
Abstract
Point mutations within the ABL kinase domain of the BCR-ABL gene are associated with clinical resistance to imatinib mesylate in chronic myeloid leukemia (CML). To obtain more information about the association between BCR-ABL mutations and type of imatinib resistance, we studied 30 early chronic phase (CP) CML patients, commencing imatinib therapy, using a conventional sequencing technique. Seven patients treated in late CP and three patients treated in the accelerated phase were included for comparison. Blood samples were collected before and every third month during imatinib therapy. Mutations were not seen in any blood sample collected before start of therapy. During imatinib treatment, 2 of the 30 early CP patients acquired point mutations and both of them had other signs of imatinib resistance. None of the five early CP patients with a complete hematologic response (HR), but no cytogenetic response at 12 months, displayed any missense mutation. Likewise, none of 12 early CP patients with detectable BCR-ABL transcripts but in complete hematologic and cytogenetic remission at 12 months displayed any mutation. We conclude that screening early CP patients for BCR-ABL mutations before start of imatinib therapy is not cost-effective. BCR-ABL kinase domain mutations do not appear to explain cytogenetic or molecular (detectable BCR-ABL transcripts by polymerase chain reaction) disease persistence in patients otherwise in stable disease. However, in patients with signs of expanding disease burden, a search for BCR-ABL mutations is warranted.
Similar content being viewed by others

Introduction
Chronic myeloid leukemia (CML) is a hematopoietic stem cell disorder characterized by the t(9;22) chromosomal translocation resulting in the formation of the BCR-ABL fusion gene, which is a prerequisite in the pathogenesis of CML. The BCR-ABL gene product has a constitutively activated tyrosine kinase activity [5, 19]. Imatinib mesylate (Gleevec®/Glivec®, Novartis, Hanover, NJ, USA), a small molecule inhibitor of the BCR-ABL tyrosine kinase, can induce complete cytogenetic remission in up to 80% of CML patients treated in first chronic phase (CP) [14]. However, primary refractoriness or relapse after initial response to imatinib is observed in some CML patients, particularly those with late stage or advanced disease [13, 22]. The mechanism of resistance to imatinib ranges from nonspecific multidrug resistance [20] to BCR-ABL inherent genetic alterations [22], including overexpression of BCR-ABL due to gene amplification [18] and point mutations in the ABL kinase domain [9, 27]. Point mutations, which impair imatinib binding by interrupting the critical contact point or by inducing a conformation to which imatinib binding is reduced, were identified as an important mechanism of acquired imatinib resistance [2, 3, 11]. To date, more than 50 different mutations were found to be associated with resistance to imatinib [2, 3, 9, 11, 27]. Some mutants, such as T315I and E255K, are insensitive to imatinib at clinically achievable doses, whereas others, such as M351T or Y253F, retain intermediate levels of sensitivity to imatinib [11]. The probability of finding a mutation increases with disease duration and with advanced disease stage [2]. Nevertheless, it was reported that mutations may be detected even before initiation of treatment with imatinib [24, 25, 27]. There is a large variation in the previously reported frequency of mutations found in association with the imatinib resistance, ranging from 26 to 90% of the patients [1–3, 9, 11, 12, 21, 25–27]. This difference probably reflects the heterogeneity in patient populations studied. The definition of resistance also varies from one study to another and most work has focused on patients with disease relapse while on imatinib treatment. Few data are available to date on the incidence of mutations in newly diagnosed CML patients commencing imatinib therapy [11, 27].
To evaluate the frequency of BCR-ABL kinase domain mutations and their association with response to imatinib treatment, we sequentially studied 30 newly diagnosed CML patients in CP commencing imatinib therapy. Seven patients treated in late CP and three patients treated in accelerated phase (AP) were included for comparison.
