
Abstract
The sequencing of genomes of the pathogenic Mycobacterial species causing pulmonary and extrapulmonary tuberculosis, leprosy and other atypical mycobacterial infections, offer immense opportunities for discovering new therapeutics and identifying new vaccine candidates. Enhanced RV, which uses additional algorithms to Reverse Vaccinology (RV), has increased potential to reduce likelihood of undesirable features including allergenicity and immune cross reactivity to host. The starting point for MycobacRV database construction includes collection of known vaccine candidates and a set of predicted vaccine candidates identified from the whole genome sequences of 22 mycobacterium species and strains pathogenic to human and one non-pathogenic Mycobacterium tuberculosis H37Ra strain. These predicted vaccine candidates are the adhesins and adhesin-like proteins obtained using SPAAN at Pad > 0.6 and screening for putative extracellular or surface localization characteristics using PSORTb v.3.0 at very stringent cutoff. Subsequently, these protein sequences were analyzed through 21 publicly available algorithms to obtain Orthologs, Paralogs, BetaWrap Motifs, Transmembrane Domains, Signal Peptides, Conserved Domains, and similarity to human proteins, T cell epitopes, B cell epitopes, Discotopes and potential Allergens predictions. The Enhanced RV information was analysed in R platform through scripts following well structured decision trees to derive a set of nonredundant 233 most probable vaccine candidates. Additionally, the degree of conservation of potential epitopes across all orthologs has been obtained with reference to the M. tuberculosis H37Rv strain, the most commonly used strain in M. tuberculosis studies. Utilities for the vaccine candidate search and analysis of epitope conservation across the orthologs with reference to M. tuberculosis H37Rv strain are available in the mycobacrvR package in R platform accessible from the “Download” tab of MycobacRV webserver. MycobacRV an immunoinformatics database of known and predicted mycobacterial vaccine candidates has been developed and is freely available at http://mycobacteriarv.igib.res.in.
Similar content being viewed by others

Introduction
Mycobacterial infections have emerged as a major health problem worldwide (Lienhardt et al. 2012; Mayer and Dukes 2010). Among the mycobacterial infections, Tuberculosis (TB) and Leprosy are ranked as the most dreaded diseases affecting mankind (Stone et al. 2009). In the year 2012, according to World Health Organization, there were about 8.6 million incident cases of TB, 1.3 million deaths from TB including 320,000 deaths from HIV-associated TB (Baddeley et al. 2013). These statistics establish the present scenario of devastating impact of TB. Leprosy also known as Hansen’s disease, caused by Mycobacterium leprae has ravaged humans for years and continues to be severe health problem in many developing countries (Dogra et al. 2013). Mycobacterium ulcerans, a member of Non Tuberculosis Mycobacteria (NTM), causes buruli ulcer and is considered the third most common mycobacterial disease of non-immunocompromised individuals after tuberculosis and leprosy. Among other mycobacterial infections, Mycobacterium avium complex (MAC), a member of NTM, consisting of Mycobacterium avium and Mycobacterium intracellulare is responsible for majority of these infections (Waller et al. 2006). MAC causes life-threatening opportunistic infections in immunosuppressed Acquired Immune Deficiency Syndrome (AIDS) patients. Another member of MAC complex, namely, Mycobacterium avium subsp. paratuberculosis causes Crohn’s disease and ulcerative colitis in humans. In immunocompromised patients, Mycobacterium abscessus can cause chronic lung disease, post-traumatic wound infections, and disseminated cutaneous diseases (Katoch 2004).
Currently the only licensed vaccine against tuberculosis is Mycobacterium bovis Bacille Calmette-Guérin (BCG), but it is known to confer highly variable protection (McShane 2011). Other mycobacterial infections caused by NTM are difficult to treat and do not respond to commonly used antituberculous drugs (Griffith 2010). BCG, though found effective against two other mycobacterial diseases Leprosy and Buruli ulcer, its effect remains skeptical (Nackers et al. 2006; Merle et al. 2010). Therefore new vaccines are needed to combat mycobacterial infections.
In this regard, various attempts have been made, resulting in development of new vaccine candidates, which are in different stages of clinical trials (Kaufmann 2011; Lockwood 2007; Marinova et al. 2013). Twelve potential vaccine candidates against tuberculosis targeting different stages of infection are being tested. Among these, two are canonical vaccines aiming to prevent active tuberculosis, two are therapeutic vaccines aiming to treat immunocompromised patients and the rest are preventive vaccines (Lockwood 2007). A few vaccine candidates against leprosy are also undergoing clinical trials for evaluation of their efficacy (Lockwood 2007).
