
Abstract
Gene therapies are classified into two major categories, namely, in vivo and ex vivo. Clinical trials of human gene therapy began with the ex vivo techniques. Based on the initial successes of gene-therapy clinical trials, these approaches have spread worldwide. The number of gene therapy trials approved worldwide increased gradually starting in 1989, reaching 116 protocols per year in 1999, and a total of 2210 protocols had been approved by 2015. Accumulating clinical evidence has demonstrated the safety and benefits of several types of gene therapy, with the exception of serious adverse events in several clinical trials. These painful experiences were translated backward to basic science, resulting in the development of several new technologies that have influenced the recent development of ex vivo gene therapy in this field. To date, six gene therapies have been approved in a limited number of countries worldwide. In Japan, clinical trials of gene therapy have developed under the strong influence of trials in the US and Europe. Since the initial stages, 50 clinical trials have been approved by the Japanese government. In this review, the history and current status of clinical trials of ex vivo gene therapy for hematological disorders are introduced and discussed.
Similar content being viewed by others


Introduction
Clinical trials of human gene therapy initially began with ex vivo techniques. The earliest efforts included a gene-marking study of transfusion of autologous tumor-infiltrating lymphocytes, modified by retroviral transduction of a neomycin resistance gene, in advanced melanoma patients, starting in 1989, and a clinical trial of transfusion of peripheral autologous lymphocytes modified by retroviral transduction of the adenosine deaminase (ADA) cDNA, for treatment of patients with severe combined immunodeficiency (SCID) with ADA deficiency, starting in 1990 [1, 2]. The results of these studies demonstrated the feasibility and safety of retroviral transduction of target genes into peripheral T cells. In addition, the second study showed that transfusion of ADA gene-transduced T cells could confer clinical benefits and save the lives of ADA-SCID patients, who are at high risk of infectious disease.
Based on the initial successes of gene-therapy clinical trials, these new therapeutic approaches have spread worldwide. The number of gene therapy trials approved worldwide increased gradually starting in 1989, reaching 116 protocols per year in 1999, and, essentially, maintaining this rate thereafter. By 2015, a total of 2210 protocols had been approved. Accumulating clinical evidence has demonstrated the safety and benefits of several types of gene therapy. Currently, 78.1 % of 2210 protocols are at phase I or I/II (http://www.wiley.com/legacy/wileychi/genmed/clinical/), but six gene therapy drugs have been approved in a limited number of countries [3–7].
However, progress has decelerated due to the occurrence of serious adverse events in several clinical trials, including a fatal systemic inflammatory response syndrome in an ornithine transcarbamylase-deficient patient following intrahepatic arterial injection of adenoviral vector; acute lymphoid malignancy in four patients with X-linked severe combined immunodeficiency; and myelodysplasia with monosomy 7 in 2 patients with X-linked chronic granulomatous disease following peripheral-blood transplantation of retrovirally transduced autologous peripheral stem cells [8–10]. These painful experiences were translated backward to basic science. With the goal of preventing similar outcomes in the future, subsequent trials adopted new gene-transfer technologies, selection of appropriate patients, and close monitoring of patients after gene therapy [11–14].
In Japan, clinical trials of gene therapy have developed under the strong influence of trials in the US and Europe. Fifty clinical trials have been approved by the Japanese government, since the initial stages: gene therapy for an ADA deficiency patient was approved in 1995, and cancer gene therapies using GM-CSF and p53 genes were approved in 1998 [15–17]. There have been no remarkable adverse events reported in Japan. Of the 58 trials to date, 37 were for malignancies and 21 were for congenital or acquired non-malignant diseases (http://www.nihs.go.jp/mtgt/section-1/prtcl/prtcl-jn.html). These clinical trials have also been strongly supported by the Japan Society of Cell and Gene Therapy, which was established in 1995 (http://jsgt.jp/ABOUT-JSGT/about-jsgt1.html).
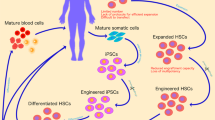
Gene therapies can be classified into two major categories, namely, in vivo and ex vivo [18]. Historically, ex vivo gene therapy has been the most widely adopted, because investigators can preliminarily check the safety and predict the efficacy of the gene-transduced target cells in vitro before administration of the therapeutic agents to patients. However, because serious adverse events have been reported, the long-term in vivo toxicity of ex vivo gene therapy must also be carefully considered [9, 10]. Ex vivo gene therapy can successfully treat many hematological disorders, including hematological malignancies. Therefore, in this review, we summarize the current status of ex vivo gene therapy for hematological disorders in Japan and around the world.
Ex vivo gene therapy clinical trials in Japan and around the world
Essentially, all congenital monogenic hematological and immunological disorders can be targeted by gene therapy. Historically, however, several hematological disorders have received the closest attention as target diseases for gene therapy clinical trials, largely because we have a great deal of biochemical information about their pathogenesis. In this review, clinical trials of gene therapy for congenital hematological and immunological disorders are introduced and discussed. Data from “Gene Therapy Clinical Trials Worldwide,” provided by the Journal of Gene Medicine (http://www.wiley.com/legacy/wileychi/genmed/clinical/), were used to generate Tables 1 and 2. Data from “List of Gene Therapy Clinical Study Protocols Approved in Japan” provided by the National Institute of Health Sciences, Japan (http://www.nihs.go.jp/mtgt/section-1/english/prtcl-e/prtcl-e.html), http://www.nihs.go.jp/mtgt/section-1/prtcl/prtcl-jn.html were used to generate Table 3.
Ex vivo gene therapy clinical trials for congenital hematological disorders in Japan and around the world
Since the first gene therapy of a monogenic disease, severe combined immunodeficiency due to ADA deficiency (ADA-SCID) was conducted safely and with clinical benefits [2]; 209 clinical protocols for monogenic diseases have been approved as of July 2015. Almost half of the approved trials involve ex vivo gene therapy. The target diseases include ADA-SCID, X-linked severe combined immunodeficiency (SCID-X1), X-linked chronic granulomatous disease (X-CGD), hemophilia, Wiskott–Aldrich syndrome (WAS), mucopolysaccharidosis, cerebral adrenoleukodystrophy, sickle cell disease, thalassemia, metachromatic leukodystrophy, familial hypercholesterolemia, Gaucher disease, Fanconi anemia, purine nucleoside phosphorylase deficiency, leukocyte adherence deficiency, gyrate atrophy, JAK3 deficiency, and epidermolysis bullosa. In Table 1, the outlines of clinical trials of primary immunodeficiencies (PIDs) and hemoglobinopathies are presented.
PIDs are a heterogeneous group of monogenic conditions caused by altered innate and/or adaptive immune responses. More than 260 disorders involving more than 300 gene mutations have been reported, and these numbers continue to increase due to improved diagnostic modalities for immune system, the introduction of genome-wide association studies, and the next-generation sequencing technologies. Patients’ phenotypes range from asymptomatic to manifestation of life-threatening conditions, including various forms of severe combined immunodeficiency (SCID) [19, 20]. Below, the results of clinical trials for ADA-SCID, SCID-X1, CGD, and Wiskott–Aldrich syndrome are introduced and discussed.