Materials and methods
Patients
All CML patients treated with imatinib at Sahlgrenska University Hospital, between the years 2000 and 2004, were identified. In total, 40 consecutive patients, aged 23 to 80 years, were included in this study. Nineteen were females and 21 were males. At the start of imatinib treatment, 30 patients were in early CP, defined as <12 months from diagnosis; 7 patients were in late CP, defined as ≥12 months from diagnosis; and 3 patients were in AP. AP was defined by the presence of any of the following: at least 20% basophils in peripheral blood (PB) or between 15 and 30% blasts in PB or bone marrow (BM). The diagnosis of CP CML was confirmed before imatinib mesylate treatment and was based on typical blood and BM morphologic findings, together with a t(9;22)(q34;q11) translocation detected by routine karyotyping or fluorescence in situ hybridization for the BCR-ABL fusion gene. Patients in CP were treated with imatinib targeting a dose of 400 mg/day and patients in AP with 600 mg/day. PB samples, obtained before start of imatinib therapy and every third month thereafter, were subjected to reverse transcription polymerase chain reaction (RT-PCR) analysis for BCR-ABL mRNA quantification and for detection of BCR-ABL kinase domain mutations. Bone marrow specimens, obtained every 6 months, were subjected to morphologic evaluation and routine metaphase karyotyping. The median follow-up is 24 months (range 2–61 months).
Imatinib resistance was defined as: (1) failure to achieve complete hematologic remission (CHR; defined as complete blood counts within institutional normal limits and no signs of extramedullary involvement) after 3 months of imatinib therapy; (2) failure to achieve at least a minimal cytogenetic response (minimal CgR; 65–95% Ph-positive metaphases) after 6 months of imatinib therapy; (3) failure to achieve a major cytogenetic response (MCgR; <35% Ph-positive metaphases) at 12 months of imatinib therapy; and (4) loss of an earlier obtained CHR or CgR. The first three defined primary resistance and the fourth defined acquired resistance.
The study was approved by local ethical committee.
Cytogenetic analysis
Cytogenetic studies were performed on 24- and/or 48-h BM cell cultures, using standard methods for preparations and G-banding. Chromosome identification and karyotype designation were made according to the International System for Human Cytogenetic Nomenclature. Cytogenetic responses were defined using standard criteria [7, 29]: no Ph-positive metaphases = complete cytogenetic response (CCgR), 1–35% Ph-positive metaphases = partial cytogenetic response (PCgR), 36–65% Ph-positive metaphases = minor cytogenetic response, 66–95% Ph-positive metaphases = minimal CgR, and above 95% Ph-positive metaphases = no cytogenetic response (no CgR).
Quantitative measurements of BCR-ABL mRNA
The real-time RT-PCR technique (qRT-PCR) for measurement of BCR-ABL fusion gene transcripts was previously described in detail [10]. Briefly, the nucleated cell fraction was isolated from EDTA-anticoagulated whole blood and mRNA was extracted using an automated poly-A RNA purification method, Genom-48 Robotic workstation (Genovision, Norway). The mRNA from 105 nucleated cells was extracted into a volume of 60 μl. Fifty microliters of the extracted mRNA (0.1 μg) were used for RT in a final reaction volume of 100 μl. Complementary DNA (cDNA) was generated by RT with random primers (Hexanucleotidemix, Roche, Sweden; f.c. 20 pmol/μl) using the Superscript II enzyme (InVitrogen, Sweden; f.c. 2 U/μl). The cDNA was stored at −20°C. BCR-ABL cDNA was quantified by real-time PCR with the RotorGene 2000 instrument (Corbett Research, Mortlake, NSW, Australia) using the TaqMan probe system with primers and probe as previously reported [10]. The housekeeping gene GAPDH, quantified by the TaqMan probe system, was used to control for differences in mRNA quality between samples and was coamplified in parallel reactions. To monitor for mRNA quality and RT efficacy only samples and reactions giving a cycle threshold (CT) value below 24 for GAPDH were accepted. This cut-off value (95% confidence interval) was established by analyzing 40 normal and 40 CML samples collected in our laboratory.
