
Abstract
Gene transfer into the hematopoietic stem cell has shown curative potential for a variety of hematological disorders. Primary immunodeficiency diseases have led to the way in this field of gene therapy as an example and a model. Clinical results from the past 15 years have shown that significant improvement and even cure can be achieved for diseases such as X-linked severe combined immunodeficiency, adenosine deaminase deficiency, chronic granulomatous disease and Wiskott–Aldrich syndrome. Unfortunately, with the initial clear clinical benefits, the first serious complications of gene therapy have also occurred. In a significant number of patients treated using vectors based on murine gamma-retroviruses and carrying powerful viral enhancer elements, insertional oncogenesis events have resulted in acute leukemias that, in some cases, have had fatal outcomes. These serious adverse events have sparked a revision of the assessment of risks and benefits of integrating gene transfer for hematological diseases and prompted the development and application of new generations of viral vectors with recognized superior safety characteristics. This review summarizes the clinical experience of gene therapy for primary immunodeficiencies and discusses the likely avenues of progress in the future development of this expanding field of clinical investigations.
Similar content being viewed by others

Introduction
In the past two decades, technical progress has allowed gene transfer into hematopoietic stem cells (HSCs) to reach levels of efficiency compatible with clinical benefit for an increasing number of human conditions, including hemoglobinopathies, primary immunodeficiencies (PIDs), and degenerative neurological disorders [1–10]. PIDs have played a major role in this process as they have provided the first pieces of evidence that gene therapy can be curative for inherited genetic diseases [11, 12]. A series of advantageous characteristics have made PIDs initial attractive target diseases for gene therapy. First, they are often curable by allogeneic hematopoietic stem cell transplantation (HCT), which provides a testing platform for the prospects of efficacy of treatments based on transplantation of autologous, gene-corrected HSCs. In addition, the target HSCs are readily accessible and amenable to in vitro culture and ex vivo gene transfer, a procedure that currently offers the best chance of success. Finally, for several PIDs, gene-corrected progenitors and mature cells are expected to show survival advantage over the unmodified, affected cell populations, which translates into the possibility that even low gene transfer efficiency may have therapeutic effects.
At the same time, performing gene therapy for PIDs involves a series of specific difficulties, starting with the challenging task of targeting the HSC. These cells are extremely infrequent in the hematopoietic tissues and their specific cellular phenotype is yet undefined, thus making their collection and enrichment laborious and inefficient. In addition, the biological process of differentiation of HSCs into mature hematopoietic progenies is characterized by several rounds of cell divisions, during which the transferred gene needs to be duplicated and passed on to daughter cells. This necessity requires stable integration of the therapeutic gene in the host genome, which can be achieved with current technologies, but that also exposes target cells to insertional oncogenesis complications. Finally, gene transfer into HSCs is facilitated by active cell division, which, however, can also induce cell differentiation and loss of long-term repopulating ability. The identification of appropriate combinations of cytokines and growth factors [e.g., interleukin (IL)-3, IL-6, stem cell factor (SCF), thrombopoietin, and Flt-3 ligand], together with culture supports such as fibronectin, has resulted in major improvements in our ability to efficiently introduce genes into HSCs in large animal models [13, 14] that have translated successfully in the first successful clinical protocols for lethal forms of PIDs, such as X-linked severe combined immunodeficiency (SCID) and adenosine deaminase (ADA) deficiency [11, 12]. As anticipated, the notions gathered by the early experimentations in PIDs have posed the bases for extending gene correction procedures to more frequent genetic disorders of hematopoiesis, such as thalassemia [3], and are expected to be applied in the near future to a variety of common disease, including malignancies and HIV infection.
Results of recent gene therapy trials for PIDs
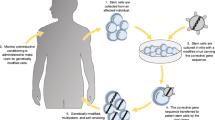
A general design for a gene therapy trial for PIDs is illustrated in Fig. 1 and, briefly, it includes collection of HSCs through bone marrow harvest or leukoapheresis after pharmacological mobilization with granulocyte colony-stimulating factor (G-CSF). HSCs are usually enriched by recovery of CD34-expressing cells that are then cultured in vitro in the presence of cytokines and growth factors, such as stem cells factor (SCF), thrombopoietin (TPO), interleukin-3 (IL-3), and flt-3 ligand. Activated cells are exposed to viral gene transfer vectors that are able to stably integrate their genome into the target cell DNA and subsequently infused intravenously to the patients, who may or may not have previously received myeloreductive chemotherapy, in most cases with busulfan [5, 8, 9, 11, 12, 15–18].

Schematic representation of a gene therapy protocol for PIDs. Bone marrow cells are collected from patients and CD34+ hematopoietic progenitors enriched by immune-magnetic separation. Enriched progenitors are cultured in the presence of cytokines and growth factors before being exposed to viral vector preparations. After several cycles of transduction, gene-corrected cells are collected for infusion to the patients who may/may not have received prior myelo-reduction chemotherapy
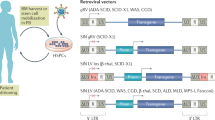
One of the important distinctive features of any clinical trial is the type of viral vector used (Fig. 2). Vectors based on murine gamma-retroviruses were used in the most successful protocols to date. These vectors, however, have led to the occurrence of hematological malignancies in several X-SCID, CGD and Wiskott–Aldrich syndrome (WAS) patients treated with gene therapy [19–22], which has prompted the field to introduce safety modifications in these constructs [23] or adopt vectors based on different retroviruses with increased safety profile, such as HIV-1 and Foamy virus [9, 24].

