that continual exposure to modest increases in blood glucose over a long period of time could have adverse effects on beta cell glucose sensitivity and hence function [30–32]. We found no difference in the deterioration in glucose sensitivity with age among GCK mutation carriers compared with controls. This is in keeping with the stable glycaemia seen on prolonged follow up of patients with heterozygous mutations [33] and the modest decline with age seen in cross-sectional studies of patients with GCK mutations [34]. This suggests that continuous exposure to the level of mild hyperglycaemia experienced by GCK mutation carriers has no significant progressive glucose toxic effect on the beta cell.
We acknowledge limitations of our study. First, we measured incretin hormones at five time-points only, so subtle differences in early incretin hormone release may not have been detected by our protocol. However, the trajectory of incretin hormone release as measured by more frequent sampling is captured reasonably well by the five time-points we chose to study. Second, we used only one 75 g oral dose of glucose. This was not a saturating dose of oral glucose in triggering maximal incretin hormone release [35], hence we cannot exclude an impact at higher levels of glucose. However, in other situations where glucokinase is the glucose sensor it operates at all concentrations of glucose and not just at maximal stimuli. Third, our study of 77 participants had 80% power to detect a mean difference of 0.14 in log GLP-1 and 0.13 in log GIP secretion, so we cannot exclude differences smaller than this.
In conclusion, there was no reduction in incretin hormone secretion in GCK mutation carriers; however, they had reduced beta cell glucose sensing and potentiation of insulin secretion as modelled using data from a five-sample OGTT. This supports the belief that although glucokinase is the key pancreatic beta cell glucose sensor, it is not the main gut glucose sensor for the incretin hormones GIP and GLP-1.
Abbreviations
- BSR:
-
basal insulin secretion rate
- EIR:
-
early insulin response
- GIP:
-
glucose-dependent insulinotrophic peptide
- GLP-1:
-
glucagon-like peptide 1
- HOMA(B):
-
homeostasis model assessment of beta cell function
- HOMA(S):
-
homeostasis model assessment of insulin sensitivity
- IGI:
-
insulinogenic index
- ISIcomp:
-
index of composite whole-body insulin sensitivity
- ISR:
-
insulin secretion rate
- QUICKI:
-
quantitative insulin sensitivity check index
- SGLT:
-
sodium–glucose cotransporter
- TIS:
-
total insulin secretion
References
Jetton TL, Liang Y, Pettepher CC et al (1994) Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J Biol Chem 269:3641–3654
Gribble FM, Williams L, Simpson AK, Reimann F (2003) A novel glucose-sensing mechanism contributing to glucagon-like peptide-1 secretion from the GLUTag cell line. Diabetes 52:1147–1154
Sternini C, Anselmi L, Rozengurt E (2008) Enteroendocrine cells: a site of ‘taste’ in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes 15:73–78
Theodorakis MJ, Carlson O, Michopoulos S et al (2006) Human duodenal enteroendocrine cells: source of both incretin peptides, GLP-1 and GIP. Am J Physiol Endocrinol Metab 290:E550–E559
Cheung AT, Dayanandan B, Lewis JT et al (2000) Glucose-dependent insulin release from genetically engineered K cells. Science 290:1959–1962
Moates JM, Nanda S, Cissell MA, Tsai MJ, Stein R (2003) BETA2 activates transcription from the upstream glucokinase gene promoter in islet beta-cells and gut endocrine cells. Diabetes 52:403–408
Han J, Lee HH, Kwon H, Shin S, Yoon JW, Jun HS (2007) Engineered enteroendocrine cells secrete insulin in response to glucose and reverse hyperglycemia in diabetic mice. Mol Ther 15:1195–1202
Reimann F, Gribble FM (2002) Glucose-sensing in glucagon-like peptide-1-secreting cells. Diabetes 51:2757–2763
Simpson AK, Ward PS, Wong KY et al (2007) Cyclic AMP triggers glucagon-like peptide-1 secretion from the GLUTag enteroendocrine cell line. Diabetologia 50:2181–2189
Liu M, Seino S, Kirchgessner AL (1999) Identification and characterization of glucoresponsive neurons in the enteric nervous system. J Neurosci 19:10305–10317