Plasmids containing BCR-ABL and GAPDH cDNA were used in serial dilutions to construct calibration curves. The CT values for BCR-ABL and the control gene were determined in triplicates from the patient samples and the copy numbers were calculated from the respective calibration curves. The estimated amount of BCR-ABL mRNA was normalized by dividing with the amount of GAPDH mRNA. The normalized values were multiplied by the constant 104. Duplicates of the three calibrators were included in each PCR run and the mean normalized BCR-ABL values for each calibrator were used for validation of the efficiency and accuracy of the PCR amplification using the KUSE software. The sensitivity of our quantitative RT-PCR is five copies of BCR-ABL plasmid or one K562 cell in 105 normal cells. Significant molecular response (SMolR) was defined as a ≥3 log (base 10) reduction in BCR-ABL transcript number, related to the individual baseline value.
Sequencing of BCR-ABL kinase domain
The blood samples and cDNA preparations that were initially assessed for BCR-ABL transcript level were used for detection of BCR-ABL kinase domain mutations. Mutation screening was performed on all samples obtained before start of imatinib treatment. Thereafter, the mutation analysis was repeated every 6 months until the BCR-ABL transcripts were undetectable. If a patient acquired imatinib resistance during treatment, i.e., loss of an earlier obtained CHR or CgR, the mutation analysis was performed on the immediate preceding sample and was repeated every 3 months afterward.
Analysis of mutations was performed using a method modified from Shah et al. [27]. The BCR-ABL kinase domain was amplified using a two-step RT-PCR procedure. With cDNA as template, the forward and reverse primers located in BCR exon b2 and ABL exon 9, respectively, the first PCR step generated a 1.3-kb fragment containing BCR-ABL junction and ABL kinase domain. In the second PCR step, using a forward primer annealing in ABL exon 4 and the same reverse primer as in the first step, an 858-bp fragment was generated. After purification, the 858-bp fragment was sequenced in the forward and reverse direction using BigDye Terminator Cycle Sequencing Ready Reaction Kit version 3.1 (Applied Biosystems, Stockholm, Sweden) and ABI Prism 3100 Genetic Analyzer system (ABI 3100, Foster City, CA, USA). Using GenBank accession no. M14752 as reference, sequences were aligned and analyzed with the CodonCode sequence analysis software (CodonCode Corporation, Dedham, MA, USA). Translated into protein, the fragment covered an amino acid sequence from position 237 to 486, which includes the P-loop and the downstream activation loop. A mutation was considered to be present in a sample if it was detected on both strands in two independent reactions. In our hands and from subcloning experiments, direct sequencing of BCR-ABL kinase domain will reveal mutant clones once they represent more than 20–30% of the leukemic clones.
Results
Hematologic and cytogenetic response/resistance to imatinib
All 30 CML patients treated in early CP achieved CHR within 3 months. Twenty-five of them achieved a MCgR (22 reached CCgR and 3 reached PCgR) within 12 months of treatment. Thirteen (43%) of the 30 patients treated in early CP were in SMolR at 12 months of imatinib therapy as evaluated by qRT-PCR analysis. Six early CP patients were or became imatinib-resistant; five patients had no CgR (>95% Ph-positive metaphases) at 6 months imatinib therapy (primary resistance) where one transformed into blast crisis (BC) at 10 months, and one patient lost an earlier obtained MCgR (acquired resistance). Five of these six imatinib-resistant patients underwent allogeneic stem cell transplantation; one patient continued on imatinib and lost CHR at 17 months of imatinib therapy. Patient characteristics and details about treatment responses are given in Tables 1 and 2.
Seven patients were in late CP when imatinib treatment started. All of them achieved CHR within 3 months and four of them achieved CCgR at 12 months imatinib therapy. However, one patient lost CHR and one patient lost CCgR while on continued imatinib therapy. Three patients treated in late CP did not achieve MCgR at 12 months and one of them lost CHR during continued imatinib therapy (Table 1).