As genome sequences of many mycobacterial species have become available, Reverse Vaccinology (RV) could be used for rapid vaccine candidate identification (Mora et al. 2003). The RV approach initiates vaccine target prediction by bioinformatics analysis of microbial genome sequences. Compared with the traditional methods, RV provides efficient alternative method for vaccine investigation saving both time and cost (Pizza et al. 2000; Rappuoli 2000; Sette and Rappuoli 2010). RV approach was first applied by Rino Rappuoli group to develop vaccine against serogroup B Neisseria meningitidis (MenB), the major cause of sepsis and meningitis in children and young adults. The initial step was the prediction of sub-cellular location, which aided in the identification of potential vaccine candidates (Pizza et al. 2000; Yu et al. 2010). Subsequently, the RV approach has been applied successfully to Streptococcus pneumonia and Chlamydia pneumonia (Maione et al. 2005; Thorpe et al. 2007) and it has been used for screening vaccine candidates for Bacillus anthracis, Porphyromonas gingivalis and Helicobacter pylori (Ariel et al. 2002; Ross et al. 2001; Chakravarti et al. 2000). Recently, immunoinformatics databases have been developed offering data to facilitate RV approach in designing towards new vaccines for malaria and fungal diseases (Chaudhuri et al. 2008, 2011). These resources use enhancements to the original RV approach by incorporating additional algorithms such as probability of a protein being an adhesin, topology of the protein (transmembrane regions) and similarity of the protein to host (human) proteins for selection of potential vaccine candidates for testing (Vivona et al. 2006, 2008; Sachdeva et al. 2005; Ansari et al. 2008). Some of these considerations were based on the initial experience gained while applying the RV approach at the experimental stage (Pizza et al. 2000).
In this work, we have used the enhanced RV approach to construct MycobacRV immunoinformatics datasets and database with potential vaccine candidates to facilitate rapid vaccine development against Mycobacteria. The database also houses the list of epitopes in the known vaccine candidates currently being tested (Vita et al. 2010) and the list of potential epitopes from the list of predicted adhesins and extracellular/surface localized proteins obtained using various epitope prediction servers. We have also included information on conservation of potential epitopes across orthologs towards facilitating epitope based vaccine development using the R platform approach described previously (Ramachandran et al. 2011). The datasets and the database house rich information and multiple features and provide researchers with a user-friendly interface for ease of navigation both in R platform and through web.
Materials and methods
Rationale
Immunogenicity is an important criterion for vaccine candidate selection. The selected candidates for immunization must elicit sufficiently high and sustained immune response in host. In the RV approach, selection of potential vaccine candidates in the first step is carried out using Bioinformatics analysis of protein sequences encoded in the pathogen genomes (Mora et al. 2003; Pizza et al. 2000; Rappuoli 2000; Sette and Rappuoli 2010). In accordance with this principle, the starting point of MycobacRV database construction was the collection of known vaccine candidates and a set of predicted vaccine candidates. These predicted vaccine candidates are proteins predicted as adhesins and adhesin-like proteins using SPAAN at P ad > 0.6 (Sachdeva et al. 2005) and screening for putative extracellular or surface localization characteristics using PSORTb v.3.0 (Yu et al. 2010). A slightly lower P ad value of 0.6 instead of 0.7 (Sachdeva et al. 2005) was used with SPAAN to include the well known mycobacterial adhesin Heparin Binding Hemagglutinnin (HBHA) across the selected pathogenic mycobacterial genomes (Menozzi et al. 1998) along with other characterized host cell binding proteins such as ESAT-6, Antigen 85A, Antigen 85B and Antigen 85C (Kinhikar et al. 2010; Armitige et al. 2000). The allowance of this slight relaxation was favored to increase the likelihood of mycobacterial adhesins being predicted. However, a stringent screening was set using PSORTb v.3.0 to screen for proteins with “Extracellular” or “Cell Wall” location predictions to facilitate selection of highly probable surface proteins with putative adhesin like characteristics. These protein sequences were subsequently analyzed by various algorithms of enhanced RV.
Homology
The Homology information component includes exhaustive search for orthologs, paralogs, conserved domains and similarity to the host proteins.
-
1.
Orthologs are genes present in different species that evolved from a common ancestor gene by the event of speciation (Koonin 2005). These genes usually retain the same function during the course of evolution. Ortholog information for a vaccine candidate hints at a similar function in the corresponding orthologous species and perhaps an equivalent immunogenic response from host. This knowledge is useful in the development of broad spectrum vaccines covering a wide range of species. In this work, we have used Reciprocal Best Hits (RBH) method (Altschul et al. 1990; Moreno-Hagelsieb and Latimer 2008) to fetch the orthologs. The RBH principle holds that two genes from different genomes are orthologous if they find each other as the best hit in BLAST search in the other genome. The BLASTP runs were carried out and results were screened at a maximum E-value threshold of 1 × 10−6, including Smith-Water algorithm and Soft-filtering (Altschul et al. 1990; Moreno-Hagelsieb and Latimer 2008).
