
Abstract
Aims/hypothesis
Heterozygous activating mutations in the pancreatic ATP-sensitive K+ channel cause permanent neonatal diabetes mellitus (PNDM). This results from a decrease in the ability of ATP to close the channel, which thereby suppresses insulin secretion. PNDM mutations that cause a severe reduction in ATP inhibition may produce additional symptoms such as developmental delay and epilepsy. We identified a heterozygous mutation (L164P) in the pore-forming (Kir6.2) subunit of the channel in three unrelated patients and examined its functional effects.
Methods
The patients (currently aged 2, 8 and 20 years) developed diabetes shortly after birth. The two younger patients attempted transfer to sulfonylurea therapy but were unsuccessful (up to 1.1 mg kg−1 day−1). They remain insulin dependent. None of the patients displayed neurological symptoms. Functional properties of wild-type and mutant channels were examined by electrophysiology in Xenopus oocytes.
Results
Heterozygous (het) and homozygous L164P KATP channels showed a marked reduction in channel inhibition by ATP. Consistent with its predicted location within the pore, L164P enhanced the channel open state, which explains the reduction in ATP sensitivity. HetL164P currents exhibited greatly increased whole-cell currents that were unaffected by sulfonylureas. This explains the inability of sulfonylureas to ameliorate the diabetes of affected patients.
Conclusions/interpretation
Our results provide the first demonstration that mutations such as L164P, which produce a severe reduction in ATP sensitivity, do not inevitably cause developmental delay or neurological problems. However, the neonatal diabetes of these patients is unresponsive to sulfonylurea therapy. Functional analysis of PNDM mutations can predict the sulfonylurea response.
Similar content being viewed by others


Introduction
ATP-sensitive potassium (KATP) channels link cellular metabolism to membrane electrical activity by regulating K+ fluxes across the plasma membrane [1]. They are found in multiple tissues but are of particular importance in regulating insulin secretion from pancreatic beta cells [1, 2]. At substimulatory glucose concentrations, KATP channels are open so that the membrane potential is hyperpolarised and Ca2+ influx and insulin secretion are prevented [3]. Glucose metabolism enhances ATP production, resulting in closure of KATP channels, stimulation of electrical activity, opening of voltage-gated Ca2+ channels and exocytosis of insulin granules.
KATP channels are hetero-octamers of Kir6.x and sulfonylurea receptor (SUR) subunits [4–6]. Four inwardly rectifying subunits (Kir6.2 in pancreatic beta cells) form the pore of the channel and four auxiliary SUR subunits (SUR1 in pancreatic beta cells) associate with the tetrameric pore and regulate its gating [7–9]. Binding and/or hydrolysis of Mg-nucleotides by the intracellular nucleotide-binding domains (NBDs) of SUR produces channel opening [10–13]. It is believed that reciprocal changes in the intracellular concentrations of ATP and MgADP are involved in the metabolic regulation of KATP channels.
Over the last 4 years, many different missense mutations in the genes encoding Kir6.2 (KCNJ11) and SUR1 (ABCC8) have been shown to cause permanent neonatal diabetes mellitus (PNDM). This is a rare disorder characterised by high blood glucose levels that manifests within the first 6 months of life. A subgroup of mutations were associated with a more severe clinical profile characterised by Delayed development of motor, intellectual and social skills, muscle weakness, Epilepsy, facial dysmorphism and Neonatal Diabetes (DEND syndrome) [14–16]. Mutations in Kir6.2 have also been found to cause a remitting relapsing form of neonatal diabetes that resembles transient neonatal diabetes mellitus [17, 18].
All Kir6.2 mutations analysed to date were heterozygous and most were de novo mutations [14, 19–21]. In most cases, the diabetes they caused could be successfully treated with sulfonylureas [14, 20, 22], which directly close KATP channels by binding to the SUR1 subunit of the channel [23]. All PNDM mutations result in a reduced KATP channel sensitivity to inhibition by MgATP in vitro [15, 24]. This is expected to cause an increased KATP current amplitude and reduced insulin secretion. Studies to date suggest that the severity of the clinical phenotype reflects the extent of the reduction of the channel ATP sensitivity. Thus, mutations that produce a small increase in KATP current in the presence of physiological concentrations of MgATP (1–5 mmol/l) lead to PNDM, whereas mutations that cause a larger increase in KATP current give rise to DEND syndrome [15].