Schematic representation of viral transfer vectors used for gene therapy of PIDs: GCsapM-ADA gamma-retroviral vector [8], the SRS11 EFS γc self-inactivating gamma-retroviral vector [23], and the CL20-I4-EF1a-hgc-OPT HIV-1-based lentiviral vector [58]. LTR long terminal repeat; 3 U3 LTR region, R R LTR region, 5 U5 LTR region, hADA human ADA cDNA, SFFV spleen focus-forming virus, Δ3 U3 LTR region deleted of promoter and enhancer sequences, EF1α promoter of elongation factor 1-α gene, hγc human common γc cDNA, WPRE woodchuck hepatitis virus post-transcriptional regulatory element, HIV-1 human immunodeficiency virus type-1, Ins 400-bp insulator fragment from the chicken β-globin locus, hγc-OPT codon optimized human common γc cDNA
Results of gene therapy trials for ADA deficiency
Genetic deficiency of ADA results in an extreme reduction of lymphocyte numbers and impairment of immune functions that can lead to SCID and early death of affected individuals from overwhelming infections [25]. HCT and enzyme replacement therapy (ERT) are available forms of treatment for this disease, but each has drawbacks that limit their efficacy [26] [27]. For these reasons, in the mid-1980s, ADA deficiency was identified as an ideal candidate disorder for trials of gene therapy. A series of pioneering clinical trials explored the feasibility of gamma-retroviral vector-mediated transfer of the ADA cDNA sequence into patients’ peripheral blood T lymphocytes [28–33], bone marrow or cord blood HSCs [34, 35], but failed at providing long-term improvements of the disease in treated patients.
The first conclusive evidence that gene therapy can cure ADA deficiency was obtained when the experimental protocols were changed to add steps aimed at increasing the initial advantage of gene-corrected HSC. These included the administration of mild myeloreductive chemotherapy (e.g., 4 mg/kg Busulfan or 140 mg/m2 Melphalan) and the withholding of ERT. For Aiuti and collaborators [1, 12] in Italy, this approach revealed extremely effective with nine out of ten treated patients showing immune reconstitution with increases in T-cell counts and normalization of T-cell function, five patients demonstrating restoration of B-cell function after vaccination, and eight patients remaining off ERT in the long term.
While the Italian trial was ongoing, our own investigations were proceeding in a two-site trial (Children’s Hospital Los Angeles and NIH) that aimed at comparing the relative efficacy of two different gamma-retroviral vectors. In the first phase of this trial, patients were maintained on ERT and no chemotherapy was administered, which resulted in long-term, low-level marking in two of four treated patients, but no immunological improvement. In its second phase, the trial involved low-dose busulfan chemotherapy (75–90 mg/m2) and withdrawal of ERT. Six patients were enrolled, with three achieving adequate immune reconstitution. These results demonstrated that the use of reduced-intensity conditioning favored engraftment of gene-modified stem cells and the generation of ADA-expressing lymphocytes, which, in turn, resulted in immune reconstitution [8].
Concomitantly, a similar gene therapy trial was conducted in the UK and enrolled six patients who were treated following withdrawal of ERT and administration of nonmyeloablative chemotherapy with melphalan (140 mg/m2) or busulfan (4 mg/kg). In two of these patients, PEG-ADA was restarted due to the absence of measurable gene marking in peripheral blood cells. The other patients showed increase in T-cell and B-cell numbers, with normalization of in vitro lymphocyte responses and adequate immunoglobulin production in three patients [7, 16].
In summary, based on cumulative data from more than 40 ADA deficient patients treated with gene therapy, it can be concluded that gene therapy approaches can be effective and to date maintain a record of safety. We have also learned that, in this disease, selective advantage of gene-corrected cells alone is not sufficient to provide immune reconstitution in the absence of preparative chemotherapy. This observation is corroborated by the results of a Japanese clinical trial started in 2003 in which two patients who received gene-corrected CD34+ bone marrow cells after discontinuation of PEG-ADA in the absence of chemotherapy did not achieve full immune reconstitution [36].
Future applications of gene therapy for ADA deficiency will use vectors based on HIV-1, which are expected to afford more efficient and safer gene transfer into HSC.
Results of gene therapy trials for chronic granulomatous disease
Genetic defects affecting the expression of the gp91phox, p22phox, p47phox, or p67phox molecules can cause the impaired superoxide production in phagocytic cells that is characteristic of CGD and that can result in severe and life-threatening abscesses and/or granuloma formations in the skin, liver, lungs, or bone of affected patients. Current management options include antibiotic prophylaxis, administration of IFN-γ, and HCT [37]. A gene therapy option, however, would be a beneficial additional alternative for many patients.
Early clinical trials were performed by the groups of Malech at the NIH and Dinauer at the University of Indiana Medical School. In these trials, gamma-retroviral vectors were used to transfer the p47phox or gp91phox cDNA sequences into G-CSF-mobilized peripheral blood CD34+ hematopoietic progenitors that were then reinfused into the patients without prior myelosuppression. A total of eleven patients were treated under these studies that unfortunately only resulted in the transitory functional correction of ≤0.5 % of peripheral blood granulocytes [38–40].