Severe combined immunodeficiency due to ADA deficiency (ADA-SCID)
Inherited ADA deficiency causes a range of phenotypes, the most severe of which is SCID presenting in infancy, usually resulting in early death. Ten-to-fifteen percent of patients have a delayed clinical onset starting at 6–24 months of age, and a smaller percentage of patients have later onset, diagnosed between the age of 4 years and adulthood. The latter patients exhibit less severe infections and more gradual immunologic deterioration. ADA deficiency accounts for approximately 15 % of all SCID cases and one-third of cases of autosomal recessive SCID. ADA is an enzyme that catalyzes the irreversible deamination of adenosine and deoxyadenosine in the purine catabolic pathway (http://www.omim.org/) [20]. Enzyme activity is highest in lymphoid tissues, particularly the thymus. The lack of ADA results in accumulation of toxic purine metabolites, causing lymphocytes to undergo apoptosis during differentiation and selection. Enzyme replacement therapy (ERT) with polyethylene glycol-modified bovine ADA is clinically useful but non-curative. To achieve a definitive cure, hematopoietic stem cell transplantation (HSCT) from an unaffected, matched sibling donor is the treatment of choice. For those without well-matched donors, ex vivo gene therapy with autologous peripheral T cells is considered to be the treatment of choice. The first clinical trial of gene therapy for ADA-SCID, involving two children, started in 1990. Infusions of retrovirally ADA gene-transduced T cells were achieved safely, and normalized blood T cells (including many cellular and humoral immune responses) and integrated vector and ADA gene expression in T cells persisted after the discontinuation of therapy. The usefulness of T-cell-directed gene transfer in the treatment of ADA-SCID was also demonstrated by another clinical trial; however, the effects required multiple rounds of T-cell infusions and were not sufficient to alleviate the requirement for ERT, although the dose could be reduced [2, 22]. To develop a longer lasting source of ADA-expressing T cells, the ADA gene was retrovirally transduced into hematopoietic stem cells, including bone marrow cells, BM CD34+ cells, and umbilical cord blood CD34+ cells, and infused into patients. These results demonstrated the feasibility of hematopoietic cell gene therapy using a gamma retrovirus vector. The level of ADA expression, however, was not sufficient to confer significant clinical benefits [21–24]. Several strategies were adopted to overcome these problems: optimization of gene transduction methods, non-myeloablative preconditioning with busulfan or melphalan to create space in the bone marrow, and discontinuation of ERT around the time of reinfusion of gene-modified stem cells [19, 21]. Nine out of ten ADA-SCID patients treated with gene therapy after non-myeloablative preconditioning with busulfan maintained lasting immune reconstitution, and eight and five of the ten were able to discontinue ERT or intravenous immune globulin, respectively, emphasizing the importance of non-myeloablative preconditioning for discontinuation of ERT [25–27].
More than 40 ADA-SCID patients have been with gamma-retroviral ADA gene therapy in the US and Europe, and 100 % of these patients survived. Furthermore, unlike other PIDs, such as X-SCID, X-CGD, and WAS, no severe adverse events related to insertional mutagenesis have been reported in any of these patients, despite equivalent vector design and integration profiles similar to those seen in other trials [21, 28]. Clonal analysis of long-term repopulating cell progeny in vivo revealed highly polyclonal T-cell populations and shared retroviral integration sites among multiple lineages, demonstrating the engraftment of multipotent HSCs [29]. ADA enzyme production may cross-rescue ADA-deficient lymphocytes, thus placing less replicative stress on the gene-corrected cells [21]. To achieve safer gene therapy for ADA-SCID patients, current trials have adopted self-inactivating HIV-1-based lentiviral vectors. Lentiviral vectors are pseudotyped with the vesicular stomatitis virus glycoprotein, which uses the broadly expressed low-density lipoprotein receptor for cell entry and thus achieves broad-cell tropism. At least 30 ADA-SCID patients have been treated with lentiviral vectors in the US and the UK. Excellent immune reconstitution has been achieved in these patients, with no vector-related complications [21].
In Japan, the first gene therapy for an ADA-SCID patient was initiated in 1995. The patient, a 5-year-old Japanese male, received periodic infusions of retrovirally ADA gene-transduced autologous T cells. The percentage of peripheral-blood lymphocytes carrying the transduced ADA gene remained stable at 10–20 % during the 12 months since the fourth infusion. ADA enzyme activity in the patient’s circulating T cells, which was barely detectable before gene transfer, increased to levels comparable to those of a heterozygous carrier individual, and was associated with increased T-cell counts and improvement in the patient’s immune function. In 2003–2004, two ADA-SCID patients, including one who received T-cell gene therapy, safely received retrovirally ADA gene–transduced autologous bone marrow CD34+ cells without any cytoreductive conditioning after discontinuation of ERT. Partial recovery of immunity due to moderate systemic detoxification was achieved, allowing temporary discontinuation of ERT for 6 and 10 years in patients 1 and 2, respectively. Vector integration-site analyses confirmed that hematopoiesis was reconstituted by a limited number of clones, some of which had myelo-lymphoid potential. These results emphasize that cytoreductive conditioning is important for achieving optimal benefits from SCGT [30] (Table 3).
X-linked severe combined immunodeficiency (SCID-X1)
X-linked severe combined immunodeficiency (SCID-X1) is caused by mutation in the gene encoding the gamma subunit of the interleukin-2 receptor (γc), a key subunit of the cytokine receptor complex for interleukin (IL)2, IL4, IL7, IL9, IL15, and IL21. Common γc deficiency is the most frequent genetic form of SCID, accounting for 50–60 % of cases, and is typically characterized by an absence of mature T cells and natural killer (NK) lymphocytes due to a complete blockade in their development [31]. SCID-X1 is a lethal condition that can be cured by allogeneic stem cell transplantation. Between 1999 and 2002, the first ex vivo gene therapy was conducted in France, using retrovirally common γc gene-transduced CD34+ bone marrow cells. The patients in this trial were 10 SCID-X1 boys without HLA-identical stem cell donors. Immune functions, including normalization of the numbers and phenotypes of T cells, the repertoire of T-cell receptors, and the in vitro proliferative responses of T cells to several antigens after immunization, persisted for up to 2 years after treatment [32]. Subsequently, a similar study was initiated in the UK. A total of 20 patients were enrolled and successfully treated with ex vivo-transduced CD34+ cells without any conditioning regime [33]. In the US, five older patients were treated with a similar protocol. Functional reconstitution, however, was not achieved in those patients, probably due to loss of thymic function by the time gene therapy was initiated. The promising results in France and the UK were subsequently hampered by the occurrence of lymphoid malignancies. Five patients developed acute T-cell leukemia; four entered remission after standard chemotherapy, but one died. All of the leukemias were associated with viral integration in oncogenes; in four of the cases, the integration occurred in LMO2. The enhancer activity of the viral LTR was the most likely cause of the initial aberrant expression of these oncogenes and dysregulation of the cell cycle; subsequent accumulation of additional genetic abnormalities, such as associated rearrangements in BMI1, CCND2, and NOTCH1, led to frank leukemic transformation [34]. In the French cases, transduced T cells were detected for up to 10.7 years after gene therapy. Seven patients, including three of the leukemia survivors, exhibited sustained immune reconstitution, whereas the three others required immunoglobulin-replacement therapy. Sustained thymopoiesis was established by the persistent presence of naive T cells, even after chemotherapy, in three patients. The T-cell receptor repertoire was diverse in all patients. Transduced B cells were not detected. Correction of immunodeficiency improved the patients’ health. After nearly 10 years of follow-up, gene therapy had corrected the immunodeficiency associated with SCID-X1.
Gene therapy is considered to be an option for patients who do not have an HLA-identical donor for HSCT and for whom the risks are deemed acceptable; the treatment is associated with a risk of acute leukemia [35, 36]. Patients who underwent gene therapy also demonstrated a clear advantage in terms of T-cell development relative to patients who underwent HSCT with a mismatched donor; moreover, patients treated with gene therapy exhibited faster T-cell reconstitution and better long-term thymic output. Interestingly, this advantage of gene therapy relative to haploidentical HSCT seemed to be independent of the presence of clinical graft-versus-host disease in the latter condition [37]. Parallel trials in Europe and the US evaluated treatment with a self-inactivating (SIN) γ-retrovirus vector containing deletions in viral enhancer sequences expressing γc (SIN-γc). All of the patients, nine boys with SCID-X1, received bone marrow-derived CD34+ cells transduced with the SIN-γc vector, without cytoreductive conditioning. After 12.1–38.7 months of follow-up, seven of the boys exhibited recovery of peripheral-blood T cells that were functional and led to resolution of infections. The patients remained healthy thereafter. The kinetics of CD3+ T-cell recovery was not significantly different from those observed in previous trials. Assessment of insertion sites in peripheral blood from patients in the current trial revealed significantly less clustering of insertion sites within LMO2, MECOM, and other lymphoid proto-oncogenes than in the previous trials. Thus, the modified γ-retrovirus vector is a safer method for treatment of SCID-X1 than the first-generation Moloney murine leukemia virus vector expressing γc [13].