Three AP patients were treated with imatinib; two of them obtained HR that lasted for 2 months and one was in CHR for 12 months. However, they all lost their HR/CHR while on imatinib treatment.
BCR-ABL kinase domain mutations
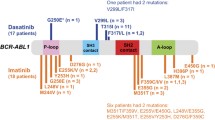
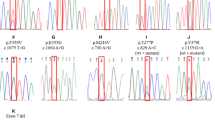
No BCR-ABL kinase domain mutation was detected in any sample collected before start of imatinib therapy, irrespective of disease phase. During imatinib treatment, mutations in BCR-ABL kinase domain were found in 2 of the 30 (7%) early CP patients, 4 of 7 (57%) late CP patients, and in all 3 (100%) AP patients. Six different point mutations were detected (Table 2). Three mutations (G250E, Y253H, and E255K) were clustered in the P-loop, one mutation (T315I) was located in the SH3 contact area, one mutation (E355K) was located in the activation loop, and one mutation (E450G) was located in the C-terminal part of the kinase domain. The E450G was the most frequently observed mutation, detected in 4 of our 40 patients.
Five of the 30 early CP patients had a primary cytogenetic resistance, defined as failure to achieve at least a minimal CgR at 6 months or MCgR at 12 months, and none of them displayed any BCR-ABL kinase domain mutation up to 12 months after start of imatinib therapy. Also, none of the patients with molecular disease persistence, i.e., detectable BCR-ABL transcripts by qRT-PCR, but without other signs of imatinib resistance, had any kinase domain mutation at 12 months; these patients were followed with mutation screening every 6 months and no mutations have evolved during a median follow-up of 31 months (range from 12 to 59 months). Thus, it appears as if primary imatinib resistance is hardly ever explained by mutations. Conversely, acquired imatinib resistance is frequently associated by mutations. Three of our 30 patients treated in early CP lost an earlier obtained MCgR and CHR. Two of these patients were found to have acquired kinase domain mutations at the time of resistance detection; in one of them the mutation was discernible in a sample collected 3 months before clinical signs of imatinib resistance. Furthermore, 8 patients out of our whole material of 40 patients developed an acquired imatinib resistance, either hematologic resistance (n=6), defined as loss of CHR or transformation into BC, or cytogenetic resistance (n=2), defined as loss of MCgR or CCgR; BCR-ABL domain mutations were found in 7 of them.
In two patients treated in late CP, a temporary mutation was detected in a single sample, at 6 months (E255K and E450G) and 9 months (E450G) after start of imatinib therapy, respectively. Both these patients were in CHR and CCgR at the time of mutation detection; over a follow-up of 54 and 23 months, respectively, the mutations had not reappeared and no imatinib resistance had evolved.
Discussion
Because the response to imatinib therapy in CML seems to be the best predictor of prognosis, monitoring for unsatisfactory response has become routine to identify patients at risk of disease progression. Such imatinib resistance can either be primary or acquired. There is no consensus on the definition of primary imatinib resistance, but the following landmarks are generally agreed on as primary imatinib failure that warrants a change in therapy: (1) not achieving CHR by 3 months, (2) no evidence of cytogenetic response by 6 months, and (3) no MCgR by 12 months. Conversely, acquired resistance can be defined as progression to blast phase, progression to AP, loss of HR, loss of MCgR, or loss of CCgR with a tenfold rise in BCR-ABL [8]. Mutations in the BCR-ABL kinase domain that interfere with imatinib binding and lead to the reactivation of kinase activity appear to be the most common mechanism of acquired resistance and more than 50 mutations have now been described [1, 3, 9, 11, 12, 21, 25–27]. It is not known at this stage what causes primary resistance but point mutations in BCR-ABL are believed to be unusual.