In this paper, we identify a KCNJ11 mutation (L164P) that causes neonatal diabetes without obvious neurological complications. In functional studies, we show that the L164P mutation produces a large increase in the resting whole-cell current and a marked reduction in KATP channel sensitivity to inhibition by ATP. These effects are a secondary consequence of an increase in the channel open probability (P o) produced by the mutation. Surprisingly, other mutations that produce a similar increase in P o cause DEND syndrome. The L164P mutant channel was also far less blocked by the sulfonylurea tolbutamide, which explains why the patients were unable to transfer to glibenclamide therapy.
Methods
Participants
Informed consent was obtained from all individuals investigated (or from their parents if they were children).
Molecular genetic analysis
Genomic DNA was extracted from peripheral lymphocytes using standard procedures. The KCNJ11 gene was amplified and sequenced as described [25]. Other family members were also tested for the novel mutation. Family relationships were confirmed using a combination of six microsatellites on chromosome 11: D11S902, D11S419, D11S1397, D11S1901, D11S921 and D11S1888.
Oocyte preparation
Female Xenopus laevis were anaesthetised with ethyl 3-aminobenzoate methanesulfonate salt (MS222; 2 g/l added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Subsequently, animals were operated on for a second time, but under terminal anaesthesia. Immature stage V–VI oocytes were incubated for 60 min with 1 mg/ml collagenase (Type V; Sigma, Poole, UK) and manually defolliculated. All procedures were carried out in accordance with UK Home Office Legislations and the University of Oxford ethical guidelines. Oocytes were coinjected with ∼0.8 ng wild-type or mutant Kir6.2 mRNA and ∼4 ng mRNA encoding SUR. The final injection volume was 50 nl per oocyte. Isolated oocytes were maintained in Barth’s solution and studied 1–4 days after injection.
Electrophysiology
Wild-type or mutant Kir6.2 (GenBank D50581) were coexpressed with SUR1 in Xenopus oocytes (GenBank L40624) as described [26]. Because all the patients were heterozygous for the L164P mutation, their pancreatic beta cells will contain a mixture of wild-type and mutant Kir6.2 subunits. To simulate this heterozygosity, we coinjected Xenopus oocytes with a 1:1 mixture of mutant and wild-type Kir6.2 together with SUR1 mRNA. This is expected to give rise to a mixed population of channels composed of homomeric wild-type channels, homomeric mutant channels and heteromeric channels containing between one and three mutant subunits [21]. We refer to this global channel population as heterozygous channels.
Whole-cell currents were recorded using a two-electrode voltage clamp in response to voltage steps of ±20 mV from a holding potential of −10 mV, filtered at 1 kHz and digitised at 4 kHz. Oocytes were perfused with a solution containing (in mmol/l): 90 KCl, 1 MgCl2, 1.8 CaCl2 and 5 HEPES (pH 7.4 with KOH). Metabolic inhibition was produced by 3 mmol/l sodium azide.
Macroscopic currents were recorded from giant inside-out patches using an EPC10 amplifier (List Medical Electronics, Darmstadt, Germany) controlled with Pulse v8.74 software (Heka Electronik, Lambrecht, Germany). Macroscopic currents were elicited by 3 s voltage ramps from −110 to +100 mV (holding potential 0 mV), or recorded at a constant potential of −60 mV. They were filtered at 0.5 kHz and digitised at 1 kHz. The pipette solution contained (mmol/l): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 HEPES (pH 7.4 with KOH). The internal (bath) solution contained (mmol/l): 107 KCl, 1 K2SO4, 2 MgCl2, 10 EGTA, 10 HEPES (pH 7.2 with KOH) and Mg-nucleotides as indicated. Experiments were conducted at 20–22°C. Solutions were changed using a local perfusion system consisting of tubes of 200 μm diameter into which the tip of the patch pipette was inserted.