More recent trials have introduced nonmyeloablative conditioning. In one trial, the use of low-dose busulfan (10 mg/kg) prior to infusion of gamma-retroviral vector-transduced, mobilized CD34+ cells in three X-CGD patients resulted in 4–24 % of functionally neutrophils. However, the effects remained detectable in the long term at levels of around 1 % in only one subject [41].
A trial performed in Germany in 2004 used a gamma-retroviral vector expressing gp91phox under the transcriptional control of the spleen focus-forming virus LTR and targeted G-CSF-mobilized CD34+ progenitor cells of two patients with X-CGD who received 8 mg/kg of busulfan before the infusion of gene-corrected cells. Around 15 % of neutrophils were found to be functionally corrected early after treatment. This fraction increased due to insertional activation of the PRDM16 and MDS1/EVI1 genes in clonal cell populations that expanded with time. Ultimately, epigenetic inactivation of the vector gradually reduced the NAPDH-positive neutrophils to less than 5 % by 1–2 years after treatment. Unfortunately, however, activation of the EVI1 gene due to retroviral insertion resulted in myelodysplasia with monosomy 7 that had lethal complications for both treated patients [17, 21, 42]. The same protocol was used in Switzerland to enroll a X-CGD boy with severe Aspergillus infection. NAPDH-positive neutrophils were detected at levels up to 30 %, and the procedure resulted in eradication of the fungal infection [43]. Unfortunately, also this patient developed MDS and has undergone allogeneic stem cell transplantation [42].
Another gene therapy trial using a gamma-retroviral vector was conducted in South Korea and enrolled two X-CGD patients. Mobilized peripheral blood CD34+ cells were transduced and infused after a conditioning regimen of fludarabine and busulfan. The level of NAPDH-positive cells reached 6–14 % of neutrophils, but the correction was short term and declined to less than 1 % in both patients 3 years after treatment [44].
Additional, unpublished experience of gene therapy for CGD has been generated in the UK where conditioning with melphalan (140 mg/m2) and either the vector employed in the German trial or the NIH construct were used. Similar to other trials, only in the short term, low levels of gp91phox expression were detected in treated patients [45].
In summary, the current experience with gene therapy of CGD points to a yet unexplained difficulty in achieving long-term engraftment of significant levels of transduced cells. The lack of a strong selective advantage of gene-corrected populations in this disease may play a major role and may indicate that more significant levels of HSC transduction and engraftment will be needed to obtain clinical benefit.
More efficient methods of gene transfer are also being developed based on HIV-1 vectors [46, 47] that are hoped to improve the outcome and safety of future clinical trials.
Results of gene therapy trials for X-linked severe combined immunodeficiency
Mutations of the common gamma chain (γc) of the receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 cause X-SCID, a profound immunodeficiency with a combined impairment of T- and B-cell immunity. HCT can be curative for affected boys; however, especially in the case of haploidentical donors, transplanted patients often achieve only partial chimerism of hematopoietic lineages, with persistent impairment of humoral immune function [48]. Such unsatisfactory results have fostered the development of gene therapy approaches for this disease.
Based on successful preclinical in vivo experiments in mouse models of X-SCID [49–53], the first gene therapy trial for X-SCID, launched in 1999 in France, followed by a very similar experimentation carried out in the UK. Both trials involved gamma-retroviral γc gene transfer into CD34+ bone marrow cells harvested from X-SCID patients who lacked HLA-identical bone marrow donors. Ten typical X-SCID patients were treated at each site. The results from these trials have been impressive for the quick kinetics of reconstitution of normal levels of circulating, γc-expressing, polyclonal and functional T lymphocytes in the majority of treated patients. Improvement of humoral immunity was also observed allowing discontinuation of prophylactic immunoglobulin replacement treatment in eleven of the twenty patients. Although the restoration was incomplete with regard to the numbers of gene-corrected B and NK cells, the effects of the treatment were sufficient to provide protective immunity and to allow most patients to return home and lead a normal life [4, 6, 11, 15, 54, 55]. Altogether, these data demonstrate that gene therapy can be curative for X-SCID with long-lasting (>10 years) beneficial effects.
Unfortunately, four patients in the French trial and one patient in the British cohort developed T-cell leukemia between 2 and 5 years after gene therapy. In all cases, the adverse event was the result of insertional oncogenesis due to the aberrant expression of the LMO2 (LIM domain only-2) or CCND2 (cyclin D2) oncogenes induced by the integration of the γc retroviral vector in the proximity of the gene regulatory regions. Conventional treatment for T acute lymphoblastic leukemia was administered, in one case followed by matched unrelated HCT. Despite this aggressive therapy, one patient succumbed to the disease, while the others remain in remission up to 10 years after the occurrence of the severe adverse events [19, 20, 56].
One additional trial was begun at the NIH in 2003 to offer a rescue treatment option for older X-SCID patients who had failed to respond to HCT. Three patients (11, 10, and 14 years old) were enrolled in this trial that used a gamma-retroviral vector and targeted G-CSF-mobilized peripheral blood CD34+ cells. T-cell numbers and function significantly improved in the youngest subject, but no immunological improvement was observed in the other two subjects, perhaps due to age-dependent loss of thymic function [18].
Our understanding of the mechanisms leading to LMO2- and CCND2-mediated lymphoproliferation in the first French and British trials remains incomplete. However, it is widely accepted that the enhancer activity of the gamma-retroviral vector promoter was responsible for the activation of the LMO2 and CCND2 promoters and consequent deregulated LMO2 and CCND2 gene expression. Vigorous cell proliferation of LMO2- and CCND2-expressing cells due to the selective advantage conferred to them by the concomitant expression of the γc gene may have provided a “second hit” and favored secondary changes leading to the onset of the overt malignancies.