It was initially assumed that detection of any BCR-ABL kinase domain mutation was the immediate cause of imatinib resistance. However, it has become clear that different mutations are associated with a different degree of resistance, some of which can be overcome by escalating the imatinib dose or by the use of the second-generation tyrosine kinase inhibitors. Moreover, a specific subgroup of mutations, i.e., those falling within the P-loop, is considered to be associated with a particularly poor prognosis in terms of survival in late CP and AP CML patients [3, 28]. However, not all investigators agree with the invariably bad prognosis of patients with P-loop mutations [17].
We studied 40 CML patients commencing imatinib therapy. The majority of our patients were in early CP and received imatinib as first line therapy. Sequencing of the BCR-ABL kinase domain did not detect mutations in any of the pretreatment samples analyzed. During imatinib therapy, six different mutations were detected in the 9 patients; two out of 30 treated in early CP, four out of 7 treated in late CP, and three out of 3 treated in AP. Seven of these patients had developed imatinib resistance. The remaining two patients displayed temporary mutations, detected at a single sampling point and without other signs of imatinib resistance. Besides these two patients, mutations could not be detected in any patient in hematologic remission at 12 months of imatinib therapy, irrespective of their cytogenetic or BCR-ABL mRNA status. Eight patients developed an acquired resistance to imatinib defined as loss of HR, loss of cytogenetic response, or transformation to BC; seven of them revealed an acquired BCR-ABL kinase domain mutation. Thus, our results suggest that cytogenetic resistance and molecular persistence, in an otherwise stable disease, are rarely caused by point mutations in BCR-ABL. By contrast point mutations were seen in the majority of cases of acquired resistance. Also, in 2 out of 40 patients, a provocative observation of a mutation as an isolated and temporary event was made, which suggest that mutations should be considered with caution outside the context of a trigger for mutation analysis, e.g., unsatisfactory response or loss of earlier obtained response.
Our data are in line with those of other investigators in that mutations are mostly seen in patients with secondary acquired rather than primary intrinsic imatinib resistance [3, 28]. Branford et al. [3] reported that 61% of patients with a single rise of at least twofold in the BCR-ABL mRNA level had detectable mutations. This contrasted with only one mutation among 158 cases with stable or decreasing levels of BCR-ABL transcripts. However, a recent study by Wang et al. [31] failed to confirm this stringent association between rise in BCR-ABL mRNA and mutation detection. They found that a single twofold or greater rise in BCR-ABL mRNA was a poor predictor of mutation detection, but confirmation of the rise in a subsequent test was highly predictive of mutations. It is not clear what underlies this discrepancy. One possibility is fluctuations in assay performance, varying from laboratory to laboratory. Nevertheless, because most imatinib-treated CML patients will obtain CCgR, the definition of molecular relapse has become clinically important. It was suggested that a reasonable compromise would be to consider a five- to tenfold rise of BCR-ABL transcripts that are significant and would necessitate a repeat test within a short time frame [6].
In some cases BCR-ABL kinase domain mutations preceded the progression to advanced phase disease and could even be detected in the pretreatment samples [25, 32]. These studies were performed using sensitive techniques, e.g., allele-specific oligonucleotide PCR, for mutation detection and they could imply that mutations should be identified as early as possible because they may indicate the need to reconsider the treatment strategy. Our study and other reports have failed to detect mutations in pretreatment samples, possibly due to lack of sufficient sensitivity of the techniques used for mutation screening. However, it can be argued that mutant clones at low levels may not have the same clinical significance as clones that are detected in the context of rising disease burden [16, 32]. It has to be kept in mind that cytogenetic nonresponders in late CP, as many as 50%, had evidence of BCR-ABL kinase domain mutations by denaturing high-performance liquid chromatography analysis [28]; the vast majority of these patients had a sustained HR at the time of mutation detection.