Nucleotide concentration–inhibition curves were fit with the Hill equation:
where G is the K ATP conductance in the presence of ATP, G c is the K ATP conductance in the absence of the nucleotide, [ATP] is the ATP concentration, IC50 is the nucleotide concentration at which inhibition is half maximal and h is the slope factor (Hill coefficient).
Single-channel currents were measured at −60 mV, filtered at 5 kHz and digitised at 20 kHz. Unitary amplitude and P o were measured from the Gaussian fit to all-points amplitude histograms of tracts of current of 30–90 s duration.
Data were analysed with in-house routines developed in the IgorPro platform (Wavematrics, Portland, OR, USA). Data are given as means ± SEM in the text and in the figures. Statistical significance was evaluated using a two-tailed Student t test and p < 0.05 taken to indicate a significant difference.
Results
Patient characteristics and genetics
Three unrelated probands with permanent neonatal diabetes were shown to be heterozygous for the KCNJ11 gene mutation L164P (c.491T>C, p.Leu164Pro). All were female and had unaffected parents of different ethnic origin. Two patients have been reported previously [27, 28]. Mutation testing and microsatellite analysis of DNA from both parents and the child for two of the cases confirmed that the mutation had arisen de novo (the parents of the third case were not available for testing). None of the patients had any neurological complications or obvious developmental delay.
The first patient is currently 8 years old. She was born in Singapore from Sri Lankan parents, at 38 weeks of gestation with a birthweight of 2.6 kg. She developed diabetes at 30 weeks of age. When she came to Australia at the age of 6.5 years, her HbA1c level was elevated (7.4%) and her diabetes required insulin treatment (0.45 U kg−1 day−1). Glibenclamide treatment (1.0 mg kg−1 day−1) was trialled for 2 months (at 7 years of age) but did not produce a decrease in her insulin requirement. She continues to require insulin (up to 0.7 U kg−1 day−1) with an HbA1c ranging from 7.2 to 8.2%. She has normal developmental milestones, her neurological examination is normal and she has no evidence of diabetic complications.
The second patient, born in Australia of Afghan origin, is currently 2.7 years old. She was born at term following an uneventful pregnancy, with a birthweight of 2.7 kg. She presented at 8 weeks in diabetic ketoacidosis and was subsequently treated (at 2.5 months for 4 weeks, then at 5 months for 3 months) with glibenclamide (up to 1.1 mg kg−1 day−1, regimens of two times per day or three times per day), but had no reduction in insulin requirement (0.4 U kg−1 day−1) [28]. Her mean HbA1c since diagnosis has been 7.6% (range 7.2–8.7%). Assessment at 5 months by a neurologist was entirely normal, with a normal EEG. Subsequently, she has had normal developmental milestones and her neurological examinations have been normal at all follow up visits every 3 months. Diabetic complications have not been assessed due to the young age of the patient.
The third patient is from Slovakia [27]. She was born at term with a birthweight of 2.6 kg, and was diagnosed with diabetes at 5 weeks of age. She is currently 20 years old and is treated with insulin (1.24 U kg−1 day−1). Glibenclamide transfer was not attempted because the patient also suffers from hepatitis C. She has poor glycaemic control, with an HbA1c level of 15.2%, and various diabetic complications including retinopathy and nephropathy. Psychomotoric development in childhood was normal. A neurological examination suggested the presence of diabetic motoric polyneuropathy in the lower extremities but there were no further neurological findings. Although her IQ was not tested, she successfully completed a specialised business school training suggesting normal mental development.
Functional analysis: effects on whole-cell KATP currents
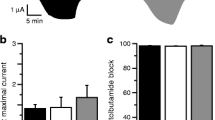
We examined the functional effects of the L164P mutation by heterologous expression in Xenopus oocytes. When wild-type Kir6.2/SUR1 channels are expressed in oocytes they are normally closed because they are inhibited by resting intracellular ATP concentrations ([ATP]i). They can be opened, however, by azide (3 mmol/l; Fig. 1), a metabolic inhibitor that lowers [ATP]i [29]. Heterozygous L164P (hetL164P) currents were about 18-fold larger at rest than wild-type currents, but increased twofold on metabolic inhibition indicating that the channel is only partially closed at resting ATP levels (Fig. 1). In contrast, homomeric L164P (homL164P) channels displayed a much greater resting current and were little affected by metabolic inhibition (Fig. 1).