Alternative gene transfer constructs have been developed to improve the safety of gene therapy for X-SCID. An enhancer-less gamma-retroviral vector was found to be effective in the mouse model of X-SCID [23] and is used in a consortium study including centers in London, Paris, Boston, Cincinnati, and Los Angeles that had enrolled nine patients as of December 2013. Data published in abstract form indicate that 5 of 7 evaluable patients achieved significant numbers of corrected, diverse and functional circulating T lymphocytes. The kinetics of reconstitution appear similar to the earlier French and British trials. Importantly, preliminary analysis of retroviral integration sites does not show clustering near LMO-2, EVI1 or other lymphoid oncogenes [57]. An alternative vector based on HIV-1 has been developed by Sorrentino and colleagues [58] and is used in a two-site clinical trial open at the St. Jude Children’s Research Center in Memphis, where typical X-SCID patients will be enrolled, and at NIH, where atypical older patients are treated. The latter arm of the trial uses nonmyeloablative conditioning to improve the efficacy of engraftment of gene-corrected cells and has enrolled one patient with encouraging preliminary results [59].
Results of gene therapy trials for WAS
Wiskott–Aldrich syndrome is a multifaceted disorder that, in its severe presentation, is characterized by eczema, thrombocytopenia, recurrent infections, autoimmune disorders, and a high incidence of lymphomas. Abnormalities of lymphoid and myeloid cell function contribute to the often heterogeneous and medically challenging clinical presentation of affected patients [60]. The unsatisfactory results of haploidentical HCT in WAS patients [48, 61–63] justify efforts to test the safety and efficacy of gene therapy for this disease.
In vivo models of gene therapy using Was gene knockout mice have shown that T-cell numbers and responses can be improved in animals treated with different viral vector-mediated gene correction systems [24, 64–71]. In addition, competitive repopulation experiments demonstrated that wild-type cells have a selective advantage of over Was knockout populations [72], which is consistent with the observations of in vivo selective survival advantage of revertant cells in WAS patients [73–75] and suggests that gene-corrected cells could similarly have a selective advantage over unmodified populations after gene therapy.
The first clinical trial of gene therapy for WAS started in Germany in 2006 and used a gamma-retroviral vector to transduce mobilized peripheral blood CD34+ cells of ten patients who received conditioning with busulfan (8 mg/kg) prior to infusion of gene-corrected cells. Nine of ten treated subjects showed a significant increase of platelet counts and normalization of T-cell numbers and function, NK-cell function, immunoglobulin production, and responses to vaccines. Patients also markedly improved their clinical status, including bleeding tendency, susceptibility to infections, and autoimmunity. Unfortunately, insertional oncogenesis mediated by LMO-2 activation occurred in five patients and resulted in T-cell leukemia. In addition, one subject developed acute myeloid leukemia [5, 76]. Therefore, while gamma-retroviral vector-mediated gene therapy can correct WAS, it seems to carry an unacceptable level of risk for application in this disease.
More recently, a multicenter gene therapy trial opened in Milan, London, Paris, and Boston using the HIV-based gene transfer construct extensively tested in the mouse models described above [77]. The results from the first series of three patients treated in Milan have recently been published. Similar to the German trial, improvement of platelet counts, immune function and clinical manifestations of the disease was observed at 1+ year after gene therapy. Importantly, however, comparison of retroviral and lentiviral vector integration sites in samples from the German and Italian studies showed lack of overrepresentation of sites targeting oncogenes in the Italian patient group, while demonstrating early enrichment of oncogenic targets in patients from the German trial [9]. While long-term observation is warranted to confirm the superior safety of lentiviral gene transfer, it is hoped that use of this class of vectors will allow to capitalize on the potential of gene therapy as an alternative treatment option for WAS, while protecting patients from the occurrence of leukemic adverse events.
Future directions and concluding remarks
Preclinical experiments are underway for a number of other forms of PID that would benefit from gene therapy approaches. Several of such diseases represent significant challenges and will require additional progress beyond the current available technologies (Table 1). Ultimately, a general shift from the current “gene addition” approaches to “gene editing” strategies will be required and some of these efforts have been already tested on PID models, such as the early demonstrations of the IL2RG gene repair with zinc-finger nuclease technology [78] and the more recent development of site-directed gene addition strategies for CGD in induced pluripotent stem cells (iPSc) [79]. The current successes obtained in the field of PIDs indicate a sign of maturity of the field of gene therapy where clinical experimentation is evolving from anectodal reports to execution of sound empirical studies informed by previous clinical results. While still distant from the gold standard of randomized studies, the lively activity that is witnessed in this area of gene therapy will undoubtedly continue to foster the progress of this form of clinical investigation.
References
Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–58.
Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–23.
Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–22.
Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355–64.
Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, et al. Stem-cell gene therapy for the Wiskott–Aldrich syndrome. N Engl J Med. 2010;363:1918–27.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3:97ra79.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3:97ra80.
Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635–46.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science. 2013. doi:10.1126/science.1233151 (Epub ahead of print).
Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158.
Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–72.
Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–3.
Kiem HP, Andrews RG, Morris J, Peterson L, Heyward S, Allen JM, et al. Improved gene transfer into baboon marrow repopulating cells using recombinant human fibronectin fragment CH-296 in combination with interleukin-6, stem cell factor, FLT-3 ligand, and megakaryocyte growth and development factor. Blood. 1998;92:1878–86.