-
2.
Paralogs are genes evolved through the event of gene duplication within a genome (Koonin 2005). In contrast to orthologs, paralogs evolve new functions, though they evolved from a common ancestor gene. Paralog information of the vaccine candidates in the same species illuminates on the total repertoire of related vaccine candidate genes of a given family. This information was obtained using BLASTCLUST run at a similarity threshold of 0.8 and minimum length coverage of 0.95 on the individual genomes of the selected mycobacterial species and strains (Kondrashov et al. 2002).
-
3.
Domains are conserved autonomously folding, functional unit of a protein (Marchler-Bauer et al. 2005). Conserved domain data of vaccine candidates provides information on functional domains. This information where available, is useful along with other complementary data. Conserved Domain Database Search (CDD) of National Center for Biotechnology Information (NCBI) was used to obtain the conserved domain information (Marchler-Bauer et al. 2005).
-
4.
An ideal vaccine candidate is desirable not to have any observable similarity to human proteins to avoid generation of potential auto immune response. An initial assessment of this feature can help in the avoidance of expensive dead-ends where a vaccine candidate having studied extensively is discovered to be toxic to the host. In this work, we have assessed the similarity of the individual candidate proteins to the human reference proteins RefSeq Release 54 by performing BLASTP using a maximum E-value threshold of 0.01, which borders on the limits of threshold similarity (Altschul et al. 1990). Setting this threshold will likely avoid collecting even remotely similar proteins in the database.
Motif and topology
-
1.
Betawrap motifs are right-handed parallel beta-helix supersecondary structural motifs present in some bacterial and fungal protein sequences such as toxins, virulence factors and adhesins. These motifs are present in virulence factors of various pathogens (Bradley et al. 2001). Therefore the presence of these motifs in a vaccine candidate value adds to their probable role in virulence. This information will be useful when prioritizing vaccine candidates. Betawrap predictions were obtained for the selected candidate proteins using the BetaWrap server based on three-dimensional dynamic profile method which generates interstrand pairwise correlations from a processive sequence wrap (Bradley et al. 2001).
-
2.
For topology we predicted the transmembrane domains of the candidate proteins. Transmembrane domains are the regions of membrane proteins, which traverse in and out, looping through the membrane. It has been observed that proteins with multiple transmembrane domains are generally difficult to express and purify (Vivona et al. 2006). Therefore this information also facilitates in prioritizing for vaccine candidates. We used TMHMM Server v. 2.0 to predict transmembrane helices (Krogh et al. 2001).
Subcellular location
-
1.
Subcellular location defines the putative location of the protein in the cell. This information forms important criteria for vaccine candidate selection because of the established fact that extracellular or cell surface located proteins are accessible to antibodies and the components of the immune system and hence could be useful. We used subcellular localization prediction server PSORTb v.3.0 for this purpose (Yu et al. 2010).
-
2.
Additionally, signal peptides were also predicted using SignalP 3.0 server (Bendtsen et al. 2004). Signal Peptide is a short stretch of sequence present at the N-terminus of the protein directing it to the secretory pathway. Membrane proteins destined for secretion are targeted to the appropriate intracellular membrane by their signal peptide (Rehm et al. 2001). Hence an assessment of presence of signal peptide in vaccine candidate would provide additional claim for extracellular location of the protein.
Immunoinformatics
Immunoinformatics deals with applying bioinformatics principles and tools to the molecular activities of the immune system. The focus of immunoinformatics has been to enable identification of antigens or epitopes capable of eliciting immune response. Immunoinformatics provides databases and predictive tools, which are used in discovering novel vaccines (Vivona et al. 2008). Epitope, also known as ‘antigenic determinant’ is a surface localized part of antigen capable of eliciting an immune response (Vivona et al. 2008).
We used various epitope prediction algorithms for prediction of B cell epitopes, discotopes (discontinuous B cell epitopes) and T cell (MHC Class I epitope and MHC Class II epitopes). The prediction algorithms are based on different computational approaches, each having an associated success rate of prediction. We therefore used multiple algorithms to fetch epitope predictions, to obtain enriched information.
-
1.
Linear B cell epitopes: Linear epitope constitutes a single continuous stretch of amino acids within a protein sequence antigen recognized by soluble or membrane bound antibodies (Vivona et al. 2008). Among the algorithms used for linear B cell epitope prediction, ABCPred is based on artificial neural networks and BcePred uses physico-chemical properties for epitope prediction (Kolaskar and Tongaonkar 1990; Saha and Raghava 2006a, b, 2007).
-
2.