Schuit FC, Huypens P, Heimberg H, Pipeleers DG (2001) Glucose sensing in pancreatic beta-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes 50:1–11
Byrne MM, Sturis J, Clement K et al (1994) Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 93:1120–1130
Krarup T, Madsbad S, Moody AJ et al (1983) Diminished immunoreactive gastric inhibitory polypeptide response to a meal in newly diagnosed type I (insulin-dependent) diabetics. J Clin Endocrinol Metab 56:1306–1312
Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ (1994) Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 43:535–539
Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ (1996) Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. Am J Physiol 271:E458–E464
Levy JC, Matthews DR, Hermans MP (1998) Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21:2191–2192
Matthews D, Hosker J, Rudenski A, Naylor B, Treacher D, Turner R (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Wareham N, Phillips D, Byrne C, Hales C (1995) The 30 minute insulin incremental response in an oral glucose tolerance test as a measure of insulin secretion (Letter). Diabet Med 12:931
Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E (2002) Meal and oral glucose tests for the assessment of β-cell function: modeling analysis in normal subjects. Am J Physiol Endocrinol Metab 283:E1159–E1166
Mari A, Tura A, Gastaldelli A, Ferrannini E (2002) Assessing insulin secretion by modeling in multiple-meal tests: role of potentiation. Diabetes 51(Suppl 1):S221–S226
Tura A, Mari A, Winzer C, Kautzky-Willer A, Pacini G (2006) Impaired beta-cell function in lean normotolerant former gestational diabetic women. Eur J Clin Invest 36:22–28
Muscelli E, Mari A, Natali A et al (2006) Impact of incretin hormones on beta-cell function in subjects with normal or impaired glucose tolerance. Am J Physiol Endocrinol Metab 291:E1144–E1150
Matschinsky F, Liang Y, Kesavan P et al (1993) Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest 92:2092–2098
Velho G, Petersen K, Perseghin G et al (1996) Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY 2) subjects. J Clin Invest 98:1755–1761
Guenat E, Seematter G, Philippe J, Temler E, Jequier E, Tappy L (2000) Counterregulatory responses to hypoglycemia in patients with glucokinase gene mutations. Diabetes Metab 26:377–384
Margolskee RF, Dyer J, Kokrashvili Z et al (2007) T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc Natl Acad Sci USA 104:15075–15080
Jang HJ, Kokrashvili Z, Theodorakis MJ et al (2007) Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci USA 104:15069–15074
Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA (2005) Beta-cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab 90:493–500
Muscelli E, Mari A, Casolaro A et al (2008) Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 57:1340–1348
Leahy JL, Bonner-Weir S, Weir GC (1988) Minimal chronic hyperglycemia is a critical determinant of impaired insulin secretion after an incomplete pancreatectomy. J Clin Invest 81:1407–1414
Robertson RP, Olson LK, Zhang HJ (1994) Differentiating glucose toxicity from glucose desensitization: a new message from the insulin gene. Diabetes 43:1085–1089
Rossetti L, Giaccari A, DeFronzo RA (1990) Glucose toxicity. Diabetes Care 13:610–630
Martin D, Bellanne-Chantelot C, Deschamps I, Froguel P, Robert JJ, Velho G (2008) Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care 31:1321–1323
Stride A, Vaxillaire M, Tuomi T et al (2002) The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 45:427–435
Nauck MA, Homberger E, Siegel EG et al (1986) Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 63:492–498
Acknowledgements
This work was funded by Diabetes UK (BDA:RD03/0002684), University Hospital Birmingham Charities and the Danish Medical Research Council.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Murphy, R., Tura, A., Clark, P.M. et al. Glucokinase, the pancreatic glucose sensor, is not the gut glucose sensor. Diabetologia 52, 154–159 (2009). https://doi.org/10.1007/s00125-008-1183-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1183-9