
Abstract
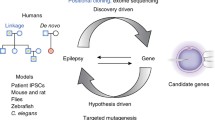
Research in genetics of epilepsy represents an area of great interest both for clinical purposes and for understanding the basic mechanisms of epilepsy. Most mutations in epilepsies without structural brain abnormalities have been identified in ion channel genes, but an increasing number of genes involved in a diversity of functional and developmental processes are being recognized through whole exome or genome sequencing. Targeted molecular diagnosis is now available for different forms of epilepsy. The identification of epileptogenic mutations in patients before epilepsy onset and the possibility of developing therapeutic strategies tested in experimental models may facilitate experimental approaches that prevent epilepsy or decrease its severity. Functional analysis is essential for better understanding pathogenic mechanisms and gene interactions. In vitro experimental systems are either cells that usually do not express the protein of interest or neurons in primary cultures. In vivo/ex vivo systems are organisms or preparations obtained from them (e.g., brain slices), which should better model the complexity of brain circuits and actual pathophysiological conditions. Neurons differentiated from induced pluripotent stem cells generated from the skin fibroblasts of patients have recently allowed the study of mutations in human neurons having the genetic background of a given patient. However, there is remarkable complexity underlying epileptogenesis in the clinical dimension, as reflected by the fact that experimental models have not provided yet results having clinical translation and that, with a few exceptions concerning rare conditions, no new curative treatment has emerged from any genetic finding in epilepsy.



Similar content being viewed by others


References
Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2011;51:676–685.
Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in idiopathic epilepsy. Neurotherapeutics 2007;4:295–304.
Helbig I, Scheffer IE, Mulley JC, Berkovic SF. Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol 2008;7:231–245.
Marini C, Scheffer IE, Crossland KM, et al. Genetic architecture of idiopathic generalized epilepsy: clinical genetic analysis of 55 multiplex families. Epilepsia 2004;45:467–478.
Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins. In: Wolf P (ed.) Epileptic seizures and syndromes. John Libbey, London, 1994, pp. 157–164.
Greenberg DA, Delgado-Escueta AV, Widelitz H, et al. Juvenile myoclonic epilepsy (JME) may be linked to the BF and HLA loci on human chromosome 6. Am J Med Genet 1988;31:185–192.
Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor a4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 1995;11:201–203.
Mantegazza M, Rusconi R, Scalmani P, Avanzini G, Franceschetti S. Epileptogenic ion channel mutations: from bedside to bench and, hopefully, back again. Epilepsy Res 2010;92:1–29.
Hirose S, Scheffer IE, Marini C, et al. SCN1A testing for epilepsy: application in clinical practice. Epilepsia 2013;54:946–952.
Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 2007;16:2892–2899.
Dravet C, Bureau M, Roger J. Severe myoclonic epilepsy in infants. In: Roger J, Dravet C, Bureau M, Dreifuss FE, Wolf P (eds) Epileptic syndromes in infancy, childhood and adolescence. John Libbey Eurotext, London, 1985, pp. 58–104.
Guerrini R, Dravet Ch. Severe epileptic encephatopathies of infancy. In: Engel J and Pedley TA (eds) Epilepsy. Raven Press, New York, 1997, pp. 2285–2302.
Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia 2011;52(Suppl. 2):30–34.
Catarino CB, Liu JY, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain 2011;134:2982–3010.
Suls A, Claeys KG, Goossens D, et al. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum Mutat 2006;27:914–920.
Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–1678.
Patino GA, Claes LR, Lopez-Santiago LF, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 2009;29:10764–10778.
Guerrini R, Oguni H. Borderline Dravet syndrome: a useful diagnostic category? Epilepsia 2011;52(Suppl. 2):10–12.
Fujiwara T, Sugawara T, Mazaki-Miyazaki E, et al. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 2003;126:531–546.
Wallace RH, Scheffer IE, Barnett S, et al. Neuronal sodium channel a1 subunit (SCN1A) mutations in generalised epilepsy with febrile seizures plus. Am J Hum Genet 2001;68:859–865.
Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130:843–852.
Depienne C, Arzimanoglou A, Trouillard O, et al. Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum Mutat 2006;27:389.
Marini C, Mei D, Helen Cross J, Guerrini R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia 2006;47:1737–1740.
Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 2011;53:87–100.
Ogiwara I, Iwasato T, Miyamoto H, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 2007;22:4784–4804.
Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006;9:1142–1149.
Han S, Tai C, Westenbroek RE, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012;489:385–390.
Higurashi N, Uchida T, Lossin C, et al. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol Brain 2013;6:19.
Jiao J, Yang Y, Shi Y, et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet 2013;22:4241–4252.
Liu Y, Lopez-Santiago LF, Yuan Y, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol 2013;74:128–139.
Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120:479–490.
Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na +-channel beta1 subunit gene SCN1B. Nat Genet 1998;19:366–370.
Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24:343–345.
Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–1685.
Wallace RH, Marini C, Petrou S, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 2001;28:49–52.
Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 2001;28:46–48.
Tian M, Mei D, Freri E, et al. Impaired surface alphabetagamma GABA(A) receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol Dis 2013;50:135–141.
Cestele S, Labate A, Rusconi R, et al. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 2013;54:927–935.
Rusconi R, Scalmani P, Cassulini RR, et al. Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J Neurosci 2007;27:11037–11046.
Rusconi R, Combi R, Cestele S, et al. A rescuable folding defective Nav1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum Mutat 2009;30:E747–760.
Thompson CH, Porter JC, Kahlig KM, Daniels MA, George AL, Jr. Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J Biol Chem 2012;287:42001–42008.
Sugiura Y, Ogiwara I, Hoshi A, Yamakawa K, Ugawa Y. Different degrees of loss of function between GEFS+ and SMEI Nav 1.1 missense mutants at the same residue induced by rescuable folding defects. Epilepsia 2012;53:e111–114.
Cestele S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci U S A 2013;110:17546–17551.
Cestele S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M. Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J Neurosci 2008;28:7273–7283.
Tang B, Dutt K, Papale L, et al. A BAC transgenic mouse model reveals neuron subtype-specific effects of a generalized epilepsy with febrile seizures plus (GEFS+) mutation. Neurobiol Dis 2009;35:91–102.
Chiu C, Reid CA, Tan HO, et al. Developmental impact of a familial GABAA receptor epilepsy mutation. Ann Neurol 2008;64:284–293.
Zara F, Specchio N, Striano P, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia 2013;54:425–436.
Plouin P. Benign familial neonatal convulsions. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R (eds) Idiopathic generalized epilepsies; clinical, experimental and genetic aspects. John Libbey, London, 1994, pp. 39–44.
Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res 2007;73:245–249.
Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science 1998;279:403–406.
Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet 1998;18:53–55.
Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 1998;18:25–29.
Heron SE, Cox K, Grinton BE, et al. Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet 2007;44:791–796.
Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 2003;126:2726–2737.
Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 2005;6:850–862.
Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 1998;396:687–690.
Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch 2010;460:277–288.
Weber YG, Lerche H. Genetic mechanisms in idiopathic epilepsies. Dev Med Child Neurol 2008;50:648–654.
Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 2005;8:51–60.
Singh NA, Otto JF, Dahle EJ, et al. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J Physiol 2008;586:3405–3423.
Berkovic SF, Heron SE, Giordano L, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 2004;55:550–557.
Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002;360:851–852.
Scalmani P, Rusconi R, Armatura E, et al. Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 2006;26:10100–10109.
Liao Y, Deprez L, Maljevic S, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010;133:1403–1414.
Vigevano F. Benign familial infantile seizures. Brain Dev 2005;27:172–177.
Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet 1997;61:889–898.
Marini C, Conti V, Mei D, et al. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 2012;79:2109–2114.
Scheffer IE, Grinton BE, Heron SE, et al. PRRT2 phenotypic spectrum includes sporadic and fever-related infantile seizures. Neurology 2012;79:2104–2108.