Discontinuous B cell Epitopes: Epitopes whose residues are distantly placed in the sequence brought together by physico-chemical folding, recognized by soluble or membrane bound antibodies, constitute discontinuous epitopes (Vivona et al. 2008). Discontinuous epitopes were predicted using Discotope 1.2, CEP and BEPro servers based on available crystal structures of antigens (Andersen et al. 2006; Kulkarni-Kale et al. 2005; Sweredoski and Baldi 2008).
-
3.
The immune response against M. tuberculosis infection is mainly cell mediated response with involvement of MHC Class I and MHC Class II molecules (Kaufmann 2002).
MHC Class I T cell epitopes: These are short regions presented on the surface of an antigen-presenting cell, where they are bound to MHC Class I molecules. Among the algorithms used to predict T cell epitopes belonging to MHC Class I, NetMHC 3.0 uses artificial neural networks (ANNs) and weight matrices, Bimas is based on a predicted half-time of dissociation to HLA class I molecules, ARB (Average Relative Binding Method) of Immune Epitope Database (IEDB) uses half maximal inhibitory concentration calculation and IEDB-consensus Method combines NetMHC, Stabilized matrix method (SMM), Scoring Matrices derived from Combinatorial Peptide Libraries (CombLib) algorithms to predict epitopes (Zhang et al. 2008; Bui et al. 2005; Wang et al. 2010; Moutaftsi et al. 2006; Parker et al. 1994; Lundegaard et al. 2008).
-
4.
MHC Class II T cell epitopes: These are short regions presented on the surface of an antigen-presenting cell, where they are bound to MHC Class II molecules. T cell epitopes belonging to MHC Class II were predicted using Propred, which uses quantitative matrices derived from published literature for epitope prediction, IEDB-ARB (Average Relative Binding Method) uses half maximal inhibitory concentration calculations and IEDB-consensus Method combines NN-align, SMM-align, and CombLib algorithms to predict epitopes (Zhang et al. 2008; Bui et al. 2005; Wang et al. 2010; Moutaftsi et al. 2006; Singh and Raghava 2001).
-
5.
Allergens: We also fetched potential allergen information, as it is desirable for a vaccine candidate to be non-allergic in a general sense. For this purpose Algpred, an allergen prediction algorithm, was used with combined approach. This combined approach included finding similarity to known allergic epitopes, searching Multiple EM for Motif Elicitation (MEME)/Motif Alignment and Search Tool (MAST) allergen motifs using MAST, search based on SVM modules and BLAST search against 2890 allergen-representative peptides obtained from Bjorklund et al. 2005 (Saha and Raghava 2006a, 2006b). Additionally, Allermatch, an allergen prediction algorithm based on “Codex alimentarius and FAO/WHO Expert consultation on allergenicity of foods derived through modern biotechnology” was used (Fiers et al. 2004).
Data layout
The whole proteome sequences of 22 selected pathogenic mycobacterial strains and species and a non-pathogenic Mycobacterium tuberculosis H37Ra strain, were sourced from various databases (Table 1) (Cooper et al. 2010; McCarthy 2005; Ioerger et al. 2009). The 742 adhesin and adhesin like protein sequences from 23 strains and species of the selected mycobacteria were analyzed with 20 algorithms of enhanced RV listed in Table 2. The data emerging from these algorithms were structured into “First Layer” and “Second Layer”. “Motif and topology”, “Subcellular location” and “Homology” data were organized into “First Layer” and “Immunoinformatics” data (epitopes and allergens) were organized into “Second Layer”. This strategy was adopted to limit exhaustive epitope analysis to only selected proteins by users. Also the experimentally known epitopes of these proteins were characterized (Vita et al. 2010) and arranged into “Second Layer”. Researchers can interrogate with optimal selection criteria to screen for potential vaccine candidates and the list of conserved epitopes. As an example to this selection process we show suitable decision criteria with the help of well structured decision trees to select a set of most probable vaccine candidates. The implementation process is summarized below.
Potential vaccine candidates identification (top candidates using enhanced RV)
This list of most probable vaccine candidates can be accessed through the “Probable Vaccine Candidate” checkbox in the “Vaccine Candidate Search” tab of MycobacRV Database.
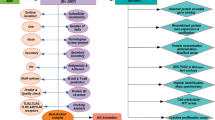
The decision trees describing these processes are presented in Figs. 1, 2 and 3. All analysis following the decision trees can be carried out through scripts in R in object oriented mode. R is a programming language integrated with an R environment, facilitating easy and rapid data analysis with the help of its integrated suite of software facilities (R Core Team 2013). We have developed a package mycobacrvR containing utilities for the vaccine candidate search using various criteria such as SPAAN score, localization of protein, number of transmembrane helix, human reference hits and allergen property as well as for the epitope conservation study. A list of all the function of this package is available in supplementary Table 1.