In conclusion, most imatinib resistance in CML is not caused by BCR-ABL mutations if cytogenetic and molecular (i.e., detectable BCR-ABL transcripts by PCR) persistent disease is included in the concept. Monitoring imatinib-treated patients for BCR-ABL kinase domain mutations provides a guide for clinical management in patients with signs of imatinib resistance. There is evidence in the majority of patients with acquired resistance of either increased expression of BCR-ABL or, more frequently, mutations in the kinase domain of BCR-ABL. Both types of resistance can be overcome by alternative ABL inhibitors that exhibit increased potency or capture additional conformations of the ABL kinase. Two of these compounds, dasatinib and nilotinib (AMN107), are in phase I/II trials and have demonstrated very encouraging clinical activity [15, 30]. Both agents are active against most imatinib-resistant ABL kinase mutants with the notable exception of the T315I mutant, which is completely resistant to imatinib, nilotinib, and dasatinib [23, 26]. However, monitoring all patients for mutations at regular time points is not feasible or cost-effective. It was shown that a rising level of BCR-ABL mRNA measured by quantitative RT-PCR [4] is tightly linked to the emergence of a mutant clone. Therefore, patients with stable or decreasing BCR-ABL levels may not require mutation screening, but for patients with signs of an expanding disease burden, a search for BCR-ABL mutations is warranted.
References
Al-Ali HK, Heinrich MC, Lange T, Krahl R, Mueller M, Muller C, Niederwieser D, Druker BJ, Deininger MW (2004) High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib. Hematol J 5:55–60
Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP, Hughes TP (2002) High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 99:3472–3475
Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, Taylor K, Herrmann R, Seymour JF, Arthur C, Joske D, Lynch K, Hughes T (2003) Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 102:276–283
Branford S, Rudzki Z, Parkinson I, Grigg A, Taylor K, Seymour JF, Durrant S, Browett P, Schwarer AP, Arthur C, Catalano J, Leahy MF, Filshie R, Bradstock K, Herrmann R, Joske D, Lynch K, Hughes T (2004) Real-time quantitative PCR analysis can be used as a primary screen to identify patients with CML treated with imatinib who have BCR-ABL kinase domain mutations. Blood 104:2926–2932
Daley GQ, Van Etten RA, Baltimore D (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247:824–830
Deininger M, Buchdunger E, Druker BJ (2005) The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 105:2640–2653
Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M (2001) Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 344:1038–1042
Goldman JM (2004) Chronic myeloid leukemia—still a few questions. Exp Hematol 32:2–10
Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL (2001) Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293:876–880
Hardling M, Wei Y, Palmqvist L, Swolin B, Stockelberg D, Gustavsson B, Ekeland-Sjoberg K, Wadenvik H, Ricksten A (2004) Serial monitoring of BCR-ABL transcripts in chronic myelogenous leukemia (CML) treated with imatinib mesylate. Med Oncol 21:349–358
Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, Hanfstein B, Schoch C, Cross NC, Berger U, Gschaidmeier H, Druker BJ, Hehlmann R (2002) Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 16:2190–2196
Hofmann WK, Komor M, Wassmann B, Jones LC, Gschaidmeier H, Hoelzer D, Koeffler HP, Ottmann OG (2003) Presence of the BCR-ABL mutation Glu255Lys prior to STI571 (imatinib) treatment in patients with Ph+ acute lymphoblastic leukemia. Blood 102:659–661
Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML, Gathmann I, Bolton AE, van Hoomissen IC, Goldman JM, Radich JP (2003) Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 349:1423–1432
Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, O’Brien SG, Russell N, Fischer T, Ottmann O, Cony-Makhoul P, Facon T, Stone R, Miller C, Tallman M, Brown R, Schuster M, Loughran T, Gratwohl A, Mandelli F, Saglio G, Lazzarino M, Russo D, Baccarani M, Morra E (2002) Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 346:645–652
Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG (2006) Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 354:2542–2551
Khorashad JS, Anand M, Marin D, Saunders S, Al-Jabary T, Iqbal A, Margerison S, Melo JV, Goldman JM, Apperley JF, Kaeda J (2006) The presence of a BCR-ABL mutant allele in CML does not always explain clinical resistance to imatinib. Leukemia 20:658–663
Kreil S, Mueller M, Hanfstein B, La Rosee P, Lahaye T, Hehlmann R, Hochhaus A (2003) Management and clinical outcome of CML patients after imatinib resistance associated with ABL kinase domain mutations. Blood 102:238a
le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L, Cabrita G, Marchesi E, Supino R, Gambacorti-Passerini C (2000) Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood 95:1758–1766
Lugo TG, Pendergast AM, Muller AJ, Witte ON (1990) Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 247:1079–1082
Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM, Melo JV (2000) Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood 96:1070–1079
Miething C, Feihl S, Mugler C, Grundler R, von Bubnoff N, Lordick F, Peschel C, Duyster J (2006) The Bcr-Abl mutations T315I and Y253H do not confer a growth advantage in the absence of imatinib. Leukemia 20:650–657
Muller MC, Gattermann N, Lahaye T, Deininger MW, Berndt A, Fruehauf S, Neubauer A, Fischer T, Hossfeld DK, Schneller F, Krause SW, Nerl C, Sayer HG, Ottmann OG, Waller C, Aulitzky W, le Coutre P, Freund M, Merx K, Paschka P, Konig H, Kreil S, Berger U, Gschaidmeier H, Hehlmann R, Hochhaus A (2003) Dynamics of BCR-ABL mRNA expression in first-line therapy of chronic myelogenous leukemia patients with imatinib or interferon alpha/ara-C. Leukemia 17:2392–2400
O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, Druker BJ (2005) In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res 65:4500–4505
Roche-Lestienne C, Preudhomme C (2003) Mutations in the ABL kinase domain pre-exist the onset of imatinib treatment. Semin Hematol 40:80–82
Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, Fenaux P, Preudhomme C (2002) Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood 100:1014–1018
Roumiantsev S, Shah NP, Gorre ME, Nicoll J, Brasher BB, Sawyers CL, Van Etten RA (2002) Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc Natl Acad Sci U S A 99:10700–10705
Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2:117–125
Soverini S, Martinelli G, Rosti G, Bassi S, Amabile M, Poerio A, Giannini B, Trabacchi E, Castagnetti F, Testoni N, Luatti S, de Vivo A, Cilloni D, Izzo B, Fava M, Abruzzese E, Alberti D, Pane F, Saglio G, Baccarani M (2005) ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on chronic myeloid leukemia. J Clin Oncol 23:4100–4109
Talpaz M, Kantarjian HM, McCredie K, Trujillo JM, Keating MJ, Gutterman JU (1986) Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N Engl J Med 314:1065–1069
Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, Huang F, Decillis AP, Sawyers CL (2006) Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 354:2531–2541
Wang L, Knight K, Lucas C, Clark RE (2006) The role of serial BCR-ABL transcript monitoring in predicting the emergence of BCR-ABL kinase mutations in imatinib-treated patients with chronic myeloid leukemia. Haematologica 91:235–239
Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, Stoffregen EP, McWeeney S, Kovacs I, Park B, Druker BJ, Deininger MW (2005) High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood 106:2128–2137
Acknowledgements
This study was supported by grants from the Swedish Research Council (project K2002-71X-11630-07B), FoU Västra Götaland, the Swedish Society for Medical Research, “JK Foundation” Sahlgrenska University Hospital, and “Volvo Assar Gabrielssons Foundation.”
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wei, Y., Hardling, M., Olsson, B. et al. Not all imatinib resistance in CML are BCR-ABL kinase domain mutations. Ann Hematol 85, 841–847 (2006). https://doi.org/10.1007/s00277-006-0171-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-006-0171-8