The gamma chain of the human IL-2 receptor was originally cloned in Japan in 1992 [38]. Gene therapy using a γ-retrovirus vector expressing the gamma chain of the human IL-2 receptor was approved in Japan in 2002. The clinical trial was not started, however, because lymphoid malignancy arose in French patients in 2002.
X-linked chronic granulomatous disease (X-CGD)
Chronic granulomatous disease is a genetically heterogeneous immunodeficiency disorder resulting from an inability of phagocytes to kill microbes that they have ingested. This impairment in killing is caused by any of several defects in the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (phox) complex, which generates the microbicidal ‘respiratory burst’, patients carrying mutations in subunits of NADPH oxidase suffer from defects in neutrophil function (http://www.omim.org/). The fully assembled NADPH oxidase, which consists of the cytosolic phox proteins (p47phox, p67phox, and p40phox, respectively, encoded by NCF1, NCF2, and NCF4), translocated to the membrane-bound flavocytochrome (gp91phox and p22phox, respectively, encoded by CYBB and CYBA), leads through a series of reactions to the production of reactive oxygen species (ROS), which are essential for phagocytic killing of invading microbes. Mutations in CYBB, which encodes gp91phox, lead to the X-linked form of this disease; these mutations account for 60–80 % of all CGD patients in most European countries, North America and Japan. The next most common form, representing about one-third of cases, is an autosomal recessive disease resulting from mutations in NCF1 (encoding p47phox) on chromosome 7. CGD patients are susceptible to recurrent and severe bacterial and fungal infections, notably Staphylococcus and Aspergillus, and are also affected by inflammatory complications in the lungs and gastrointestinal and genitourinary tracts. Conventional therapy involves a broad regimen of antifungals, antibiotics, anti-inflammatory medications, and HSCT, but its effectiveness is limited [34, 39, 40].
Initial trials of ex vivo gene therapy for X-CGD were conducted in the US in 1995 and 1998, targeting p47phox and gp91phox deficiency, respectively, using gamma-retroviral vectors to transduce granulocyte colony stimulating factor (G-CSF)-mobilized CD34+ HSCs without any cytoreductive conditioning. A total of 10 patients were involved. In both trials, only transient production of ROS-producing neutrophils was detected, and no remarkable long-term clinical benefits accrued [34, 39–41].
Subsequent clinical trials in Germany used gamma-retroviral vectors to transduce granulocyte colony stimulating factor (G-CSF)-mobilized CD34+ HSCs after a non-myeloablative conditioning regimen. Two adult patients exhibited substantial gene transfer to neutrophils, leading to a large number of functionally corrected phagocytes and notable clinical improvement. Large-scale analysis of the distribution of retroviral integration sites in both individuals revealed activating insertions in MDS1-EVI1, PRDM16, or SETBP1 that influenced regulation of long-term hematopoiesis by enhancing gene-corrected myelopoiesis 3–4-fold. Thus, gene therapy in combination with bone marrow conditioning was successfully used to treat CGD, an inherited disease affecting the myeloid compartment [42]. Subsequently, a total of 13 patients were treated in similar manner, and transient clinical benefits due to gene transfer were observed [10, 34, 43–47]. Three patients, including two adults, exhibited a temporary increase in gene-marked neutrophils followed by the clearance of infections. However, insertional mutagenesis occurred due to integration into the MECOM (MDS/EVI1 complex) and PRDM16 oncogene loci, and transactivation by the viral SFFV LTR was similar to that observed in SCID-X1 patients. Two adults with fatal outcomes exhibited silencing of transgene expression due to methylation of the viral promoter, and myelodysplasia with monosomy 7 due to insertional activation of ecotropic viral integration site 1 (EVI1). Forced overexpression of EVI1 in human cells disrupts normal centrosome duplication, linking EVI1 activation to the development of genomic instability, monosomy 7, and clonal progression toward myelodysplasia [34, 46]. The frequency of these adverse events highlighted the fact that only gp91phox-transduced cells with gain-of-function events could persist in patients who underwent ex vivo gene therapy using LTR-driven retrovirus vectors [18]. Moreover, ectopic expression of the gp91phox transgene and the highly activated bone marrow environment caused by X-CGD patients’ chronic infections was thought to promote the loss of gene-corrected cell engraftment. To increase the safety and efficacy of the gene therapy for X-CGD patients, subsequent trials adopted self-inactivating gamma retrovirus (SIN-γRV) and lentiviral vectors (SIN-LVs) with an internal promoter and a fully myeloablative conditioning regimen (12–16 mg/kg busulfan), prior to gene therapy. The vector currently under evaluation in multicenter trials is an SIN-LV configuration with a chimeric promoter consisting of myeloid-specific Cathepsin G and c-Fes regulatory elements; this promoter element allows preferential expression in myeloid cells and differentiated granulocytes [28, 48].
In Japan, a clinical trial for gene therapy to treat X-CGD was first performed in 2014. This trial, in a 27-year-old man, used a gp91phox gamma-retroviral vector in autologous G-CSF-mobilized CD34+ HSCs after busulfan conditioning. The patient received 3.9 × 108 CD34+ cells, including 76.8 % of gp91phox-expressing cells. Although clinical symptoms of lymphadenitis and CGD-colitis improved until day 95 of transplantation and no genotoxicity was observed, the number of gene-transduced cells was decreased to about 0.1 % of neutrophils at day 182 probably due to the reduction of gene-modified CD34+ stem cells. At present, gene therapy was considered to be useful as a short-term treatment for CGD patients who had no HLA-matched donor for HSCT [49].
Other ex vivo gene therapies for hematological and non-hematological congenital disorders
Wiskott–Aldrich syndrome (WAS) is a rare, complex, X-linked recessive PID disorder caused by mutations in the WAS gene that encodes the WAS protein (WASp). WAS is characterized by recurrent infections, microthrombocytopenia, eczema, and elevated risk of autoimmune manifestations and tumors. WASp is a key regulator of actin polymerization in hematopoietic cells, and contains domains involved in signaling, cell locomotion, and immunologic-synapse formation. The complex biological features of this disease result from multiple dysfunctions in different subgroups of leukocytes, including functional defects in T and B cells, disturbed formation of the NK-cell immunologic synapse, and impaired migratory responses in all leukocyte subgroups. Severe WAS leads to early death from infection or bleeding. Currently, the only curative therapy involves allogeneic HSCT, which is itself associated with considerable risk of death or complications related to transplantation. Therapy with WAS gene-corrected autologous HSCs, thus, represents a valid alternative approach for patients lacking a suitable donor or those who are more than 5 years old [18, 50].
The first gene therapy for WAS was conducted in Germany in 2006; the trial involved two patients. Sustained expression of WASp in HSCs, lymphoid and myeloid cells, and platelets after ex vivo gene therapy was achieved using autologous CD34+ HSCs transduced with gamma retroviral vector after non-myeloablative preconditioning. T and B cells, natural killer (NK) cells, and monocytes were functionally corrected. After treatment, the patients’ clinical condition markedly improved, with resolution of hemorrhagic diathesis, eczema, autoimmunity, and predisposition to severe infection. Comprehensive insertion-site analysis revealed vector integration that targeted multiple genes controlling growth and immunologic responses in a persistently polyclonal hematopoiesis [50]. Subsequently, 10 patients with severe WAS were treated using the HSC gene therapy. Nine of the ten patients exhibited sustained engraftment and correction of WASp expression in lymphoid and myeloid cells and platelets. Gene therapy resulted in partial or complete resolution of immunodeficiency, autoimmunity, and bleeding diathesis. Analysis of retroviral insertion sites revealed >140,000 unambiguous integration sites and a polyclonal pattern of hematopoiesis in all patients soon after gene therapy. Seven patients developed acute leukemia [one with acute myeloid leukemia (AML), four with T-cell acute lymphoblastic leukemia (T-ALL), and two with primary T-ALL with secondary AML associated with a dominant clone; vector integration occurred at LMO2 (six T-ALL), MDS1 (two AML), or MN1 (one AML) locus]. Thus, LMO2-driven leukemogenesis is not specific for X-SCID gene therapy, but is also seen in WAS gene therapy. Cytogenetic analysis revealed additional genetic alterations, including chromosomal translocations. This study showed that hematopoietic stem cell gene therapy for WAS is feasible and effective; however, the use of γ-retroviral vectors was associated with a substantial risk of leukemogenesis [51].