Decision tree to identify non-allergen proteins fulfilling all first layer conditions. The ‘union’ operator was used for combining data and the ‘intersect’ operator was used for extracting common elements. For example, the ‘union’ operator was used to combine all the non-allergens predicted by allergen prediction algorithms and the ‘intersect’ operator was used to acquire the common candidates of all non-allergens fulfilling “First Layer” conditions

Decision tree to identify proteins having both B cell and T cell epitopes. The set of proteins possessing both B cell and T cell epitopes were obtained using ‘intersect’ operator as shown

The final set of most probable vaccine candidates. This set was obtained by intersecting the results of the two decision trees described in Figs. 1 and 2. These candidates meet the criteria of being non-allergic, having less than two transmembrane helices, no similarity to human proteins and having both B cell and T cell epitopes from 23 strains and species of the selected mycobacteria
Case 1: First layer data and allergen prediction
Through this process we obtained a set of 426 protein sequences as most probable adhesin vaccine candidates meeting the criteria set in the scripts. A stringent criterion (S = 100, L = 1, b = T, where ‘S’ refers to similarity threshold, ‘L’ to minimum length coverage, ‘b’ to both) specified in the BLASTCLUST computer program (Cooper et al. 2010) was used to identify redundancy in the set of 426 protein sequences, thereby providing a non-redundant set of 233 most probable adhesin vaccine candidates. The non-redundant set of most probable vaccine candidates along with the example R scripts used for analysis can be obtained from the “Download” tab of the webserver. The decision criteria applied can be modified by researchers and implemented suitably by modifying the R scripts. Flow chart describing first layer data filtration and allergen prediction algorithm of mycobacrvR functions is presented in supplementary Table 1.
The “First Layer” data of 22 selected human mycobacterial pathogens was used to filter protein sequence candidates with less than two transmembrane helices and having no similarity to human reference proteins (Vivona et al. 2008). Thereafter the non-allergen candidates fulfilling “First Layer” conditions (having less than two transmembrane helices and having no similarity to human reference proteins) were selected. These candidates were further analysed for the presence of B cell epitopes and T cell epitopes. The candidates possessing both B cell and T cell epitopes were further selected.
The filtered candidates having both B cell and T cell epitopes, predicted non-allergens and fulfilling other “First Layer” criteria formed the final set of most probable vaccine candidates.
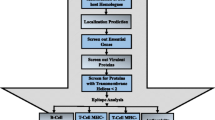
Case 2: Epitope conservation study
Mycobacterium tuberculosis H37Rv strain was chosen as reference for the epitope conservation study. This analysis was carried out using R scripts. For each of the vaccine candidate (adhesin and adhesin like proteins) from M.tuberculosis H37Rv strain, the predicted B cell and T cell epitopes were analyzed for epitope conservation. Identity score was measured as:
Number of Occurrences of the potential epitopes across all orthologs of the protein/Total number of orthologs of the protein.
Exact match approach was used for epitope conservation study. This was done so as to provide users with accurate conservation ratio for epitopes based on exact epitope sequence matches. This information was organized into MycobacRV database and can be accessed through the “Epitope Conservation Data” tab. The detailed description showing ortholog profile for presence or absence of the query epitopes in the selected mycobacterial species have also been provided. The epitope conservation data across species would aid in broad spectrum, epitope based rational vaccine development studies. The flow chart below describes the algorithm for epitope conservation across orthologs for the filtered ginumbers of a species with reference to Mycobacterium tuberculosis H37Rv strain using the epitope prediction data from B cell or T cell epitope prediction servers (e.g. ABCPred, Bcepred, Propred, NetMHC, IEDB server). The Flow chart of mycobacrvR function for epitope conservation study is presented in supplementary Table 2.
Database architecture
Database design
The GI number identification tags assigned to proteins were used as primary keys. The database was developed using MySQL version 4.1.20 at back end and operated in Red Hat Enterprise Linux ES release 4. The web interfaces have been developed in HTML and PHP 5.1.4, which dynamically execute the MySQL queries to fetch the stored data and is run through Apache2 server. The overall layout of MycobacRV is shown in Fig. 4.