Whole-cell KATP current. Mean steady state whole-cell currents evoked by voltage steps from −10 to −30 mV before (control, white bars) and after application of 3 mmol/l azide (grey bars) and in the presence of 3 mmol/l azide plus 0.5 mmol/l tolbutamide (black bars). The number of oocytes was five to seven in each case. G, KATP conductance; G c, KATP conductance expressed relative to the conductance in the absence of the nucleotide. WT, wild-type; hetL164P and homL164P channels as indicated
The sulfonylurea tolbutamide (0.5 mmol/l) blocked whole-cell KATP currents by 98% but had no effect on homL164P currents (4% block). HetL164P channels were blocked by only 54% (Fig. 1).
Functional analysis: effects on KATP channel ATP sensitivity
The increase in resting whole-cell KATP currents suggests that the L164P mutation may reduce the channel ATP sensitivity, as found for other PNDM mutations [21]. We first measured the ATP sensitivity of wild-type and mutant KATP currents in the absence of Mg2+, to isolate the effects of ATP on Kir6.2 (in the absence of Mg2+, ATP does not interact with SUR1 [11]). Both homL164P and hetL164P channels had severely impaired ATP sensitivity. HomL164P channels were not blocked at all by ATP, even at concentrations as high as 10 mmol/l. The concentration–inhibition curve for hetL164P showed a striking shift to higher ATP concentrations, with an IC50 of about 100 μmol/l, and a marked pedestal of unblocked current at very high ATP concentrations. The data were best fitted by assuming that in the heterozygous state about 20% of channels are never closed by ATP (Fig. 2a,b and Table 1).

ATP-inhibition of L164P channels is less that that of wild-type channels. a, c Currents recorded in inside-out patches excised from Xenopus oocytes expressing hetKir6.2-L164P/SUR1 (hetL164P) or homKir6.2-L164P/SUR1 (homL164P) channels, as indicated, in response to 3 s voltage ramps from −110 to +100 mV. ATP (10 mmol/l) was applied as indicated by the horizontal bars in the absence (a) or presence (c) of 2 mmol/l Mg2+. b, d, mean relationship between [ATP] and K ATP conductance (G), expressed relative to the conductance in the absence of the nucleotide (G c), for wild-type (white circles, n = 9), hetL164P (white/black circles, n = 6) or homL164P (black circles, n = 4) channels. Experiments were carried out in the absence (b) or presence (d) of 2 mmol/l Mg2+. The continuous lines through the black circles were drawn by eye. The smooth curves are the best fit to the Hill equation with IC50 of 11 μmol/l (wild-type) and 100 μmol/l (hetL164P) (b, 0 mmol/l Mg2+) or IC50 of 16 μmol/l (wild-type) and 122 μmol/l (hetL164P) (d, 2 mmol/l Mg2+)
Molecular mechanism of the reduced ATP sensitivity
Mutations that reduce the ATP sensitivity of the KATP channel can act in several ways. They may prevent ATP binding directly. They may impair the mechanism by which nucleotide binding is coupled to channel gating. They may also stabilise the intrinsic open state of the channel (i.e. that in the absence of ATP), which shifts the gating equilibrium in the presence of ATP towards channel opening and thus indirectly reduces the channel ATP sensitivity (e.g. [15, 30, 31]).
In a structural model of Kir6.2 [32], L164P lies within the channel pore, at a considerable distance from the ATP-binding site (Fig. 3). It is not predicted to interact directly with ATP. We therefore examined whether the L164P mutation alters intrinsic gating. Experiments were carried out in the absence of ATP, where intrinsic gating can be assessed. The L164P mutation had no effect on single-channel current amplitude (Table 2). However, the intrinsic P o was markedly increased, being 0.86 (n = 6) for homL164P compared with 0.4 (n = 4) for wild-type channels (Fig. 4 and Table 2). This suggests that, at least in part, the L164P mutation alters channel ATP sensitivity indirectly, via an increase in P o.