Guerrini R, Mink JW. Paroxysmal disorders associated with PRRT2 mutations shake up expectations on ion channel genes. Neurology 2012;79:2086–2088.
Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet 2011;43:1252–1255.
Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005;122:957–968.
Guipponi M, Rivier F, Vigevano F, et al. Linkage mapping of benign familial infantile convulsions (BFIC) to chromosome 19q. Hum Mol Genet 1997;6:473-477.
Vanmolkot KR, Kors EE, Hottenga JJ, et al. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol 2003;54:360–366.
Zhou X, Ma A, Liu X, et al. Infantile seizures and other epileptic phenotypes in a Chinese family with a missense mutation of KCNQ2. Eur J Pediatr 2006;165:691–695.
Striano P, Bordo L, Lispi ML, et al. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia 2006;47:218–220.
Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25.
Weckhuysen S, Ivanovic V, Hendrickx R, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 2013;81:1697–1703.
Miceli F, Soldovieri MV, Ambrosino P, et al. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci U S A 2013;110:4386–4391.
Orhan G, Bock M, Schepers D, et al. Dominant-negative Effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol 2013 Dec 7 [Epub ahead of print].
Nakamura K, Kato M, Osaka H, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 2013;81:992–998.
Touma M, Joshi M, Connolly MC, et al. Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings. Epilepsia 2013;54:e81-85.
Lossin C, Shi X, Rogawski MA, Hirose S. Compromised function in the Na(v)1.2 Dravet syndrome mutation R1312T. Neurobiol Dis 2012;47:378–384.
Planells-Cases R, Caprini M, Zhang J, et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J 2000;78:2878–2891.
Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–781.
Marini C, Mei D, Parmeggiani L, et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 2010;75:646–653.
Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–2119.
Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009;5(2):e1000381.
Ohira R, Zhang YH, Guo W, et al. Human ARX gene: genomic characterization and expression. Mol Genet Metab 2002;77:179–188.
Gecz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev 2006;16:308–316.
Guerrini R, Moro F, Kato M, et al. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 2007;69:427–433.
Marsh E, Fulp C, Gomez E, et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 2009;132:1563–1576.
Kato M, Das S, Petras K, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat 2004;23:147–159.
Stromme P, Mangelsdorf ME, Scheffer IE, Gecz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev 2002;24:266–268.
Wallerstein R, Sugalski R, Cohn L, Jawetz R, Friez M. Expansion of the ARX spectrum. Clin Neurol Neurosurg 2008;110:631–634.
Shinozaki Y, Osawa M, Sakuma H, et al. Expansion of the first polyalanine tract of the ARX gene in a boy presenting with generalized dystonia in the absence of infantile spasms. Brain Dev 2009;31:469–472.
Kato M, Saitoh S, Kamei A, et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet 2007;81:361–366.
Mari F, Azimonti S, Bertani I, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet 2005;14:1935–1946.
Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet 2003;72:1401–1411.
Buoni S, Zannolli R, Colamaria V, et al. Myoclonic encephalopathy in the CDKL5 gene mutation. Clin Neurophysiol 2006;117:223–227.
Archer HL, Evans J, Edwards S, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet 2006;43:729–734.
Elia M, Falco M, Ferri R, et al. CDKL5 mutations in boys with severe encephalopathy and early-onset intractable epilepsy. Neurology 2008;71:997–999.
Weaving LS, Christodoulou J, Williamson SL, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004;75:1079–1093.
Melani F, Mei D, Pisano T, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol 2011;53:354–360.
Mei D, Marini C, Novara F, et al. Xp22.3 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia 2010;51:647–654.
Ohtahara S, Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res 2006;70(Suppl. 1):S58-67.
Djukic A, Lado FA, Shinnar S, Moshe SL. Are early myoclonic encephalopathy (EME) and the Ohtahara syndrome (EIEE) independent of each other? Epilepsy Res 2006;70(Suppl. 1):S68–76.