Tetrapodic Layout of MycobacRV. The primary keys were the ginumbers of the proteins. In Homology we include -orthologs, paralogs, absence of similarity against Human proteins, Motif and Topology consists of beta helix supersecondary structural motifs, conserved domains, transmembrane topologies, Subcellular location consists of signal peptides, subcellular localization prediction, Immunoinformatics consists of predicted antigenic regions, epitopes and potential non-allergens. This data was further analysed through decision tree. Also epitope conservation studies were made for conservation of epitopes across orthologs
Database access and interface
The tabs provided in MycobacRV web-server are- “Home”, “Vaccine Candidate Search”, “Advanced Search”, “Epitope Conservation Data”, “Known Vaccines”, “Download”, “Help” and “Contact”. The “Vaccine Candidate Search” tab provides complete data for 742 predicted vaccine candidates, organized into “First Layer” and “Second Layer” (Fig. 5). The data for most probable vaccine candidate for a selected species can be fetched by checking the checkbox provided and then clicking the submit button. The “Advanced Search” tab provides user with facility to filter data on the basis of Protein length, number of transmembrane spanning regions, presence or absence of betawraps, paralogs, orthologs, conserved domains, similarity to Human Reference proteins (retrieved from NCBI through ftp on January 22, 2013). The “First Layer” data filter criteria can also be exercised here. The “Epitope Conservation Data” tab provides users with the epitope conservation data analysis for Mycobacterium tuberculosis H37Rv strain. The “Known Vaccines” tab takes the user to the page containing the list of known vaccine candidates provided in tabular form along with the cited references. The “Download” tab of the webserver provides the non-redundant set of most probable vaccine candidates along with Rdata and the example R scripts used for analysis. Also the utilities for the vaccine candidate search and analysis of epitope conservation across the orthologs with reference to M. tuberculosis H37Rv strain available in the mycobacrvR package in R platform is available for download. Results obtained using any of the operations can be exported by users into text files.

‘Immunoinformatics Data’ search tab of MycobacRV webserver. Searches can be performed using multiple options by clicking the checkboxes. The backend data consists of immunoinformatics data on 742 adhesin and adhesin like proteins with extracellular and surface localized characteristics. The non-redundant set of most probable vaccine candidates along with the example R scripts used for analysis can be obtained from the “Download” tab of the webserver
Results
MycobacRV provides comprehensive analysis data for 742 predicted and known adhesin and adhesin like proteins from 23 strains and species of Mycobacteria. Analysis of enhanced RV data through decision trees provided a list of 233 non-redundant set of most probable vaccine candidates from 23 strains and species of Mycobacteria. Recent trends of vaccinologists aim for epitope based vaccines (Patronov and Doytchinova 2013; Khan et al. 2007). Towards facilitating these efforts, we analyzed the information on epitopes in terms of their conservation among orthologs (Khan et al. 2007). The epitope conservation data for epitopes and its associated information including other molecular features of the protein provides facility for enablement of epitope based vaccine design.
The predicted vaccine candidates mainly include PE family, PPE family, PE-PGRS family, Mpt family, Cfp2, pstS2, ESAT-6, HBHA, Antigen 85A, Antigen 85B, Antigen 85C and hypothetical proteins. Some of these proteins (Antigen 85A, Antigen 85B, Antigen 85C and ESAT-6) are undergoing investigations for new vaccine development (Kaufmann 2011). These results show that the approach adopted by us in preparing MycobacRV will be useful for future developments in developing new vaccines for mycobacterial infections in general and tuberculosis in particular.
The development of the mycobacrvR package serves as a model for developing data on other pathogens. Because this is prepared in the open source mode, this allows further development in future by other users and developers towards expansion in order to rapidly facilititate the goal of epitope based vaccines.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andersen PH, Nielsen M, Lund O (2006) Prediction of residues in discontinuous B cell epitopes using protein 3D structures. Protein Sci 15:2358–2367
Ansari FA, Kumar N, Bala Subramanyam M, Gnanamani M, Ramachandran S (2008) MAAP: malarial adhesins and adhesin-like proteins predictor. Proteins 70:659–666
Ariel N, Zvi A, Grosfeld H, Gat O, Inbar Y, Velan B, Cohen S, Shafferman A (2002) Search for potential vaccine candidate open reading frames in the Bacillus anthracis virulence plasmid pXO1: in silico and in vitro screening. Infect Immun 70:6817–6827