In 2010, three WAS patients in Italy received bone marrow-derived CD34+ cells genetically modified with an SIN lentiviral vector encoding functional WASp after a reduced-intensity conditioning regimen. All three patients exhibited stable engraftment of WASp-expressing cells and improvements in platelet counts, immune functions, and clinical scores. Vector integration analyses revealed highly polyclonal and multilineage hematopoiesis resulting from the gene-corrected HSCs. Lentiviral gene therapy did not induce selection of integrations near oncogenes, and no aberrant clonal expansion was observed after 20–32 months [52]. Between 2010 and 2014, seven WAS patients in France and England received a single infusion of CD34+ cells genetically modified with an SIN lentiviral vector. Six of the seven patients were alive at the time of last follow-up and exhibited sustained clinical benefit. One patient died 7 months after treatment due to a preexisting drug-resistant herpes virus infection. Eczema and susceptibility to infections resolved in all six patients. Autoimmunity improved in five patients. No severe bleeding episodes were recorded after treatment, and at last follow-up, all six surviving patients were free of blood product support and thrombopoietic agonists. Hospitalization days were reduced from a median of 25 days during the 2 years before treatment to a median of 0 days during the 2 years after treatment. All six surviving patients exhibited high-level, stable engraftment of functionally corrected lymphoid cells. The degree of myeloid cell engraftment and of platelet reconstitution correlated with the dose of gene-corrected cells administered [53]. No evidence of vector-related toxicity was observed either clinically or by molecular analysis in either of these clinical trials using SIN lentiviral vectors, although long-term follow-up is still required [52, 53].
Ex vivo gene therapy for congenital diseases other than PIDs has also been developed. Sickle cell disease (SCD) and β-thalassemia major (β-TM), the latter defined clinically as transfusion-dependent cases regardless of the underlying genotype, are the most common monogenic disorders worldwide, with approximately 400,000-affected conceptions or births each year. Over the past 8 years, clinical trials have evaluated gene therapy by ex vivo lentiviral transfer of a therapeutic β-globin gene derivative (β(AT87Q)-globin) into hematopoietic stem cells, driven by cis-regulatory elements that confer high, erythroid-specific expression. β(AT87Q)-globin is used as both a strong inhibitor of HbS polymerization and a biomarker. While long-term studies are underway in multiple centers in Europe and in the US, proof-of-principle of efficacy and safety has already been obtained in multiple patients with β-TM and SCD [54].
LG001 and HGB-205 were the first gene therapy studies worldwide to treat β-TM and SCD subjects, respectively, achieving the first conversion to long-term transfusion independence of a β-TM patients and the first evidence of clinical benefit in SCD [54, 55]. An SIN lentiviral vector expressing HPV569 β(AT87Q)-globin was used in the LG001 trial. Four β-TM patients were enrolled, 3 of whom were treated between 2006 and 2011. Transduced cells successfully engrafted in two of them. The HPV drug product was well tolerated, with no non-hematologic or drug product-related serious adverse events. One of the two treated patients receiving no backup cells exhibited clinical benefits as evidenced by transfusion independence for approximately 8 years. Integration-site analysis revealed the relative dominance of a clone bearing the vector in intron 3 of the HMGA2 gene, which reached a maximum representation of 30 % of transduced myeloid cells 15 months after gene therapy. The BB305 SIN lentiviral vector was the second generation of the HPV569, lacking the cHS4 insulator, and exhibited elevated vector titers and transduction efficiency. Several clinical trials using BB305 had been initiated in France, two separate international, and USA for β-TM and SCD patients [54].
The following two ex vivo gene therapies are for non-hematological diseases, but HSCs were used as therapeutic vehicle. First, X-linked adrenoleukodystrophy (ALD) is a severe brain demyelinating disease in boys that is caused by a deficiency in the ALD protein, an adenosine triphosphate-binding cassette transporter encoded by the ABCD1 gene. ALD progression can be halted by allogeneic hematopoietic cell transplantation (HCT). An ex vivo gene therapy trial for two ALD patients without any matched donors was initiated in 2005. Lentivirally ABCD1 gene-transduced autologous CD34+ cells were reinfused into the patients after fully myeloablative preconditioning. Over a span of 24–30 months of follow-up, polyclonal reconstitution was observed, suggesting successful gene transduction into HSCs. Beginning 14–16 months after infusion of the genetically corrected cells, progressive cerebral demyelination in the two patients stopped, a clinical outcome comparable to that achieved by allogeneic HCT. Thus, lentiviral-mediated gene therapy of HSCs provided clinical benefits in ALD [56].
Second, metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disease caused by arylsulfatase A (ARSA) deficiency. Patients with MLD exhibit progressive motor and cognitive impairment and die within a few years of symptom onset. Three presymptomatic patients who exhibited genetic, biochemical, and neurophysiological evidence for late infantile MLD received lentivirally ARSA gene-transduced HSCs. After receiving the gene-corrected HSCs, the patients exhibited extensive and stable ARSA gene replacement, which led to high enzyme expression throughout hematopoietic lineages and in cerebrospinal fluid. Analyses of vector integrations revealed no evidence of aberrant clonal behavior. The disease did not manifest or progress in the three patients for 7–21 months beyond the predicted age of symptom onset. These findings suggested the clinical usefulness of lentiviral gene therapy for MLD patients [57].
Ex vivo gene therapy clinical trials for hematological malignancies in Japan and around the world
Ex vivo gene therapy clinical trials for hematological malignancies have been primarily performed to enhance host immune function in patients who could not receive stem-cell transplantation due to a lack of appropriate donors, who relapsed, or who were refractory to standard chemotherapies (Table 2). As recent advances in gene therapy using the receptor gene-modified T-cell therapy are excellently reviewed by Davila ML and Sadelain M in this special issue, in the following, we will discuss immune therapy using suicide gene-transduced donor lymphocytes and gene-modified tumor vaccines.
Suicide gene therapy
The most effective and established cell therapy approach is allogeneic HSCT, which is currently the only cure for patients with several high-risk hematological malignancies. Alloreactive T cells induce potent graft-versus-tumor effect (GvT) and also trigger detrimental graft-versus-host disease (GvHD). Gene therapy technology allows T cells to exert the GvT effect, while avoiding or controlling GvHD.
Herpes simplex virus thymidine kinase (HSV-TK) is the suicide gene most extensively tested in humans. Expression of HSV-TK in donor lymphocytes confers sensitivity to the anti-herpes drug ganciclovir (GCV). Progressive improvements in suicide genes, vector technology, and transduction protocols have ameliorated the toxicity of GvHD, while preserving the antitumor efficacy of allogeneic HSCT. Several clinical trials in the last 20 years documented the safety and the efficacy of the HSV-TK approach and established its important role in cellular therapy against cancer. Recently developed gene therapies have improved immune effector cell survival, homing, function, safety, and specificity using high-avidity tumor-reactive T-cell receptors (TCRs) or chimeric antigen receptors, and suicide gene therapy has been reappraised with the goal of avoiding or controlling the toxic effects induced by these innovative therapies [58].
After the successful administration and long-term detection of retrovirally gene-modified Epstein–Barr virus (EBV)-specific T cells in the US [59], eight Italian patients who relapsed or developed EBV-induced lymphoma after T-cell-depleted BMT were treated with donor lymphocytes retrovirally transduced with the HSV-TK suicide gene. The transduced lymphocytes survived for up to 12 months, and exerted antitumor activity in five patients. Three patients developed GvHD, which was effectively controlled by ganciclovir-induced elimination of the transduced cells. These data suggested that genetic manipulation of donor lymphocytes increased the efficacy and safety of allo-BMT and expanded its application to a larger number of patients [60]. Based on these initial clinical trials, several groups around the world have exploited suicide gene therapy to control T-cell reactivity in the context of allogeneic HSCT. The major advantage of this approach is that the donor T cells permanently acquire sensitivity to the prodrug GCV, allowing donor T-cell alloreactivity to be selectively eliminated without the administration of immunosuppressive drugs that might interfere with the natural process of post-transplant immune reconstitution. In the TK suicide gene/prodrug system, only alloreactive gene-modified T cells that proliferate actively during GVHD are killed by GCV, whereas resting transduced T cells and untransduced cells are spared. TK gene therapy has proven to be safe, with no documented adverse events related to the gene-transfer procedure, including appearance of replication-competent retroviruses or genotoxic effects of vector integration [61–71].