Tisdale JF, Hanazono Y, Sellers SE, Agricola BA, Metzger ME, Donahue RE, et al. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood. 1998;92:1131–41.
Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181–7.
Gaspar HB, Bjorkegren E, Parsley K, Gilmour KC, King D, Sinclair J, et al. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14:505–13.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9.
Chinen J, Davis J, De Ravin SS, Hay BN, Hsu AP, Linton GF, et al. Gene therapy improves immune function in preadolescents with X-linked severe combined immunodeficiency. Blood. 2007;110:67–73.
Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–42.
Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–50.
Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204.
Witzel M, Braun CJ, Boztug K, Schmidt M, Albert M, Schwarzer A, et al. Hematopoietic stem cell gene therapy for Wiskott–Aldrich syndrome. J Clin Immunol. 2012; 32:9.
Thornhill SI, Schambach A, Howe SJ, Ulaganathan M, Grassman E, Williams D, et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol Ther. 2008;16:590–8.
Uchiyama T, Adriani M, Jagadeesh GJ, Paine A, Candotti F. Foamy virus vector-mediated gene correction of a mouse model of Wiskott–Aldrich syndrome. Mol Ther. 2012;20:1270–9.
Candotti F, Grunebaum E, Roifman C, Hirschhorn R. Immunodeficiency due to defects of purine metabolism. In: Ochs HD, Smith CIE, Puck JM, editors. Primary immunodeficiency diseases: a molecular and genetic approach. 3rd ed. New York City: Oxford; 2014 (in press).
Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114:3524–32.
Hassan A, Booth C, Brightwell A, Allwood Z, Veys P, Rao K, et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012;120:3615–24 (quiz 26).
Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 1995;270:475–80.
Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA-immunodeficient patients. Science. 1995;270:470–5.
Onodera M, Ariga T, Kawamura N, Kobayashi I, Ohtsu M, Yamada M, et al. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood. 1998;91:30–6.
Misaki Y, Ezaki I, Ariga T, Kawamura N, Sakiyama Y, Yamamoto K. Gene-transferred oligoclonal T cells predominantly persist in peripheral blood from an adenosine deaminase-deficient patient during gene therapy. Mol Ther. 2001;3:24–7.
Aiuti A, Vai S, Mortellaro A, Casorati G, Ficara F, Andolfi G, et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med. 2002;8:423–5.
Muul LM, Tuschong LM, Soenen SL, Jagadeesh GJ, Ramsey WJ, Long Z, et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101:2563–9.
Hoogerbrugge PM, van Beusechem VW, Fischer A, Debree M, Le Deist F, Perignon JL, et al. Bone marrow gene transfer in three patients with adenosine deaminase deficiency. Gene Ther. 1996;3:179–83.
Kohn DB, Weinberg KI, Nolta JA, Heiss LN, Lenarsky C, Crooks GM, et al. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med. 1995;1:1017–23.
Otsu M, Onodera M, Yamada M, Kawamura N, Kobayashi R, Kobayashi E, et al. Update on a Japanese clinical trial of stem cell gene therapy for ADA-deficiency. Hum Gene Ther. 2010;21:1437.
Kang EM, Marciano BE, DeRavin S, Zarember KA, Holland SM, Malech HL. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2011;127:1319–26 (quiz 27–8).
Malech HL, Sekhsaria S, Whiting-Theobald N, Linton GL, Vowells SJ, Li F, et al. Prolonged detection of oxidase-positive neutrophils in the peripheral blood of five patients following a single cycle of gene therapy for chronic granulomatous disease. Blood. 1996;88:486a.
Malech HL, Horwitz ME, Linton GF, Theobald-Whiting N, Brown MR, Farrell CJ, et al. Extended production of oxidase normal neutrophils in X-linked chronic granulomatous disease (CGD) following gene therapy with gp91(phox) transduced CD34+ cells. Blood. 1998;92:690A.
Goebel WS, Dinauer MC. Gene therapy for chronic granulomatous disease. Acta Haematol. 2003;110:86–92.
Kang EM, Choi U, Theobald N, Linton G, Long Priel DA, Kuhns D, et al. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115:783–91.
Aiuti A, Bacchetta R, Seger R, Villa A, Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies: part 2. Curr Opin Immunol. 2012;24:585–91.
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–22.
Kang HJ, Bartholomae CC, Paruzynski A, Arens A, Kim S, Yu SS, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther. 2011;19:2092–101.
Grez M, Reichenbach J, Schwable J, Seger R, Dinauer MC, Thrasher AJ. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther. 2011;19:28–35.
Naumann N, De Ravin SS, Choi U, Moayeri M, Whiting-Theobald N, Linton GF, et al. Simian immunodeficiency virus lentivector corrects human X-linked chronic granulomatous disease in the NOD/SCID mouse xenograft. Gene Ther. 2007;14:1513–24.
Santilli G, Almarza E, Brendel C, Choi U, Beilin C, Blundell MP, et al. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther. 2011;19:122–32.
Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet. 2003;361:553–60.
Lo M, Bloom ML, Imada K, Berg M, Bollenbacher JM, Bloom ET, et al. Restoration of lymphoid populations in a murine model of X-linked severe combined immunodeficiency by a gene-therapy approach. Blood. 1999;94:3027–36.