Homology model of Kir6.2 [32]. For clarity, only two subunits are shown. ATP (yellow) is shown in its binding site. Residue L164 is shown in red

The L164P mutation enhances single-channel activity. Representative single KATP channel currents recorded at −60 mV in inside-out patches from oocytes expressing wild-type or homL164P channels, as indicated. Currents were recorded in the absence of Mg2+ and nucleotides
Effects on KATP channel ATP sensitivity in the presence of Mg2+
Previous studies have shown that Kir6.2 mutations associated with neonatal diabetes may not only decrease the sensitivity of Kir6.2 to ATP, but can also enhance channel activation by Mg-nucleotides [24]. We therefore explored the effect of the L164P mutation on the ATP sensitivity in the presence of 2 mmol/l Mg2+.
Surprisingly, there was no difference in the effect of ATP in the presence and absence of Mg2+ on homL164P channels (Fig. 2c,d). This contrasts with what has been observed for mutations in the ATP-binding site that completely abolished ATP inhibition in the absence of Mg2+, such as R50P and G334D: channels containing these mutations were activated by MgATP [33, 34]. The IC50 for ATP inhibition of hetL164P channels was also not significantly affected by Mg2+, although the pedestal of unblocked current at high ATP concentrations was increased by about 50% (Table 1). At 3 mmol/l MgATP, a concentration within the physiological range, the unblocked current was 34%, substantially greater than that found for wild-type channels (<1%; Table 1).
Functional analysis: effects on MgADP sensitivity
The lack of MgATP activation of hetL164P channels could be due to reduced functional coupling between SUR1 and Kir6.2-L164P, or to reduced MgATP binding/hydrolysis at the NBDs of SUR1. To explore the former possibility, we measured the ability of MgADP to activate hetL164P channels preblocked by 100 μmol/l MgATP. It was necessary to preblock the channels as the mutant channel has an open probability close to maximal and further activation is therefore not possible. Figure 5 shows that 30 μmol/l MgADP activates wild-type currents by 3.7 ± 1.8 fold (n = 3) and that 100 μmol/l MgADP did not produce any further increase (3.7 ± 1.2-fold, n = 3). In contrast, hetL164P channels were activated less: 1.4 ± 0.1 fold (n = 3) by 30 μmol/l MgADP and 1.5 ± 0.1 fold (n = 4) by 100 μmol/l MgADP.

Sensitivity to MgADP of wild-type and hetL164P channels. a Representative currents (I) recorded at −60 mV from inside-out excised membrane patches from Xenopus oocytes expressing wild-type or hetL164P channels, as indicated. Patches were exposed to 100 μmol/l ADP in the continuous presence of 100 μmol/l ATP: 2 mmol/l Mg2+ was present throughout. b Mean current in the presence of 100 μmol/l MgATP or 30 or 100 μmol/l MgADP plus 100 μmol/l MgATP, normalised to the current in the absence of nucleotides for wild-type (grey bars) and hetL164P (black bars) channels. Bars indicate means ± SEM. The number of patches was three to four in each case
Despite the fact that 100 μmol/l MgATP blocked mutant channels less, the lower extent of activation of hetL164P channels by 30 μmol/l MgADP is not due to the fact that channel activity is already maximal, as the amplitude is still significantly less than that in control solution (Fig. 5).
Discussion
We describe the clinical and functional effects of a KCNJ11 mutation, L164P, associated with permanent neonatal diabetes. This mutation causes a marked reduction in KATP channel inhibition by ATP primarily by stabilising the open state of the channel. This leads to an increase in the whole-cell K ATP current, and in beta cells is expected to result in a reduction in insulin secretion. Surprisingly, unlike other mutations that reduced the channel ATP sensitivity by a similar amount, no motor or mental developmental delay was associated with the L164P mutation.