Based on these promising results, the safety and efficacy of suicide gene-modified donor T cells were tested in the more challenging condition of haploidentical HSCT. In this setting, the risk of GvHD is particularly high due to the immunological disparity between donor and host [58, 61]. In a phase I/II, multicenter, non-randomized trial of haploidentical stem-cell transplantation, and retrovirally HSV-TK-transduced donor lymphocytes (TK cells) were administered after transplantation. The primary study endpoint was immune reconstitution, defined as circulating CD3+ count ≥100 cells per μL in two consecutive observations. Fifty patients received haploidentical stem-cell transplants for high-risk leukemia. Immune reconstitution was not recorded before infusion of TK cells. Starting 28 days after transplantation, 28 patients received TK cells; and 22 patients achieved immune reconstitution (median, 75 days from transplantation and 23 days from infusion). Ten patients developed acute GVHD and one developed chronic GVHD, which were controlled by the induction of the suicide gene. Overall survival at 3 years was 49 % for 19 patients who were in remission from primary leukemia at the time of stem-cell transplantation. After TK-cell infusion, the last death due to infection was at 166 days, and this was the only infectious death after day 100. No acute or chronic adverse events related to the gene-transfer procedure were observed [72].
In candidates for haploidentical stem-cell transplantation, infusion of TK cells was considered to be effective in accelerating immune reconstitution, while controlling GVHD and protecting patients from late mortality [72]. TK cells played an active role in supporting a thymic-dependent pathway of immune reconstitution, which resulted in maturation and differentiation of donor hematopoietic precursors in the recipient thymus. This process was especially remarkable, because the cohort consisted of adults with a median age of 51, a stage of life usually characterized by low thymic output. Thus, infusion of genetically modified donor T cells after HSCT can drive recovery of thymic activity in adults, leading to immune reconstitution [61, 72, 73]. The HSV-TK strategy is currently under evaluation in a phase III clinical trial in patients undergoing haploidentical HSCT for high-risk acute leukemia. Preliminary results of this ongoing trial confirmed the potential clinical benefit of the T-cell gene-transfer technology integrated with T-cell depletion haploidentical HSCT, and highlighted the role of early immune reconstitution as a surrogate endpoint for survival outcomes and the dose-related antileukemic effects of TK [58].
An inducible T-cell safety switch based on the fusion of human caspase 9 to a modified human FK-binding protein, allowing conditional dimerization, was recently developed. When exposed to a chemical inducer of dimerization (CID), the inducible caspase 9 is activated, leading to the rapid death of cells expressing this construct [74]. Alloreplete haploidentical T cells expressing the inducible caspase 9 suicide gene could reconstitute immunity post-transplant, and administration of CID could eliminate these cells from both peripheral blood and the central nervous system (CNS), leading to rapid resolution of GVHD and GVHD-associated cytokine release syndrome [75]. The safety and efficiency of repeated CID treatments for persistent or recurring toxicity from T-cell therapies have also been demonstrated [76]. This approach was considered to be useful for the rapid and effective treatment of toxicities associated with infusion of engineered T lymphocytes [75, 76].
In Japan, clinical trials designed to examine the feasibility, safety, and efficacy of donor lymphocyte infusion (DLI) of TK cells were approved in 2002 and 2009. DLI of TK cells was safely performed in a total of eight patients. However, these TK cells disappeared rapidly, probably due to their insufficient in vivo expansion in these patients [77, 78]. A comparison of onset with remission of acute GVHD confirmed that TK cells were predominantly eliminated and that proliferative CD8(+) non-TK cells were also depleted in response to ganciclovir administration. The TCR Vβ-chain repertoire of both TK cells and non-TK cells changed markedly after administration of ganciclovir. In addition, whereas the TCR repertoire of non-TK cells returned to a normal spectratype long after transplantation, that of TK cells remained skewed. With the long-term prophylactic administration of acyclovir, TK cells expanded oligoclonally, and the frequency of splice variants of TK cells increased. Known cancer-associated genes were not evident near the oligoclonally expanded HSV-TK insertion sites [78, 79].
Gene-manipulated leukemia cell vaccines
Both autologous and allogeneic whole tumor cells were clinically developed as another form of cellular vaccine for cancer. The advantage of using the whole tumor-cell approach is that the tumor antigens do not have to be prospectively identified, and multiple antigens can be simultaneously targeted. Cytokine-expressing whole tumor cell approaches, including GM-CSF-expressing cells (GVAX), have been extensively clinically investigated in the context of solid tumors [15, 80]. Here, we briefly discuss clinical trials of GVAX for hematological malignancy.
Although allogeneic HSCT affords durable clinical benefits for many patients with hematologic malignancies, mediated via the GvL effect, patients with high-risk acute myeloid leukemia (AML) or advanced myelodysplasia (MDS) often relapse, underscoring the need to intensify tumor immunity within this cohort. In a phase I clinical trial, high-risk AML or MDS patients were immunized with irradiated, autologous, GM-CSF gene-transduced cells early after allogeneic, and non-myeloablative HSCT. Despite the administration of a calcineurin inhibitor as prophylaxis against GVHD, vaccination elicited local and systemic reactions that were qualitatively similar to those previously observed in non-transplanted, immunized solid-tumor patients. While the frequencies of acute and chronic GVHD were not elevated, nine of ten who completed vaccination achieved durable complete remissions, with a median follow-up of 26 months. Six long-term responders exhibited marked reductions in the levels of soluble NKG2D ligands, and three exhibited normalization of cytotoxic lymphocyte NKG2D expression as a function of treatment. These results established the safety and immunogenicity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic HSCT, and raised the possibility that this combinatorial immunotherapy might potentiate GvL effects in patients [81]. In a phase II study of immune gene therapy, autologous leukemia cells admixed with GM-CSF-secreting K562 cells were administered to adult patients with acute myeloid leukemia. “Primed” lymphocytes were collected after a single pre-transplantation dose of immunotherapy and reinfused with the stem cell graft. Fifty-four subjects were enrolled; 85 % achieved complete remission; and 52 % received pre-transplantation immunotherapy. For all patients who achieved complete remission, the 3-year relapse-free survival (RFS) rate was 47.4 %, and overall survival was 57.4 %. For the 28 immunotherapy-treated patients, the RFS and overall survival rates were 61.8 and 73.4 %, respectively. Post-treatment induction of delayed-type hypersensitivity reactions to autologous leukemia cells was associated with a higher 3-year RFS rate (100 vs 48 %). Minimal residual disease was monitored by the quantitative analysis of Wilms tumor-1 (WT1), a leukemia-associated gene. A decrease in WT1 transcripts in blood was noted in 69 % of patients after the first immunotherapy dose, and was also associated with a higher 3-year RFS (61 vs 0 %). Immunotherapy in combination with primed lymphocytes and autologous stem cell transplantation showed encouraging signs of potential activity in acute myeloid leukemia [82].
A pilot study was performed to determine whether K562/GM-CSF immunotherapy could improve clinical responses to imatinib mesylate (IM) in patients with chronic myeloid leukemia (CML) in cytogenetic but not molecular complete remission. Nineteen patients, with a median duration of previous imatinib mesylate therapy of 37 months, were vaccinated. Mean PCR measurements of BCR-ABL for the group decreased significantly following vaccination. A progressive decline in disease burden was observed in thirteen patients, of whom eight had increasing disease burdens before vaccination. Twelve patients, including seven subjects who became PCR-undetectable, achieved their lowest tumor burden measurements following vaccination. Thus, the K562/GM-CSF vaccine improved molecular responses in patients on imatinib mesylate, including achievement of complete molecular remissions, despite long durations of previous imatinib mesylate therapy [83].