Otsu M, Anderson SM, Bodine DM, Puck JM, O’Shea JJ, Candotti F. Lymphoid development and function in X-linked severe combined immunodeficiency mice after stem cell gene therapy. Mol Ther. 2000;1:145–53.
Soudais C, Shiho T, Sharara LI, Guy-Grand D, Taniguchi T, Fischer A, et al. Stable and functional lymphoid reconstitution of common cytokine receptor gamma chain deficient mice by retroviral-mediated gene transfer. Blood. 2000;95:3071–7.
Otsu M, Sugamura K, Candotti F. Lack of dominant-negative effects of a truncated gamma(c) on retroviral-mediated gene correction of immunodeficient mice. Blood. 2001;97:1618–24.
Aviles Mendoza GJ, Seidel NE, Otsu M, Anderson SM, Simon-Stoos K, Herrera A, et al. Comparison of five retrovirus vectors containing the human IL-2 receptor gamma chain gene for their ability to restore T and B lymphocytes in the X-linked severe combined immunodeficiency mouse model. Mol Ther. 2001;3:565–73.
Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185–93.
Ginn SL, Curtin JA, Kramer B, Smyth CM, Wong M, Kakakios A, et al. Treatment of an infant with X-linked severe combined immunodeficiency (SCID-X1) by gene therapy in Australia. Med J Aust. 2005;182:458–63.
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9.
Hacein-Bey-Abina S, Bushman F, Armant M, Blanche S, Blondeau J, Caccavelli L, et al. Immune reconstitution and preliminary safety analysis of 9 patients treated with somatic gene therapy for X-linked severe combined immunodeficiency (SCID-X1) with a self-inactivating gammaretroviral vector. Blood. 2013;122:715.
Zhou S, Mody D, DeRavin SS, Hauer J, Lu T, Ma Z, et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood. 2010;116:900–8.
De Ravin SS, Choi U, Theobald N, Lee J, Wang HM, Wu XL, et al. Lentiviral gene transfer for treatment of children >2 years old with x-linked severe combined immunodeficiency. Mol Ther. 2013; 21:S118.
Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia Roncarolo M. Recent advances in understanding the pathophysiology of Wiskott–Aldrich syndrome. Blood. 2009;113:6288–95.
Filipovich AH, Stone JV, Tomany SC, Ireland M, Kollman C, Pelz CJ, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott–Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood. 2001;97:1598–603.
Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott–Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111:439–45.
Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott–Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood. 2011;118:1675–84.
Klein C, Nguyen D, Liu CH, Mizoguchi A, Bhan AK, Miki H, et al. Gene therapy for Wiskott–Aldrich syndrome: rescue of T-cell signaling and amelioration of colitis upon transplantation of retrovirally transduced hematopoietic stem cells in mice. Blood. 2003;101:2159–66.
Strom TS, Turner SJ, Andreansky S, Liu H, Doherty PC, Srivastava DK, et al. Defects in T-cell-mediated immunity to influenza virus in murine Wiskott–Aldrich syndrome are corrected by oncoretroviral vector-mediated gene transfer into repopulating hematopoietic cells. Blood. 2003;102:3108–16.
Charrier S, Stockholm D, Seye K, Opolon P, Taveau M, Gross DA, et al. A lentiviral vector encoding the human Wiskott–Aldrich syndrome protein corrects immune and cytoskeletal defects in WASP knockout mice. Gene Ther. 2005;12:597–606.
Dupre L, Marangoni F, Scaramuzza S, Trifari S, Hernandez RJ, Aiuti A, et al. Efficacy of gene therapy for Wiskott–Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation. Hum Gene Ther. 2006;17:303–13.
Blundell MP, Bouma G, Calle Y, Jones GE, Kinnon C, Thrasher AJ. Improvement of migratory defects in a murine model of Wiskott–Aldrich syndrome gene therapy. Mol Ther. 2008;16:836–44.
Marangoni F, Bosticardo M, Charrier S, Draghici E, Locci M, Scaramuzza S, et al. Evidence for long-term efficacy and safety of gene therapy for Wiskott–Aldrich syndrome in preclinical models. Mol Ther. 2009;17:1073–82.
Bosticardo M, Draghici E, Schena F, Sauer AV, Fontana E, Castiello MC, et al. Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott–Aldrich syndrome. J Allergy Clin Immunol. 2011;127(1376–84):e5.
Catucci M, Prete F, Bosticardo M, Castiello MC, Draghici E, Locci M, et al. Dendritic cell functional improvement in a preclinical model of lentiviral-mediated gene therapy for Wiskott–Aldrich syndrome. Gene Ther. 2012;19:1150–8.
Strom TS, Li X, Cunningham JM, Nienhuis AW. Correction of the murine Wiskott–Aldrich syndrome phenotype by hematopoietic stem cell transplantation. Blood. 2002;99:4626–8.
Wada T, Schurman SH, Otsu M, Garabedian EK, Ochs HD, Nelson DL, et al. Somatic mosaicism in Wiskott–Aldrich syndrome suggests in vivo reversion by a DNA slippage mechanism. Proc Natl Acad Sci USA. 2001;98:8697–702.
Wada T, Konno A, Schurman SH, Garabedian EK, Anderson SM, Kirby M, et al. Second-site mutation in the Wiskott–Aldrich syndrome (WAS) protein gene causes somatic mosaicism in two WAS siblings. J Clin Invest. 2003;111:1389–97.