Structural considerations and molecular basis for reduced ATP sensitivity
In a homology model of Kir6.2 [32], L164 lies partway along the permeation pathway, 35 Å away from the ATP-binding site (Fig. 3). It is therefore unlikely that it acts by reducing ATP binding directly. The side-chains of L164 point into the pore, forming a hydrophobic girdle that is narrow enough to prevent the passage of water and hydrated K+ ions [35], which suggests that L164 may form a hydrophobic gate within the pore. Functional studies support this idea. Following mutation of L164 to cysteine, cadmium ions were able to block the KATP channel with high affinity [36, 37], suggesting that the four cysteines (one on each subunit) come together close enough to form a high-affinity binding site for Cd2+. These data indicate that the side-chain of L164 must face into the pore, and that the pore is very narrow at this position. Substitution of a proline for L164 is expected to produce a kink in the α-helix [38] and disrupt the hydrophobic gate. It is therefore not surprising that the L164P mutation caused a dramatic effect on the P o. Mutation of L164 to cysteine, alanine, valine, threonine or glycine also produces a very large increase in P o [30, 36, 37, 39], consistent with the importance of this residue in channel gating.
The fact that the L164P mutation enhanced P o can explain, in part, the reduced ATP sensitivity of the channel. It is also possible that the mutation may have effects additional to stabilisation of the channel open state.
Effect of Mg2+ on Kir6.2-L164P/SUR1 channel ATP sensitivity
In contrast to all PNDM mutations studied to date [24], MgATP was unable to enhance the activity of either homL164P or hetL164P channels. The lack of MgATP activation of homL164P channels may be due to the fact that P o is already very high and thus there is no scope for further activation. However, little MgATP activation was also observed for hetL164P channels. This is in marked contrast to other mutations that caused enhanced P o (I296L, [16], V59G [15]) where MgATP activated both homomeric and heterozygous channels. It is therefore possible that the L164P mutation reduces the efficacy of coupling between SUR1 and Kir6.2, and thereby decreases the ability of MgATP to stimulate channel activity. In support of this idea, MgADP activation of hetL164P channels was also reduced. This may explain the inability of Mg2+ to reduce ATP inhibition of hetL164P channels, since it is well established that MgATP must be hydrolysed to MgADP to stimulate channel activity.
The reason for the lack of Mg-nucleotide activation of hetL164P channels is unclear. Because L164 lies within the pore, far away from the NBDs of SUR1, the effect must be mediated allosterically. It is possible that this is mediated by an interaction between the backbone of L164 itself, or transmembrane domain 2 (within which L164 lies), and the transmembrane domains of SUR1. In the absence of an atomic resolution structure of the KATP channel complex, however, this cannot be definitely determined.
HomL164P channels were not blocked by tolbutamide, as expected because of their high P o [40]. The lower efficacy of tolbutamide on hetL164P channels (about 50% block) may reflect the enhanced P o of channels within the heterozygous population containing mutant subunits. It could also reflect impaired coupling between SUR1 and Kir6.2, such as that found for MgADP.
Clinical implications
Previous studies of KCNJ11 mutations have suggested that there is a good correlation between the percentage of current that remains unblocked in the presence of 3 mmol/l MgATP and the clinical phenotype. Namely, currents that are >30% of maximal are associated with DEND syndrome, and those that lie between 5–10% of maximal with neonatal diabetes alone, compared with a current of <1% for wild-type channels (Fig. 6). The L164P mutation does not conform to this simple relationship between the functional effects of the mutation and the clinical phenotype. The magnitude of the hetL164P current in 3 mmol/l MgATP was 36%, yet none of the patients had extra-pancreatic symptoms.

