
Abstract
Background
Amyotrophic lateral sclerosis (ALS) is characterized by a progressive loss of motor neurons (MNs), leading to paralysis, respiratory failure and death within 2–5 years of diagnosis. The exact mechanisms of sporadic ALS, which comprises 90% of all cases, remain unknown. In familial ALS, mutations in superoxide dismutase (SOD1) cause 10% of cases.
Methods
ALS patient-derived human-induced pluripotent stem cells (ALS hiPSCs, harboring the SOD1AV4 mutation), were differentiated to MNs (ALS-MNs). The neuroprotective effects of conditioned medium (CM) of hESCs (H9), wt hiPSCs (WTC-11) and the ALS iPSCs, on MN apoptosis and viability, formation and maintenance of neurites, mitochondrial activity and expression of inflammatory genes, were examined. For in vivo studies, 200 μl of CM from the ALS iPSCs (CS07 and CS053) was injected subcutaneously into the ALS model mice (transgenic for the human SOD1G93A mutation). Animal agility and strength, muscle innervation and mass, neurological score, onset of paralysis and lifespan of the ALS mice were assayed. After observing significant disease-modifying effects, the CM was characterized biochemically by fractionation, comparative proteomics, and epigenetic screens for the dependence on pluripotency. CM of fibroblasts that were differentiated from the wt hiPSCs lacked any neuroprotective activity and was used as a negative control throughout the studies.
Results
The secretome of PSCs including the ALS patient iPSCs was neuroprotective in the H2O2 model. In the model with pathogenic SOD1 mutation, ALS iPSC-CM attenuated all examined hallmarks of ALS pathology, rescued human ALS-MNs from denervation and death, restored mitochondrial health, and reduced the expression of inflammatory genes. The ALS iPSC-CM also improved neuro-muscular health and function, and delayed paralysis and morbidity in ALS mice. Compared side by side, cyclosporine (CsA), a mitochondrial membrane blocker that prevents the leakage of mitochondrial DNA, failed to avert the death of ALS-MNs, although CsA and ALS iPSC-CM equally stabilized MN mitochondria and attenuated inflammatory genes. Biochemical characterization, comparative proteomics, and epigenetic screen all suggested that it was the interactome of several key proteins from different fractions of PSC-CM that delivered the multifaceted neuroprotection.
Conclusions
This work introduces and mechanistically characterizes a new biologic for treating ALS and other complex neurodegenerative diseases.
Similar content being viewed by others


Background
Neuronal cell death causes a plethora of neurodegenerative diseases, including age-related loss of memory and dementias (such as Alzheimer’s disease), Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). These diseases have in common the increased reactive oxygen species (ROS), mitochondrial dysfunctions, protein misfolding and aggregation, neuroinflammation, progressive loss of specific subtypes of neurons, and so far, the lack of a cure.
ALS is a progressive neurodegenerative disease with rapid onset of paralysis and respiratory failure, and morbidity within 2–5 years after diagnosis [1]. More than 5000 patients in the United States receive a diagnosis of ALS each year [1]. Mutations in superoxide dismutase 1 (SOD1), oxidative damage and protein misfolding are involved in both familial and sporadic ALS [2,3,4,5,6,7]. Transgenic mice with human SOD1G93A (B6SJL-TgN[SOD1-G93A]1Gur) are one of the most widely used models to study ALS as they recapitulate the pathophysiology in humans, such as motor neuron (MN) loss, degradation of neuromuscular junctions, axonal degeneration and limb paralysis [3, 8,9,10,11].
Various pluripotent stem cells (PSCs), such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), are used in various biomedical fields due to their capacity of unlimited self-renewal and the ability to differentiate into multiple cell types [12]. ALS iPSCs have been established and ALS iPSC-derived MNs show pathological characteristics of this disease: diminished neurite length, protein aggregation, and an enhanced apoptosis [13,14,15,16] that is reversible by over-expression of Bcl-2 [17].
Considering the limitations of tissue-specific differentiation and oncogenic side-effects of undifferentiated PSCs, stem cell-derived acellular factors might be a safer alternative to cell transplantation [18]. We previously reported the enhancement of old muscle repair by human ESC (hESC)-derived conditioned medium (CM) (hESC-CM), and the ability of this ESC-CM to absorb/neutralize toxic beta-amyloid [19, 20]. Several other studies focused on CM from mesenchymal and adipose stem cells [21,22,23,24], but none have shown meaningful reversal of the multiple, complex, and mutually enforcing pathologies of ALS.
Materials and methods
Animals
All animal experimental procedures were performed in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health, and approved by the Office of Laboratory Animal Care (OLAC), UC Berkeley.
SOD1G93A (B6SJL-Tg(SOD1 * G93A)1Gur/J) transgenic mice were purchased from Jackson Laboratory (No. 002726; Bar Harbor, ME). All mice were randomly assigned to receive ALS iPSC-CM treatment or control CM produced by hESCs differentiated into fibroblasts (dF-CM).