Conclusion
Ex vivo gene therapies have been gradually developing, and seem to lead the gene and cell therapies. The large quantities of information obtained during the translation of preclinical findings regarding ex vivo gene therapies to clinical trials facilitated the development of new modalities for the treatment of congenital intractable diseases and hematological malignancies. Particularly for congenital diseases, newly developed gene-editing technology, excellently reviewed by Voytas and Tolar in this special issue, will unquestionably change current gene therapy approaches using autologous HSCs [84]. In the case of malignancies, the recent development of gene-modified T-cell technologies, including chimeric antigen receptors and immune checkpoint inhibitors, will change the gene therapy modality for treatment of solid tumors as well as hematological malignancies [85, 86].
In Japan, expedited approval system consisting of conditional and time-limited authorizasion based on two laws known as the “The Act on the Safety of Regenerative Medicine” and the “Pharmaceuticals, Medical Devices and Other Therapeutic Products Act” has been introduced to approve new regenerative medical products, including ex vivo gene therapy since 2014. Under the traditional approval process, long-term data collection and evaluation in clinical trials for regenerative medical products were needed. New separate approval process, however, will make it possible for the regenerative, including ex vivo gene therapy medical product obtain conditional, time-limited approval, if an exploratory clinical trial predicts likely clinical benefit [87]. This new act would accelerate the development of ex vivo gene therapy in Japan.
Needless to say, further clarifications of the pathogenesis and molecular mechanisms of intractable congenital disorders and hematological malignancies, as well as further development of gene-transfer technologies, are required to achieve our goal of ideal gene therapy.
References
Rosenberg SA, Aebersold P, Cornetta K, Cornetta K, Kasid A, Morgan RA, Moen R, Karson EM, Lotze MT, Yang JC, Topalian SL, Merino MJ, Culver K, Miller D, Blaese M, Anderson WF. Gene transfer into humans—immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–8.
Blaese RM, Culver KW, Miller AD, Carter SC, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klaine H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF. T-lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 1995;270:475–813.
Ajith TA. Strategies used in the clinical trials of gene therapy for cancer. J Exp Ther Oncol. 2015;11:33–9.
Hall FL, Levy JP, Reed RA, Petchpud WN, Chua VS, Chawla SP, Gordon EM. Pathotropic targeting advances clinical oncology: tumor-targeted localization of therapeutic gene delivery. Oncol Rep. 2010;24:829–33.
Deev RV, Bozo IY, Mzhavanadze ND, Voronov DA, Gavrilenko AV, Chervyakov YV, Staroverov IN, Kalinin RE, Shvalb PG, Isaev AA. pCMV-vegf165 intramuscular gene transfer is an effective method of treatment for patients with chronic lower limb ischemia. J Cardiovasc Pharmacol Ther. 2015;20:473–82.
Gaudet D, Brisson D. Gene-based therapies in lipidology: current status and future challenges. Curr Opin Lipidol. 2015;26:553–65.
Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5:e1115641.
Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–58.
Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–6.
Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Krämer A, Schwäble J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Göhring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kühlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204.
Friedmann T. A new serious adverse event in a gene therapy study. Mol Ther. 2007;15:1899–900.
Williams D. RAC reviews serious adverse event associated with AAV therapy trial. Mol Ther. 2007;15:2053–4.
Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, Bleesing J, Blondeau J, de Boer H, Buckland KF, Caccavelli L, Cros G, De Oliveira S, Fernández KS, Guo D, Harris CE, Hopkins G, Lehmann LE, Lim A, London WB, van der Loo JC, Malani N, Male F, Malik P, Marinovic MA, McNicol AM, Moshous D, Neven B, Oleastro M, Picard C, Ritz J, Rivat C, Schambach A, Shaw KL, Sherman EA, Silberstein LE, Six E, Touzot F, Tsytsykova A, Xu-Bayford J, Baum C, Bushman FD, Fischer A, Kohn DB, Filipovich AH, Notarangelo LD, Cavazzana M, Williams DA, Thrasher AJ. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371:1407–17.
Naldini L. Gene therapy returns to centre stage. Nautre. 2015;526:351–60.
Onodera M, Ariga T, Kawamura N, Kobayashi I, Ohtsu M, Yamada M, Tame A, Furuta H, Okano M, Matsumoto S, Kotani H, McGarrity GJ, Blaese RM, Sakiyama Y. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood. 1998;91:30–6.
Tani K, Azuma M, Nakazaki Y, Oyaizu N, Hase H, Ohata J, Takahashi K, OiwaMonna M, Hanazawa K, Wakumoto Y, Kawai K, Noguchi M, Soda Y, Kunisaki R, Watari K, Takahashi S, Machida U, Satoh N, Tojo A, Maekawa T, Eriguchi M, Tomikawa S, Tahara H, Inoue Y, Yoshikawa H, Yamada Y, Iwamoto A, Hamada H, Yamashita N, Okumura K, Kakizoe T, Akaza H, Fujime M, Clift S, Ando D, Mulligan R, Asano S. Phase I study of autologous tumor vaccines transduced with the GM-CSF gene in four patients with stage IV renal cell cancer in Japan: clinical and immunological findings. Mol Ther. 2004;10:799–816.
Fujiwara T, Tanaka N, Kanazawa S, Ohtani S, Saijo Y, Nukiwa T, Yoshimura K, Sato T, Eto Y, Chada S, Nakamura H, Kato H. Multicenter phase I study of repeated intratumoral delivery of adenoviral p53 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:1689–99.
Strachan T, Read A. Human molecular genetics. 4th ed. New York: Garland Science; 2010.
Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Human Gene Ther. 2015;26:210–9.
Hershfield MS. Genotype is an important determinant of phenotype in adenosine deaminase deficiency. Curr Opin Immunol. 2003;15:571–7.
Kuo CY, Kohn DB. Gene therapy for the treatment of primary immune deficiency. Curr Allergy Asthma Rep. 2016;16(5):39. doi:10.1007/s11882-016-0615-8.
Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, Mazzolari E, Maggioni D, Rossi C, Servida P, Ugazio AG, Mavilio F. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995;270:470–5.
Kohn DB, Weinberg KI, Nolta JA, Heiss LN, Lenarsky C, Crooks GM, Hanley ME, Annett G, Brooks JS, el-Khoureiy A, Lawrence K, Wells S, Moen RC, Bastian J, Williams-Herman DE, Elder M, Wara D, Bowen T, Hershfield MS, Mullen CA, Blaese RM, Parkman R. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med. 1995;1:1017–23.
Hoogerbrugge PM, van Beusechem VW, Fischer A, Debree M, le Deist F, Perignon JL, Morgan G, Gaspar B, Fairbanks LD, Skeoch CH, Moseley A, Harvey M, Levinsky RJ, Valerio D. Bone marrow gene transfer in three patients with adenosine deaminase deficiency. Gene Ther. 1996;3(2):179–83.
Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, Morecki S, Andolfi G, Tabucchi A, Carlucci F, Marinello E, Cattaneo F, Vai S, Servida P, Miniero R, Roncarolo MG, Bordignon C. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–3.
Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, Tabucchi A, Carlucci F, Eibl M, Aker M, Slavin S, Al-Mousa H, Al Ghonaium A, Ferster A, Duppenthaler A, Notarangelo L, Wintergerst U, Buckley RH, Bregni M, Marktel S, Valsecchi MG, Rossi P, Ciceri F, Miniero R, Bordignon C, Roncarolo MG. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–58.
Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, Schurman SH, Garabedian E, Kesserwan C, Jagadeesh GJ, Fu PY, Gschweng E, Cooper A, Tisdale JF, Weinberg KI, Crooks GM, Kapoor N, Shah A, Abdel-Azim H, Yu XJ, Smogorzewska M, Wayne AS, Rosenblatt HM, Davis CM, Hanson C, Rishi RG, Wang X, Gjertson D, Yang OO, Balamurugan A, Bauer G, Ireland JA, Engel BC, Podsakoff GM, Hershfield MS, Blaese RM, Parkman R, Kohn DB. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120(18):3635–46. doi:10.1182/blood-2012-02-400937 (Epub 2012 Sep 11).