Mean K ATP current (expressed as a % of maximum) measured in the presence of 3 mmol/l MgATP from inside-out patches expressing heterozygous mutant channels as indicated. The dashed line indicates the maximal current amplitude normally associated with PNDM. iDEND, intermediate DEND syndrome (i.e. neonatal diabetes with developmental delay [21]). Data for wild-type (WT) and Kir6.2-R201H channels are from [14], for Y330C and F333I from [26], for Q52R, V59G and R201C from [15], for V59M from [24], for I296L from [16] and for G334D from [34]
The reason for this anomaly is not clear, particularly as the resting whole-cell current was also very large. Previous studies have also indicated that such large whole-cell currents are invariably associated with more severe clinical symptoms. The lack of responsiveness of the patient to sulfonylureas is consistent with the very large increase in P o and the resting whole-cell current. It is therefore likely that hetL164P channels expressed in Xenopus oocytes are a reasonable model for the pancreatic beta cell KATP channels of the patients. We are therefore forced to conclude that compensatory mechanisms must ameliorate the extra-pancreatic effects of this mutation. The fact that the patients had very different ethnic origins suggests the compensatory mechanism is unlikely to reflect a shared genetic background: however, it does appear to be specific to the L164P mutation, which is unique (to date) in having a marked functional effect in vitro despite causing neonatal diabetes without neurological complications.
Importantly, a maximally effective concentration of tolbutamide only blocked hetL164P channels by 50%. All patients to date whose channels are blocked by <65% have not been able to transfer from insulin treatment to sulfonylurea therapy. This suggested that our patients would be unable to transfer to sulfonylurea therapy, as indeed was found to be the case. This is likely to be due to the fact that L164P destabilises the long closed state of the channel, to which sulfonylureas preferentially bind, and which is rarely entered in channels with enhanced P o. Our results further suggest that not all patients with permanent neonatal diabetes will necessarily respond to sulfonylurea treatment, and indicates that knowledge of the functional effect of the mutation is helpful for predicting the drug response in patients.
Abbreviations
- [ATP]i :
-
intracellular ATP concentration
- DEND:
-
neonatal diabetes with severe neurological complications
- HetL164P:
-
heterozygous L164P
- HomL164P:
-
homomeric L164P
- KATP :
-
ATP-sensitive K+ channel
- NBD:
-
nucleotide-binding domain
- PNDM:
-
permanent neonatal diabetes mellitus
- P o :
-
channel open probability
- SUR:
-
sulfonylurea receptor
References
Seino S, Miki T (2003) Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 81:133–176
Ashcroft FM, Harrison DE, Ashcroft SJ (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312:446–448
Ashcroft F, Rorsman P (2004) Type 2 diabetes mellitus: not quite exciting enough? Hum Mol Genet 13(Spec No. 1):R21–R31
Inagaki N, Gonoi T, Clement JPT et al (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170
Clement JPT, Kunjilwar K, Gonzalez G et al (1997) Association and stoichiometry of KATP channel subunits. Neuron 18:827–838
Shyng S, Nichols CG (1997) Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110:655–664
Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387:179–183
Drain P, Li L, Wang J (1998) KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci U S A 95:13953–13958
Haider S, Tarasov AI, Craig TJ, Sansom MS, Ashcroft FM (2007) Identification of the PIP(2)-binding site on Kir6.2 by molecular modelling and functional analysis. EMBO J 26:3749–3759
Nichols CG, Shyng SL, Nestorowicz A et al (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272:1785–1787
Gribble FM, Tucker SJ, Ashcroft FM (1997) The essential role of the Walker A motifs of SUR1 in KATP channel activation by Mg-ADP and diazoxide. EMBO J 16:1145–1152
Shyng S, Ferrigni T, Nichols CG (1997) Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol 110:643–654
Zingman LV, Alekseev AE, Bienengraeber M et al (2001) Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31:233–245
Gloyn AL, Pearson ER, Antcliff JF et al (2004) Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350:1838–1849
Proks P, Antcliff JF, Lippiat J, Gloyn AL, Hattersley AT, Ashcroft FM (2004) Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci U S A 101:17539–17544
Proks P, Girard C, Haider S et al (2005) A gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep 6:470–475