Mignot C, Moutard ML, Trouillard O, et al. STXBP1-related encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia 2011;52:1820–1827.
Milh M, Villeneuve N, Chouchane M, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011;52:1828–1834.
Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788.
Lemke JR, Lal D, Reinthaler EM, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 2013;45:1067–1072.
Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 2013;45:1061–1066.
Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 2013;45:1073–1076.
Lemke JR, Hendrickx R, Geider K, et al. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann Neurol 2014;75:147–154.
Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221.
Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830.
Suls A, Jaehn JA, Kecskes A, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013;93:967–975.
Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012;90:502–510.
Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44:1255–1259.
Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 2012;44:1188–1190.
Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain 1995;118:61–73.
Picard F, Baulac S, Kahane P, et al. Dominant partial epilepsies. A clinical, electrophysiological and genetic study of 19 European families. Brain 2000;123:1247–1262.
Ryvlin P, Minotti L, Demarquay G, et al. Nocturnal hypermotor seizures, suggesting frontal lobe epilepsy, can originate in the insula. Epilepsia 2006;47:755–765.
Marini C, Guerrini R. The role of the nicotinic acetylcholine receptors in sleep-related epilepsy. Biochem Pharmacol 2007;74:1308–1314.
Steinlein OK, Magnusson A, Stoodt J, et al. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet 1997;6:943–948.
De Fusco M, Becchetti A, Patrignani A, et al. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet 2000;26:275–276.
Liu H, Lu C, Li Z, et al. The identification of a novel mutation of nicotinic acetylcholine receptor gene CHRNB2 in a Chinese patient: Its possible implication in non-familial nocturnal frontal lobe epilepsy. Epilepsy Res 2011;95:94–99.
Chen Y, Wu L, Fang Y, et al. A novel mutation of the nicotinic acetylcholine receptor gene CHRNA4 in sporadic nocturnal frontal lobe epilepsy. Epilepsy Res 2009;83:152–156.
Aridon P, Marini C, Di Resta C, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet 2006;79:342–350.
Phillips HA, Favre I, Kirkpatrick M, et al. CHRNB2 Is the Second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet 2001;68:225–231.
Steinlein OK, Hoda JC, Bertrand S, Bertrand D. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure 2012;21:118–123.
Phillips HA, Scheffer IE, Crossland KM, et al. Autosomal dominant nocturnal frontal lobe epilepsy: genetic heterogeneity and a probable second locus at 15q24. Am J Hum Genet 1998;63:1108–1116.
Bertrand D, Picard F, Le Hellard S, et al. How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia 2002;43(Suppl. 5):112–122.
Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A 2006;103:19152–19157.
Teper Y, Whyte D, Cahir E, et al. Nicotine-induced dystonic arousal complex in a mouse line harboring a human autosomal-dominant nocturnal frontal lobe epilepsy mutation. J Neurosci 2007;27:10128–10142.
Zhu G, Okada M, Yoshida S, et al. Rats harboring S284L Chrna4 mutation show attenuation of synaptic and extrasynaptic GABAergic transmission and exhibit the nocturnal frontal lobe epilepsy phenotype. J Neurosci 2008;28:12465–12476.
Ottman R, Risch N, Hauser WA, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet 1995;10:56–60.
Michelucci R, Poza JJ, Sofia V, et al. Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsia 2003;44:1289–1297.
Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341.
Ottman R, Winawer MR, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology 2004;62:1120–1126.
Chernova OB, Somerville RP, Cowell JK. A novel gene, LGI1, from 10q24 is rearranged and downregulated in malignant brain tumors. Oncogene 1998;17:2873–2881.
Scheel H, Tomiuk S, Hofmann K. A common protein interaction domain links two recently identified epilepsy genes. Hum Mol Genet 2002;11:1757–1762.
Skradski SL, Clark AM, Jiang H, White HS, Fu YH, Ptacek LJ. A novel gene causing a mendelian audiogenic mouse epilepsy. Neuron 2001;31:537–544.
Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet 2005;14:1613–1620.
Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 2006;313:1792–1795.
Schulte U, Thumfart JO, Klocker N, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron 2006;49:697–706.
Baulac S, Ishida S, Mashimo T, et al. A rat model for LGI1-related epilepsies. Hum Mol Genet 2012;21:3546–3557.
Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 2009;15:1208–1214.
Yu YE, Wen L, Silva J, et al. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet 2010;19:1702–1711.
Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551.
Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555.
Bar-Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013;340:1100–1106.
Annegers JF, Hauser WA, Anderson VE, Kurland LT. The risks of seizure disorders among relatives of patients with childhood onset epilepsy. Neurology 1982;32:174–179.
Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 1990;46:222–228.
Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol 1998;43:435–445.
Berkovic SF, Scheffer IE. Genetics of the epilepsies. Epilepsia 2001;42(Suppl. 5):16–23.
Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011;145:1036–1048.
Durner M, Keddache MA, Tomasini L, et al. Genome scan of idiopathic generalized epilepsy: evidence for major susceptibility gene and modifying genes influencing the seizure type. Ann Neurol 2001;49:328–335.
Sander T, Schulz H, Saar K, et al. Genome search for susceptibility loci of common idiopathic generalised epilepsies. Hum Mol Genet 2000;9:1465–1472.
Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet 2002;31:184–189.
D’Agostino D, Bertelli M, Gallo S, et al. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology 2004;63:1500–1502.
Chen Y, Lu J, Pan H, al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol 2003;54:239–243.
Heron SE, Phillips HA, Mulley JC, et al. Genetic variation of CACNA1H in idiopathic generalized epilepsy. Ann Neurol 2004;55:595–596.
Powell KL, Cain SM, Ng C, et al. A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J Neurosci 2009;29:371–380.
Dibbens LM, Feng HJ, Richards MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet 2004;13:1315–1319.
Greenberg DA, Cayanis E, Strug L, et al. Malic enzyme 2 may underlie susceptibility to adolescent-onset idiopathic generalized epilepsy. Am J Hum Genet 2005;76:139–146.
Pal DK, Evgrafov OV, Tabares P, Zhang F, Durner M, Greenberg DA. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet 2003;73:261–270.
Dibbens LM, Ekberg J, Taylor I, et al. NEDD4-2 as a potential candidate susceptibility gene for epileptic photosensitivity. Genes Brain Behav 2007;6:750–755.
Liautard C, Scalmani P, Carriero G, de Curtis M, Franceschetti S, Mantegazza M. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 2013;54:1251–1261.
Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun 2013;4:2410.
Delgado-Escueta AV, Bourgeois BF. Debate: Does genetic information in humans help us treat patients? PRO—genetic information in humans helps us treat patients. CON—genetic information does not help at all. Epilepsia 2008;49(Suppl. 9):13–24.
Ottman R, Hirose S, Jain S, et al. Genetic testing in the epilepsies—report of the ILAE Genetics Commission. Epilepsia 2010;51:655–670.
Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398.
Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998;39(Suppl.):508–512.
Cotton RG, Auerbach AD, Axton M, et al. GENETICS. The Human Variome Project. Science 2008;322:861–862.
Acknowledgement
Research in Renzo Guerrini’s and Carla Marini’s team and laboratories is funded by Europen Union FP7 Grant no. 602531 (“DESIRE”), and Italian Ministry of Health and Tuscany Region Grant RF 2009–1525669. Research in Massimo Mantegazza’s laboratory is funded by LabEx “ICST”, CNRS-PICS “NavRole”, and Europen Union FP7 Grant no. 602531 (“DESIRE”).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 338 kb)
Rights and permissions
About this article
Cite this article
Guerrini, R., Marini, C. & Mantegazza, M. Genetic Epilepsy Syndromes Without Structural Brain Abnormalities: Clinical Features and Experimental Models. Neurotherapeutics 11, 269–285 (2014). https://doi.org/10.1007/s13311-014-0267-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-014-0267-0