Booth C, Gaspar HB, Thrasher AJ. Treating Immunodeficiency through HSC gene therapy. Trends Mol Med. 2016;22:317–27.
Aiuti A, Cassani B, Andolfi G, Mirolo M, Biasco L, Recchia A, Urbinati F, Valacca C, Scaramuzza S, Aker M, Slavin S, Cazzola M, Sartori D, Ambrosi A, Di Serio C, Roncarolo MG, Mavilio F, Bordignon C. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J Clin Invest. 2007;117:2233–40.
Otsu M, Yamada M, Nakajima S, Kida M, Maeyama Y, Hatano N, Toita N, Takezaki S, Okura Y, Kobayashi R, Matsumoto Y, Tatsuzawa O, Tsuchida F, Kato S, Kitagawa M, Mineno J, Hershfield MS, Bali P, Candotti F, Onodera M, Kawamura N, Sakiyama Y, Ariga T. Outcomes in two Japanese adenosine deaminase-deficiency patients treated by stem cell gene therapy with no cytoreductive conditioning. J Clin Immunol. 2015;35:384–98.
Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Exper Opin Biol Ther. 2014;14:789–98.
Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, Thrasher AJ, Wulffraat N, Sorensen R, Dupuis-Girod S, Fischer A, Davies EG, Kuis W, Leiva L, Cavazzana-Calvo M. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185–93.
Chinen J, Davis J, De Ravin SS, Hay BN, Hsu AP, Linton GF, Naumann N, Nomicos EY, Silvin C, Ulrick J, Whiting-Theobald NL, Malech HL, Puck JM. Gene therapy improves immune function in preadolescents with X-linked severe combined immunodeficiency. Blood. 2007;110:67–73.
Ghosh S, Thrasher AJ, Gaspar HB. Gene therapy for monogenic disorders of the bone marrow. Br J Haematol. 2015;. doi:10.1111/bjh.13520 (Epub ahead of print).
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9.
Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debré M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355–64.
Touzot F, Moshous D, Creidy R, Neven B, Frange P, Cros G, Caccavelli L, Blondeau J, Magnani A, Luby JM, Ternaux B, Picard C, Blanche S, Fischer A, Hacein-Bey-Abina S, Cavazzana M. Faster T-cell development following gene therapy compared with haploidentical HSCT in the treatment of SCID-X1. Blood. 2015;125:3563–9.
Takeshita T, Asao H, Ohtani K, Ishii N, Kumaki S, Tanaka N, Munakata H, Nakamura M, Sugamura K. Cloning of the gamma chain of the human IL-2 receptor. Science. 1992;257:379–82.
Malech HL, Maples PB, Whiting-Theobald N, Linton GF, Sekhsaria S, Vowells SJ, Li F, Miller JA, DeCarlo E, Holland SM, Leitman SF, Carter CS, Butz RE, Read EJ, Fleisher TA, Schneiderman RD, Van Epps DE, Spratt SK, Maack CA, Rokovich JA, Cohen LK, Gallin JI. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA. 1997;94:12133–8.
Goebel WS, Dinauer MC. Gene therapy for chronic granulomatous disease. Acta Haematol. 2003;110:86–92.
Chiriaco M, Salfa I, Di Matteo G, Rossi P, Finocchi A. Chronic granulomatous disease: clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. 2015;. doi:10.1111/pai.12527.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kühlcke K, Schilz A, Kunkel H, Naundorf S, Brinkmann A, Deichmann A, Fischer M, Ball C, Pilz I, Dunbar C, Du Y, Jenkins NA, Copeland NG, Lüthi U, Hassan M, Thrasher AJ, Hoelzer D, von Kalle C, Seger R, Grez M. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9.
Bianchi M, Niemiec MJ, Siler U, Urban CF, Reichenbach J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J Allergy Clin Immunol. 2011;127:1243–52.
Kang EM, Choi U, Theobald N, Linton G, Long Priel DA, Kuhns D, Malech HL. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115:783–91.
Kang HJ, Bartholomae CC, Paruzynski A, Arens A, Kim S, Yu SS, Hong Y, Joo CW, Yoon NK, Rhim JW, Kim JG, Von Kalle C, Schmidt M, Kim S, Ahn HS. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther. 2011;19(2092–2101):2011.
Aiuti A, Bacchetta R, Seger R, Villa A, Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies: part 2. Curr Opin Immunol. 2012;24:585–91.
Candotti F. Gene transfer into hematopoietic stem cells as treatment for primary immunodeficiency diseases. Int J Hematol. 2014;99:383–92.
Santilli G, Almarza E, Brendel C, Choi U, Beilin C, Blundell MP, Haria S, Parsley KL, Kinnon C, Malech HL, Bueren JA, Grez M, Thrasher AJ. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther. 2011;19:122–32.
Kawai T, Goto F, Nakazawa Y, Uchiyama T, Watanabe N, Yagita M, Igarashi Y, Mizukami T, Nunoi H, Onodera M. A gene therapy clinical study of a patient with chronic granulomatous disease. In: OR27, page 177, in the abstract of the 21st annual meeting of Japan Society of Gene Therapy. 2015.
Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, Böhm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kühlcke K, Blasczyk R, Kondratenko I, Maródi L, Orange JS, von Kalle C, Klein C. Stem-cell gene therapy for the Wiskott–Aldrich syndrome. N Engl J Med. 2010;363:1918–27.
Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, Modlich U, Beier R, Göhring G, Steinemann D, Fronza R, Ball CR, Haemmerle R, Naundorf S, Kühlcke K, Rose M, Fraser C, Mathias L, Ferrari R, Abboud MR, Al-Herz W, Kondratenko I, Maródi L, Glimm H, Schlegelberger B, Schambach A, Albert MH, Schmidt M, von Kalle C, Klein C. Gene therapy for Wiskott–Aldrich syndrome—long-term efficacy and genotoxicity. Sci Transl Med. 2014;6:227ra33.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science. 2013;341:1233151.
Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, Picard C, Six E, Himoudi N, Gilmour K, McNicol AM, Hara H, Xu-Bayford J, Rivat C, Touzot F, Mavilio F, Lim A, Treluyer JM, Héritier S, Lefrère F, Magalon J, Pengue-Koyi I, Honnet G, Blanche S, Sherman EA, Male F, Berry C, Malani N, Bushman FD, Fischer A, Thrasher AJ, Galy A, Cavazzana M. Outcomes following gene therapy in patients with severe Wiskott–Aldrich syndrome. JAMA. 2015;313:1550–63.
Negre O, Eggimann AV, Beuzard Y, Ribeil JA, Bourget P, Borwornpinyo S, Hongeng S, Hacein-Bey S, Cavazzana M, Leboulch P, Payen E. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-globin gene. Hum Gene Ther. 2016;27:148–65.
Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, Cavallesco R, Gillet-Legrand B, Caccavelli L, Sgarra R, Maouche-Chrétien L, Bernaudin F, Girot R, Dorazio R, Mulder GJ, Polack A, Bank A, Soulier J, Larghero J, Kabbara N, Dalle B, Gourmel B, Socie G, Chrétien S, Cartier N, Aubourg P, Fischer A, Cornetta K, Galacteros F, Beuzard Y, Gluckman E, Bushman F, Hacein-Bey-Abina S, Leboulch P. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–22.
Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrère F, Blanche S, Audit M, Payen E, Leboulch P, l’Homme B, Bougnères P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–23.
Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NA, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, von Kalle C, Bordignon C, Ciceri F, Rovelli A, Roncarolo MG, Aiuti A, Sessa M, Naldini L. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158.
Greco R, Oliveira G, Stanghellini MT, Vago L, Bondanza A, Peccatori J, Cieri N, Marktel S, Mastaglio S, Bordignon C, Bonini C, Ciceri F. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol. 2015;6:95.
Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, Brenner MK, Rooney CM. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–5.
Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–24.
Cieri N, Mastaglio S, Oliveira G, Casucci M, Bondanza A, Bonini C. Adoptive immunotherapy with genetically modified lymphocytes in allogeneic stem cell transplantation. Immunol Rev. 2014;257:165–80.