Flanagan SE, Patch AM, Mackay DJ et al (2007) Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 56:1930–1937
Gloyn AL, Reimann F, Girard C et al (2005) Relapsing diabetes can result from moderately activating mutations in KCNJ11. Hum Mol Genet 14:925–934
Massa O, Iafusco D, D, Amato E et al (2005) KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum Mutat 25:22–27
Sagen JV, Raeder H, Hathout E et al (2004) Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53:2713–2718
Ashcroft FM (2005) ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115:2047–2058
Zung A, Glaser B, Nimri R, Zadik Z (2004) Glibenclamide treatment in permanent neonatal diabetes mellitus due to an activating mutation in Kir6.2. J Clin Endocrinol Metab 89:5504–5507
Gribble FM, Reimann F (2003) Sulphonylurea action revisited: the post-cloning era. Diabetologia 46:875–891
Proks P, Girard C, Ashcroft FM (2005) Functional effects of KCNJ11 mutations causing neonatal diabetes: enhanced activation by MgATP. Hum Mol Genet 14:2717–2726
Flanagan SE, Edghill EL, Gloyn AL, Ellard S, Hattersley AT (2006) Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia 49:1190–1197
Tammaro P, Girard C, Molnes J, Njolstad PR, Ashcroft FM (2005) Kir6.2 mutations causing neonatal diabetes provide new insights into Kir6.2-SUR1 interactions. EMBO J 24:2318–2330
Stanik J, Gasperikova D, Paskova M et al (2007) Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab 92:1276–1282
Pearson ER, Flechtner I, Njolstad PR et al (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355:467–477
Gribble FM, Ashfield R, Ammala C, Ashcroft FM (1997) Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J Physiol 498:87–98
Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG (2000) The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys J 78:2334–2348
Trapp S, Proks P, Tucker SJ, Ashcroft FM (1998) Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J Gen Physiol 112:333–349
Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM (2005) Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 24:229–239
Shimomura K, Girard CA, Proks P et al (2006) Mutations at the same residue (R50) of Kir6.2 (KCNJ11) that cause neonatal diabetes produce different functional effects. Diabetes 55:1705–1712
Masia R, Koster JC, Tumini S et al (2007) An ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetes. Diabetes 56:328–336
Haider S, Antcliff JF, Proks P, Sansom MS, Ashcroft FM (2005) Focus on Kir6.2: a key component of the ATP-sensitive potassium channel. J Mol Cell Cardiol 38:927–936
Loussouarn G, Makhina EN, Rose T, Nichols CG (2000) Structure and dynamics of the pore of inwardly rectifying KATP channels. J Biol Chem 275:1137–1144
Loussouarn G, Phillips LR, Masia R, Rose T, Nichols CG (2001) Flexibility of the Kir6.2 inward rectifier K+ channel pore. Proc Natl Acad Sci U S A 98:4227–4232
Cordes FS, Bright JN, Sansom MS (2002) Proline-induced distortions of transmembrane helices. J Mol Biol 323:951–960
Enkvetchakul D, Loussouarn G, Makhina E, Nichols CG (2001) ATP interaction with the open state of the KATP channel. Biophys J 80:719–728
Reimann F, Tucker SJ, Proks P, Ashcroft FM (1999) Involvement of the n-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol 518:325–336
Acknowledgements
We thank the patients and their referring clinicians. Financial support was provided by the Wellcome Trust (F. M. Ashcroft, A. T. Hattersley), the Royal Society (F. M. Ashcroft), the European Union (Integrated Project EuroDia LSHM-CT-2006–518153 in the Framework Programme 6 [FP6]) of the European-Community (F. M. Ashcroft, A. T. Hattersley), the Sir Graham Wilkins studentship (S. E. Flanagan) and research grants from the Slovak Research and Development Agency (51–014205; I. Klimes) and Slovak Ministry of Health (MZ.2005/15-NEDU-01; I. Klimes). P. Tammaro holds a Junior Research Fellowship at the Wolfson College, B. Zadek holds an OXION scholarship, A. T. Hattersley is a Wellcome Trust Research Leave Fellow and F. M. Ashcroft is a Royal Society Research Professor.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
P. Tammaro and S. E. Flanagan contributed equally to this study.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Tammaro, P., Flanagan, S.E., Zadek, B. et al. A Kir6.2 mutation causing severe functional effects in vitro produces neonatal diabetes without the expected neurological complications. Diabetologia 51, 802–810 (2008). https://doi.org/10.1007/s00125-008-0923-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-0923-1