Armitige LY, Jagannath C, Wanger AR, Norris SJ (2000) Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect Immun 68:767–778
Baddeley A, Dean A, Dias HM, Falzon D et al (2013) World Health Organization Global Tuberculosis Report. http://www.who.int/tb/publications/global_report/en/index.html. Accessed 1 November 2013
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 30. J Mol Biol 340:783–795
Bradley P, Cowen L, Menke M, King J, Berger B (2001) BETAWRAP: successful prediction of parallel beta-helices from primary sequence reveals an association with many microbial pathogens. Proc Natl Acad Sci USA 98:14819–14824
Bui HH, Sidney J, Peters B, Sathiamurthy M, Sinichi A, Purton KA, Mothé BR, Chisari FV, Watkins DI, Sette A (2005) Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics 57:304–314
Chakravarti DN, Fiske MJ, Fletcher LD, Zagursky RJ (2000) Application of genomics and proteomics for identification of bacterial gene products as potential vaccine candidates. Vaccine 19:601–612
Chaudhuri R, Ahmed S, Ansari FA, Singh HV, Ramachandran S (2008) MalVac: database of malarial vaccine candidates. Malar J 7:184
Chaudhuri R, Ansari FA, Raghunandanan MV, Ramachandran S (2011) FungalRV: adhesin prediction and immunoinformatics portal for human fungal pathogens. BMC Genom 12:192
Dogra S, Narang T, Kumar B (2013) Leprosy–evolution of the path to eradication. Indian J Med Res 137:15–35
Fiers MW, Kleter GA, Nijland H, Peijnenburg AA, Nap JP, Van RC (2004) Allermatch, a webtool for the prediction of potential allergenicity according to current FAO/WHO Codex alimentarius guidelines. BMC Bioinform 5:133
Griffith DE (2010) Nontuberculous mycobacterial lung disease. Curr Opin Infect Dis 23:185–190
Ioerger TR, Koo S, No EG, Chen X, Larsen MH, Jacobs WR Jr, Pillay M, Sturm AW, Sacchettini JC (2009) Genome analysis of multi- and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One 4:e7778
Katoch VM (2004) Infections due to non-tuberculous mycobacteria (NTM). Indian J Med Res 120:290–304
Kaufmann SH (2002) Protection against tuberculosis: cytokines, T cells, and macrophages. Ann Rheum Dis 61(Suppl 2):ii54–ii58
Kaufmann SH (2011) Fact and fiction in tuberculosis vaccine research: 10 years later. Lancet Infect Dis 11:633–640
Khan AM, Miotto O, Heiny AT, Salmon J, Srinivasan KN, Nascimento EJ, Marques ET Jr, Brusic V, Tan TW, August JT (2007) A systematic bioinformatics approach for selection of epitope-based vaccine targets. Cell Immunol 244:141–147
Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P, Hsu T, Jacobs WR Jr, Laal S (2010) Potential role for ESAT6 in dissemination of M tuberculosis via human lung epithelial cells. Mol Microbiol 75:92–106
Kolaskar AS, Tongaonkar PC (1990) A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 276:172–174
Kondrashov FA, Rogozin IB, Wolf YI and Koonin EV (2002) Selection in the evolution of gene duplications. Genome Biol 3:RESEARCH0008
Koonin EV (2005) Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet 39:309–338
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Kulkarni-Kale U, Bhosle S and Kolaskar AS (2005) CEP: a conformational epitope prediction server. Nucleic Acids Res 33(Web Server issue):W168–W171
Lienhardt C, Glaziou P, Uplekar M, Lönnroth K, Getahun H, Raviglione M (2012) Global tuberculosis control: lessons learnt and future prospects. Nat Rev Microbiol 10:407–416
Lockwood DNJ (2007) Leprosy Clin Evid (Online) Apr 1; 2007 pii: 0915
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M (2008) NetMHC-30: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res 36(Web Server):W509–W512
Maione D, Margarit I, Rinaudo CD, Masignani V, Mora M, Scarselli M, Tettelin H, Brettoni C, Iacobini ET, Rosini R et al (2005) Identification of a universal Group B streptococcus vaccine by multiple genome screen. Science 309:148–150
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D and Bryant SH (2005) CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res 192–196
Marinova D, Gonzola-Asensio J, Aguilo N, Martin C (2013) Recent developments in tuberculosis vaccines. Expert Rev Vaccines 12:1431–1438
Mayer KH, Dukes HC (2010) Synergistic pandemics: confronting the global HIV and tuberculosis epidemics. Clin Infect Dis 3:S67–S70
McCarthy AA (2005) Broad institute: bringing genomics to real-world medicine. Chem Biol 12:717–718
McShane H (2011) Tuberculosis vaccines: beyond bacille Calmette-Guerin. Philos Trans R Soc Lond B Biol Sci 366:2782–2789
Menozzi FD, Bischoff R, Fort E, Brennan MJ, Locht C (1998) Molecular characterization of the mycobacterial heparin-binding hemagglutinin, a mycobacterial adhesin. Proc Natl Acad Sci USA 13:12625–12630
Merle CS, Cunha SS, Rodrigues LC (2010) BCG vaccination and leprosy protection: review of current evidence and status of BCG in leprosy control. Expert Rev Vaccines 9:209–222
Mora M, Veggi D, Santini L, Pizza M, Rappuoli R (2003) Reverse vaccinology. Drug Discov Today 8:459–464
Moreno-Hagelsieb G, Latimer K (2008) Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24:319–324
Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui HH, Grey H, Sette A (2006) A consensus epitope prediction approach identifies the breadth of murine T(CD8 +)-cell responses to vaccinia virus. Nat Biotechnol 24:817–819
Nackers F, Dramaix M, Johnson RC, Zinsou C, Robert A, de Biurrun Bakedano E, Glynn JR, Portaels F, Tonglet R (2006) BCG vaccine effectiveness against Buruli ulcer: a case-control study in Benin. Am J Trop Med Hyg 75:768–774
Parker KC, Bednarek MA, Coligan JE (1994) Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol 152:163–175
Patronov A, Doytchinova I (2013) T-cell epitope vaccine design by immunoinformatics. Open Biol 3:120139
Cooper PS, Lipshultz D, Matten WT, McGinnis SD, Pechous S, Romiti ML, Tao T, Valjavec-Gratian M, Sayers EW (2010) Education resources of the National Center for Biotechnology Information. Brief Bioinform 11:563–569
Pizza M, Scarlato V, Masignani V, Giuliani MM, Aricò B, Comanducci M, Jennings GT, Baldi L et al (2000) Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 287:1816–1820
Ramachandran S, Chaudhuri R, Verma SP, Shah AR, Paul C, Chakraborty S, Puniya BL and Mandal RS (2011) Biological Data Modelling and Scripting in R, Systems and Computational Biology - Bioinformatics and Computational Modeling, Prof Ning-Sun Yang (Ed), InTech. http://www.intechopen.com/books/systems-and-computational-biology-bioinformatics-and-computational-modeling/biological-data-modelling-and-scripting-in-r
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. http://www.R-project.org/
Rappuoli R (2000) Reverse vaccinology. Curr Opin Microbiol 3:445–450
Rehm A, Stern P, Ploegh HL, Tortorella D (2001) Signal peptide cleavage of a type I membrane protein, HCMV US11, is dependent on its membrane anchor. EMBO J 20:1573–1582
Ross BC, Czajkowski L, Hocking D, Margetts M, Webb E, Rothel L, Patterson M, Agius C, Camuglia S, Reynolds E, Littlejohn T, Gaeta B, Ng A, Kuczek ES, Mattick JS, Gearing D, Barr IG (2001) Identification of vaccine candidate antigens from a genomic analysis of Porphyromonas gingivalis. Vaccine 19:4135–4142
Sachdeva G, Kumar K, Jain P, Ramachandran S (2005) SPAAN: a software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics 21:483–491
Saha S, Raghava GP (2006) AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res W202–W209
Saha S, Raghava GP (2006b) Prediction of continuous b-cell epitopes in an antigen using Recurrent Neural Network. Proteins 65:40–48
Saha S, Raghava GP (2007) Prediction methods for B-cell epitopes. Methods Mol Biol 409:387–394
Sette A, Rappuoli R (2010) Reverse vaccinology: developing vaccines in the era of genomics. Immunity 4:530–541
Singh H, Raghava GP (2001) ProPred: prediction of HLA-DR binding sites. Bioinformatics 17:1236–1237
Stone AC, Wilbur AK, Buikstra JE, Roberts CA (2009) Tuberculosis and leprosy in perspective. Am J Phys Anthropol 49:66–94
Sweredoski MJ, Baldi P (2008) PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 24:1459–1460
Thorpe C, Edwards L, Snelgrove R, Finco O, Rae A, Grandi G, Guilio R, Hussell T (2007) Discovery of a vaccine antigen that protects mice from Chlamydia pneumoniae infection. Vaccine 25:2252–2260
Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B (2010) The immune epitope database 20. Nucleic Acids Res 38(Database issue): D854–D862
Vivona S, Bernante F, Filippini F (2006) NERVE: new enhanced reverse vaccinology environment. BMC Biotechnol 6:35
Vivona S, Gardy JL, Ramachandran S, Brinkman FS, Raghava GP, Flower DR, Filippini F (2008) Computer-aided biotechnology: from immuno-informatics to reverse vaccinology. Trends Biotechnol 26:190–200
Waller EA, Roy A, Brumble L, Khoor A, Johnson MM, Garland JL (2006) The expanding spectrum of Mycobacterium avium complex-associated pulmonary disease. Chest 130:1234–1241
Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B (2010) Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform 11:568
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS (2010) PSORTb 30: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26:1608–1615
Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, Bui HH, Buus S, Frankild S, Greenbaum J, Lund O, Lundegaard C, Nielsen M, Ponomarenko J, Sette A, Zhu Z, Peters B (2008) Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res 36(Web Server):W513–W518
Acknowledgments
SR thanks grants (BSC0121) from Council of Scientific and Industrial Research (CSIR). RC thanks The Indian Council of Medical Research for fellowship. Funding for IT infrastructure through CSIR-Institute of Genomics and Integrative Biology resources is acknowledged.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
MycobacRV can be accessed at http://mycobacteriarv.igib.res.in. It is best viewed with Explorer 8.0 or later and Mozilla firefox version 3.0 or later.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chaudhuri, R., Kulshreshtha, D., Raghunandanan, M.V. et al. Integrative immunoinformatics for Mycobacterial diseases in R platform. Syst Synth Biol 8, 27–39 (2014). https://doi.org/10.1007/s11693-014-9135-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11693-014-9135-9