Munshi NC, Govindarajan R, Drake R, Ding LM, Iyer R, Saylors R, Kornbluth J, Marcus S, Chiang Y, Ennist D, Kwak L, Reynolds C, Tricot G, Barlogie B. Thymidine kinase (TK) gene-transduced human lymphocytes can be highly purified, remain fully functional, and are killed efficiently with ganciclovir. Blood. 1997;89:1334–40.
Tiberghien P, Cahn JY, Brion A, Deconinck E, Racadot E, Hervé P, Milpied N, Lioure B, Gluckman E, Bordigoni P, Jacob W, Chiang Y, Marcus S, Reynolds C, Longo D. Use of donor T-lymphocytes expressing herpes-simplex thymidine kinase in allogeneic bone marrow transplantation: a phase I-II study. Hum Gene Ther. 1997;8:615–24.
Tiberghien P, Ferrand C, Lioure B, Milpied N, Angonin R, Deconinck E, Certoux JM, Robinet E, Saas P, Petracca B, Juttner C, Reynolds CW, Longo DL, Hervé P, Cahn JY. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood. 2001;97:63–72.
Burt RK, Drobyski WR, Seregina T, Traynor A, Oyama Y, Keever-Taylor C, Stefka J, Kuzel TM, Brush M, Rodriquez J, Burns W, Tennant L, Link C. Herpes simplex thymidine kinase gene-transduced donor lymphocyte infusions. Exp Hematol. 2003;31:903–10.
Fehse B, Ayuk FA, Kröger N, Fang L, Kühlcke K, Heinzelmann M, Zabelina T, Fauser AA, Zander AR. Evidence for increased risk of secondary graft failure after in vivo depletion of suicide gene-modified T lymphocytes transplanted in conjunction with CD34+-enriched blood stem cells. Blood. 2004;104:3408–9.
Ciceri F, Bonini C, Marktel S, Zappone E, Servida P, Bernardi M, Pescarollo A, Bondanza A, Peccatori J, Rossini S, Magnani Z, Salomoni M, Benati C, Ponzoni M, Callegaro L, Corradini P, Bregni M, Traversari C, Bordignon C. Antitumor effects of HSV-TK-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood. 2007;109:4698–707.
Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, Bonini C, Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–15.
Mercier-Letondal P, Deschamps M, Sauce D, Certoux JM, Milpied N, Lioure B, Cahn JY, Deconinck E, Ferrand C, Tiberghien P, Robinet E. Early immune response against retrovirally transduced herpes simplex virus thymidine kinase-expressing gene-modified T cells coinfused with a T cell-depleted marrow graft: an altered immune response? Hum Gene Ther. 2008;19:937–50.
Borchers S, Provasi E, Silvani A, Radrizzani M, Benati C, Dammann E, Krons A, Kontsendorn J, Schmidtke J, Kuehnau W, von Neuhoff N, Stadler M, Ciceri F, Bonini C, Ganser A, Hertenstein B, Weissinger EM. Genetically modified donor leukocyte transfusion and graft-versus-leukemia effect after allogeneic stem cell transplantation. Hum Gene Ther. 2011;22:829–41.
Weissinger EM, Borchers S, Silvani A, Provasi E, Radrizzani M, Beckmann IK, Benati C, Schmidtke J, Kuehnau W, Schweier P, Luther S, Fernandez-Munoz I, Beutel G, Ciceri F, Bonini C, Ganser A, Hertenstein B, Stadler M. Long term follow up of patients after allogeneic stem cell transplantation and transfusion of HSV-TK transduced T-cells. Front Pharmacol. 2015;6:76.
Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, Turchetto L, Colombi S, Bernardi M, Peccatori J, Pescarollo A, Servida P, Magnani Z, Perna SK, Valtolina V, Crippa F, Callegaro L, Spoldi E, Crocchiolo R, Fleischhauer K, Ponzoni M, Vago L, Rossini S, Santoro A, Todisco E, Apperley J, Olavarria E, Slavin S, Weissinger EM, Ganser A, Stadler M, Yannaki E, Fassas A, Anagnostopoulos A, Bregni M, Stampino CG, Bruzzi P, Bordignon C. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10:489–500.
Vago L, Oliveira G, Bondanza A, Noviello M, Soldati C, Ghio D, Brigida I, Greco R, Lupo Stanghellini MT, Peccatori J, Fracchia S, Del Fiacco M, Traversari C, Aiuti A, Del Maschio A, Bordignon C, Ciceri F, Bonini C. T-cell suicide gene therapy prompts thymic renewal in adults after hematopoietic stem cell transplantation. Blood. 2012;120(9):1820–30.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83.
Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, Durett AG, Dakhova O, Savoldo B, Di Stasi A, Spencer DM, Lin YF, Liu H, Grilley BJ, Gee AP, Rooney CM, Heslop HE, Brenner MK. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125:4103–13.
Zhou X, Naik S, Dakhova O, Dotti G, Heslop HE, Brenner MK. Serial activation of the inducible caspase 9 safety switch after human stem cell transplantation. Mol Ther. 2016;24:823–31.
Onoder M. Gene and cell therapy for relapsed leukemia after allo-stem cell transplantation. Front Biosci. 2008;13:3408–14.
Hashimoto H, Kitano S, Ueda R, Ito A, Tada K, Fuji S, Yamashita T, Tomura D, Nukaya I, Mineno J, Fukuda T, Mori S, Takaue Y, Heike Y. Infusion of donor lymphocytes expressing the herpes simplex virus thymidine kinase suicide gene for recurrent hematologic malignancies after allogeneic hematopoietic stem cell transplantation. Int J Hematol. 2015;102:101–10.
Hashimoto H, Kitano S, Yamagata S, Miyagi Maeshima A, Ueda R, Ito A, Tada K, Fuji S, Yamashita T, Tomura D, Nukaya I, Mineno J, Fukuda T, Mori S, Takaue Y, Heike Y. Donor lymphocytes expressing the herpes simplex virus thymidine kinase suicide gene: detailed immunological function following add-back after haplo-identical transplantation. Cytotherapy. 2015;17:1820–30.
Le DT, Pardoll DM, Jaffee EM. Cellular vaccine approaches. Cancer J. 2010;16:304–10.
Ho VT, Vanneman M, Kim H, Sasada T, Kang YJ, Pasek M, Cutler C, Koreth J, Alyea E, Sarantopoulos S, Antin JH, Ritz J, Canning C, Kutok J, Mihm MC, Dranoff G, Soiffer R. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc Natl Acad Sci USA. 2009;106:15825–30.
Borrello IM, Levitsky HI, Stock W, Sher D, Qin L, DeAngelo DJ, Alyea EP, Stone RM, Damon LE, Linker CA, Maslyar DJ, Hege KM. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood. 2009;114:1736–45.
Smith BD, Kasamon YL, Kowalski J, Gocke C, Murphy K, Miller CB, Garrett-Mayer E, Tsai HL, Qin L, Chia C, Biedrzycki B, Harding TC, Tu GH, Jones R, Hege K. Levitsky HI.K562/GM-CSF immunotherapy reduces tumor burden in chronic myeloid leukemia patients with residual disease on imatinib mesylate. Clin Cancer Res. 2010;16:338–47.
Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol Ther. 2016;24(430–446):2016.
Fujiwara H. Adoptive T-cell therapy for hematological malignancies using T cells gene-modified to express tumor antigen-specific receptors. Int J Hematol. 2014;99:123–31.
Rivera GA, Saramipoor Behbahan I, Greenberg PL. Immune checkpoint pathways: perspectives on myeloid malignancies. Leuk Lymphoma. 2016;57:995–1001.
Konomi K, Tobita M, Kimura K, Sato D. New Japanese initiatives on stem cell therapies. Cell Stem Cell. 2015;16:350–2.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Tani, K. Current status of ex vivo gene therapy for hematological disorders: a review of clinical trials in Japan around the world. Int J Hematol 104, 42–72 (2016). https://doi.org/10.1007/s12185-016-2030-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2030-2