Culture of human pluripotent stem cells (hPSCs) and collection of CM
hESCs (H9, WiCell Research Institute, Madison, WI) and hiPSC (WTC-11) were obtained through the UC Berkeley Cell Culture Facility. The SOD1-mutated familial ALS-iPSCs, CS07 and CS53, were obtained from Cedars-Sinai Medical Center (Los Angeles, CA) (Additional file 1: Table S1). All hPSCs were maintained on plates coated with Vitronectin (Life Technologies, Carlsbad, CA) and cultured in Essential-8 medium (Life Technologies) at 37 °C in a 5% CO2 atmosphere. All hPSCs were passaged every 7 days by 0.5 mM EDTA (Life Technologies). For collection of CM, hiPSCs were cultured to 80%–90% of confluence. Then, the cells were washed twice (5 min each) with Opti-MEM (ThermoFisher, Carlsbad, CA) and cultured in Opti-MEM for 24 h and the CM was collected as hESC-CM, hiPSC-CM, and ALS iPSC-CM, as published [20]. The collected CMs were centrifuged at 10,000g for 30 min at 4 °C, and filtered through a 0.22-μm syringe filter (Millipore-Sigma, Temecula, CA). CMs from the same groups were pooled (7–10 dishes) and stored in aliquots at − 80 °C before use. To obtain CM from differentiated cells, fibroblasts were derived by first differentiating hESCs and hiPSCs into embryoid bodies in 100-mm Petri dishes in Essential 6 (Life Technologies) at 37 °C in 5% CO2 for 5 days, and then into fibroblasts for 7 days, followed by two passages on 0.1% gelatin-coated tissue culture dishes (BD Falcon, San Jose, CA) in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal calf serum (FCS), 1% (v/v) non-essential amino acid, and 1% (v/v) penicillin–streptomycin. CM from the fibroblasts was collected and used identically to the hPSC CM.
Culture of human fibroblast cell line
Human lung fibroblast cells, IMR-90 (CCL-186, ATCC, Manassas, VA), were maintained in DMEM supplemented with 10% FCS and 1% penicillin–streptomycin (Invitrogen, Carlsbad, CA) and maintained in a humid atmosphere at 37 °C containing 5% CO2. The cells were grown to 70% confluence and subcultured at 1:5 split when passaging.
Motor neuron precursor (MNP) and MN differentiation
The process of MNP and MN differentiation was as published [25]. Briefly, hPSCs were detached by 0.5 mM EDTA and split 1:10 on Matrigel-coated plates (1:50 dilution). After 1 day, the Essential 8 was changed to neural-inducing medium containing Neurobasal medium (NBM) plus 3 μM CHIR99021 (Tocris, Tracy, CA), 2 μM LDN-193189 (Tocris) and 3 μM SB431542 (Stemgent, Beltsville, MD). The NBM was composed of DMEM/F12 and Neurobasal medium (ThermoFisher) at 1:1, N2, B27, 0.1 mM ascorbic acid (Millipore-Sigma, Temecula, CA), 1× Glutamax and 1× penicillin/streptomycin (all from Invitrogen, Carlsbad, CA). The inducing medium was maintained for 5 days and changed every other day. On day 5, the cultured cells were split at 1:6 with Accutase and cultured in NBM containing 3 μM CHIR99021, 2 μM SB431542, 0.1 μM retinoic acid (Stemgent) and 0.5 μM purmorphamine (Stemgent). After 7 days, MNPs were generated and expanded with MNP medium, which consisted of NBM supplemented with 3 μM CHIR99021, 2 μM SB431542, 0.1 μM retinoic acid, 0.5 μM purmorphamine and 0.5 mM valproic acid (Stemgent). MNPs were passaged every 7 days by exposure to Accutase and cryopreserved in liquid nitrogen in DMEM/F12, 10% Knockout serum replacement and 10% DMSO. To induce MN differentiation, MNPs were dissociated with Accutase and cultured in suspension in the above NBM with 1 μM retinoic acid and 0.1 μM purmorphamine. The medium was changed every other day. After 7 days, MNPs differentiated into MNs. The MNs were then dissociated with Accutase into single cells and plated onto PLO/laminin-coated plates. The MNs were cultured into mature neurons in NBM plus 1 μM retinoic acid, 0.1 μM Compound E (Calbiochem, San Diego, CA), 10 ng/ml insulin-like growth factor 1, 10 ng/ml glial cell-derived neurotrophic factor (ThermoFisher), and 10 ng/ml brain-derived neurotrophic factor (ThermoFisher).