Wada T, Schurman SH, Jagadeesh GJ, Garabedian EK, Nelson DL, Candotti F. Multiple patients with revertant mosaicism in a single Wiskott–Aldrich syndrome family. Blood. 2004;104:1270–2.
Braun CJ, Boztug K, Paruzynski A, Albert MH, Schwarzer A, Modlich U, et al. Hematopoietic stem cell gene therapy for Wiskott–Aldrich syndrome. Blood. 2013;122:718.
Dupre L, Trifari S, Follenzi A, Marangoni F, Lain de Lera T, Bernad A, et al. Lentiviral vector-mediated gene transfer in T cells from Wiskott–Aldrich syndrome patients leads to functional correction. Mol Ther. 2004;10:903–15.
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–51.
Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117:5561–72.
Benjelloun F, Garrigue A, Demerens-de Chappedelaine C, Soulas-Sprauel P, Malassis-Seris M, Stockholm D, et al. Stable and functional lymphoid reconstitution in artemis-deficient mice following lentiviral artemis gene transfer into hematopoietic stem cells. Mol Ther. 2008;16:1490–9.
Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci USA. 2006;103:16406–11.
Sun JY, Pacheco-Castro A, Borroto A, Alarcon B, Alvarez-Zapata D, Regueiro JR. Construction of retroviral vectors carrying human CD3 gamma cDNA and reconstitution of CD3 gamma expression and T cell receptor surface expression and function in a CD3 gamma-deficient mutant T cell line. Hum Gene Ther. 1997;8:1041–8.
Candotti F, Oakes SA, Johnston JA, Notarangelo LD, O’Shea JJ, Blaese RM. In vitro correction of JAK3-deficient severe combined immunodeficiency by retroviral-mediated gene transduction. J Exp Med. 1996;183:2687–92.
Oakes SA, Candotti F, Johnston JA, Chen YQ, Ryan JJ, Taylor N, et al. Signaling via IL-2 and IL-4 in JAK3-deficient severe combined immunodeficiency lymphocytes: JAK3-dependent and independent pathways. Immunity. 1996;5:605–15.
Bunting KD, Sangster MY, Ihle JN, Sorrentino BP. Restoration of lymphocyte function in Janus kinase 3-deficient mice by retroviral-mediated gene transfer. Nat Med. 1998;4:58–64.
Bunting KD, Flynn KJ, Riberdy JM, Doherty PC, Sorrentino BP. Virus-specific immunity after gene therapy in a murine model of severe combined immunodeficiency. Proc Natl Acad Sci USA. 1999;96:232–7.
Bunting KD, Lu T, Kelly PF, Sorrentino BP. Self-selection by genetically modified committed lymphocyte precursors reverses the phenotype of JAK3-deficient mice without myeloablation. Hum Gene Ther. 2000;11:2353–64.
Sorrentino BP, Lu TH, Ihle J, Buckley RH, Cunningham JM. A clinical attempt to treat JAK3-deficient SCID using retroviral-mediated gene transfer to bone marrow CD34+ cells. Mol Ther. 2003;7:S449.
Wilson JM, Ping AJ, Krauss JC, Mayo-Bond L, Rogers CE, Anderson DC, et al. Correction of CD18-deficient lymphocytes by retrovirus-mediated gene transfer. Science. 1990;248:1413–6.
Bauer TR Jr, Miller AD, Hickstein DD. Improved transfer of the leukocyte integrin CD18 subunit into hematopoietic cell lines by using retroviral vectors having a gibbon ape leukemia virus envelope. Blood. 1995;86:2379–87.
Bauer TR, Schwartz BR, Conrad Liles W, Ochs HD, Hickstein DD. Retroviral-mediated gene transfer of the leukocyte integrin CD18 into peripheral blood CD34+ cells derived from a patient with leukocyte adhesion deficiency type 1. Blood. 1998;91:1520–6.
Yorifuji T, Wilson RW, Beaudet AL. Retroviral mediated expression of CD18 in normal and deficient human bone marrow progenitor cells. Hum Mol Genet. 1993;2:1443–8.
Bauer TR, Hickstein DD. Gene therapy for leukocyte adhesion deficiency. Curr Opin Mol Ther. 2000;2:383–8.
Bauer TR Jr, Hai M, Tuschong LM, Burkholder TH, Gu YC, Sokolic RA, et al. Correction of the disease phenotype in canine leukocyte adhesion deficiency using ex vivo hematopoietic stem cell gene therapy. Blood. 2006;108:3313–20.
Bauer TR Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM, et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat Med. 2008;14:93–7.
Nelson EJ, Tuschong LM, Hunter MJ, Bauer TR Jr, Burkholder TH, Hickstein DD. Lentiviral vectors incorporating a human elongation factor 1alpha promoter for the treatment of canine leukocyte adhesion deficiency. Gene Ther. 2010;17:672–7.
Hunter MJ, Tuschong LM, Fowler CJ, Bauer TR Jr, Burkholder TH, Hickstein DD. Gene therapy of canine leukocyte adhesion deficiency using lentiviral vectors with human CD11b and CD18 promoters driving canine CD18 expression. Mol Ther. 2011;19:113–21.
Hunter MJ, Zhao H, Tuschong LM, Bauer TR Jr, Burkholder TH, Persons DA, et al. Gene therapy for canine leukocyte adhesion deficiency with lentiviral vectors using the murine stem cell virus and human phosphoglycerate kinase promoters. Hum Gene Ther. 2011;22:689–96.
Bradley MB, Fernandez JM, Ungers G, Diaz-Barrientos TA, Steimle V, Mach B, et al. Correction of defective expression in MHC class II deficiency (bare lymphocyte syndrome) cells by retroviral transduction of CIITA. J Immunol. 1997;159:1086–95.
Nelson DM, Butters KA, Markert ML, Reinsmoen NL, McIvor RS. Correction of proliferative responses in purine nucleoside phosphorylase (PNP)-deficient T lymphocytes by retroviral-mediated PNP gene transfer and expression. J Immunol. 1995;154:3006–14.
Liao P, Toro A, Min W, Lee S, Roifman CM, Grunebaum E. Lentivirus gene therapy for purine nucleoside phosphorylase deficiency. J Gene Med. 2008;10:1282–93.
Lagresle-Peyrou C, Yates F, Malassis-Seris M, Hue C, Morillon E, Garrigue A, et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood. 2006;107:63–72.
Lagresle-Peyrou C, Benjelloun F, Hue C, Andre-Schmutz I, Bonhomme D, Forveille M, et al. Restoration of human B-cell differentiation into NOD-SCID mice engrafted with gene-corrected CD34 +cells isolated from Artemis or RAG1-deficient patients. Mol Ther. 2008;16:396–403.
Pike-Overzet K, Rodijk M, Ng YY, Baert MR, Lagresle-Peyrou C, Schambach A, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011;25:1471–83.
Yates F, Malassis-Seris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P, et al. Gene therapy of RAG-2−/− mice: sustained correction of the immunodeficiency. Blood. 2002;100:3942–9.
Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, et al. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 2009;41:106–11.
Brown MP, Topham DJ, Sangster MY, Zhao J, Flynn KJ, Surman SL, et al. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat Med. 1998;4:1253–60.
Tahara M, Pergolizzi RG, Kobayashi H, Krause A, Luettich K, Lesser ML, et al. Trans-splicing repair of CD40 ligand deficiency results in naturally regulated correction of a mouse model of hyper-IgM X-linked immunodeficiency. Nat Med. 2004;10:835–41.
Romero Z, Torres S, Cobo M, Munoz P, Unciti JD, Martin F, et al. A tissue-specific, activation-inducible, lentiviral vector regulated by human CD40L proximal promoter sequences. Gene Ther. 2011;18:364–71.
Yu PW, Tabuchi RS, Kato RM, Dang VK, Lansigan E, Hernandez R, et al. Correction of X-linked immunodeficiency by retroviral mediated transfer of Bruton’s tyrosine kinase. Blood. 2000;96:896.
Yu PW, Tabuchi RS, Kato RM, Astrakhan A, Humblet-Baron S, Kipp K, et al. Sustained correction of B-cell development and function in a murine model of X-linked agammaglobulinemia (XLA) using retroviral-mediated gene transfer. Blood. 2004;104:1281–90 (Epub 2004 May 13).
Moreau T, Barlogis V, Bardin F, Nunes JA, Calmels B, Chabannon C, et al. Development of an enhanced B-specific lentiviral vector expressing BTK: a tool for gene therapy of XLA. Gene Ther. 2008;15:942–52.
Kerns HM, Ryu BY, Stirling BV, Sather BD, Astrakhan A, Humblet-Baron S, et al. B cell-specific lentiviral gene therapy leads to sustained B-cell functional recovery in a murine model of X-linked agammaglobulinemia. Blood. 2010;115:2146–55.
Ng YY, Baert MR, Pike-Overzet K, Rodijk M, Brugman MH, Schambach A, et al. Correction of B-cell development in Btk-deficient mice using lentiviral vectors with codon-optimized human BTK. Leukemia. 2010;24:1617–30.
Rivat C, Booth C, Alonso-Ferrero M, Blundell M, Sebire NJ, Thrasher AJ, et al. SAP gene transfer restores cellular and humoral immune function in a murine model of X-linked lymphoproliferative disease. Blood. 2013;121:1073–6.
Taylor N, Bacon KB, Smith S, Jahn T, Kadlecek TA, Uribe L, et al. Reconstitution of T cell receptor signaling in ZAP-70-deficient cells by retroviral transduction of the ZAP-70 gene. J Exp Med. 1996;184:2031–6.
Steinberg M, Swainson L, Schwarz K, Boyer M, Friedrich W, Yssel H, et al. Retrovirus-mediated transduction of primary ZAP-70-deficient human T cells results in the selective growth advantage of gene-corrected cells: implications for gene therapy. Gene Ther. 2000;7:1392–400.
Otsu M, Steinberg M, Ferrand C, Merida P, Rebouissou C, Tiberghien P, et al. Reconstitution of lymphoid development and function in ZAP-70-deficient mice following gene transfer into bone marrow cells. Blood. 2002;100:1248–56.
Adjali O, Marodon G, Steinberg M, Mongellaz C, Thomas-Vaslin V, Jacquet C, et al. In vivo correction of ZAP-70 immunodeficiency by intrathymic gene transfer. J Clin Invest. 2005;115:2287–95.
Irla M, Saade M, Kissenpfennig A, Poulin LF, Leserman L, Marche PN, et al. ZAP-70 restoration in mice by in vivo thymic electroporation. PLoS ONE. 2008;3:e2059.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Candotti, F. Gene transfer into hematopoietic stem cells as treatment for primary immunodeficiency diseases. Int J Hematol 99, 383–392 (2014). https://doi.org/10.1007/s12185-014-1524-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-014-1524-z