Isolation of heparin-binding proteins from CM
The heparin-binding proteins of PSC-CM were prepared as published [20]. Briefly, heparin-agarose type I beads (H 6508, Sigma-Aldrich, St. Louis, MO) were washed with molecular-grade water and incubated with CM from the various cells for 2 h with agitation at 4 °C. Then the samples were centrifuged at 10,000g for 20 min. The collected heparin beads were washed twice for 10 min at 4 °C in washing buffer (1 ml phosphate-buffered saline [PBS] + 0.05% Tween-20). The heparin-binding proteins were eluted twice for 15 min at 4 °C in 400 μl of elution buffer (0.01 M Tris–HCl pH 7.5 + 1.5 M NaCl + 0.1% BSA). The collected proteins were re-equilibrated to the cell culture medium by dialysis for 2 h at 4 °C in 500 ml of McCoy's 5A Medium (Gibco, Carlsbad, CA) followed by overnight dialysis at 4 °C in 200 ml of Opti-MEM (Gibco). The eluted heparin-bound proteins were diluted to 800 μl with Opti-MEM and stored at − 80 °C before use.
Isolation of exosomes from CM
For the isolation of exosomes, 100-ml samples of hESC-CM were centrifuged at 2000g for 10 min to remove cells and debris, followed by centrifugation at 10,000g for 30 min at 4 °C to remove microvesicles [26]. The supernatant was filtered through a 0.22-μm filter (Millipore-Sigma, Temecula, CA) and then the filtered CM was sedimented by ultra-centrifugation at 120,000g for 90 min in an i70 rotor (Beckman Coulter, Brea, CA). The pellet was washed once with PBS and re-sedimented at 120,000g, 4 °C for 90 min. Finally, the pellet was resuspended in 200 μl of PBS and stored at − 80 °C for further use.
Treatment of cyclosporine (CsA)
MNs were cultured with 10 μM CsA (11–011-00, Tocris) for 3 or 5 days. Then, cell viability, apoptosis, real-time polymerase chain reaction (PCR), and enzyme-linked immunosorbent assay (ELISA) were performed.
Cell viability assay
After treatment with H2O2 and/or CM or CM-derived factors, the number of viable cells was determined by a resorufin-based assay using the commercially available CellTiter-blue® cell viability assay (G8080, Promega, Sunnyvale, CA), according to the manufacturer’s protocol. MNs were cultured in a 96-well plate at a cell density of 2 × 104 and treated with H2O2 and/or CMs for 24 h before performing the viability assay. A SpectraMax iD3 Multi‐Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) was used to measure fluorescence intensity (560/590 nm).
Antioxidant assay
The antioxidant assay was performed according to the manufacturer’s instructions (DTAC-100, BioAssay systems, Hayward, CA). Briefly, each sample (20 μl) was mixed with kit reagent (100 μl). Then the mixed sample was incubated for 10 min at room temperature. The color intensity at 570 nm was measured by a SpectraMax iD3 Multi‐Mode Microplate Reader. The Trolox values ranging 0–1000 μM were used as a reference.
Real-time PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA), and the SuperScript III First‐Strand Synthesis System (Invitrogen, Carlsbad, CA) was used to synthesize cDNA according to the manufacturer’s instructions. Real‐time PCR was performed on a Bio‐Rad iQ5 real‐time PCR machine. The primers used for PCR are listed in Additional file 1: Table S1.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.25% Triton X-100 and blocked with 5% FCS in PBS for 1 h. The fixed cells were incubated overnight at 4 °C in PBS + 1% FCS with antibodies mouse anti-OCT4 (1:500, ab18976, Abcam, Cambridge, MA), rabbit anti-SOX2 (1:500, MA516399, ThermoFisher), rabbit anti-TUJ1 (1:500, MAB1195, R&D system, Minneapolis, MN), rabbit anti-OLIG2 (1:500, NBP128667, Novus), rabbit anti-HB9 (1:500, ABN174, Millipore-Sigma, Temecula, CA), rabbit anti-CHAT (1:500, AB144P, Abcam, Cambridge, MA), and rabbit anti-cleaved caspase-3 (CC3, 1:500, 9669S, Cell Signaling Technology) followed by incubation with secondary antibodies: FITC-conjugated anti-mouse IgG (1:1000, A21202, Life Technologies), FITC-conjugated anti-rabbit IgG (1:1000, A11034, Life Technologies), Cy3-conjugated anti-mouse IgG (1:1000, A11003, Life Technologies), and Cy3-conjugated anti-rabbit IgG (1:1000, A11035, Life Technologies). The treated cells were covered with Slowfade antifade with DAPI (Life Technologies) for nuclear staining and covered with a glass coverslip. Images were captured with a fluorescence microscope (DM5000B, Leica, Wetzlar, Germmany).
Apoptosis assay
The apoptosis assay of fibroblasts was performed using Annexin V–CF Blue/7-amino-actinomycin D (7-AAD) Apoptosis Detection Kit (ab214663, Abcam) according to the manufacturer’s protocol. Briefly, cells were detached using 0.05% trypsin and washed twice with PBS. Then, samples were resuspended in 1× annexin-binding buffer and incubated with 5 μl of Annexin V-FITC and 5 μl of7-AAD for 15 min at 37 °C, avoiding light. Finally, the stained samples were analyzed on a Guava Easycyte Flowcytometer (Millipore-Sigma, Temecula, CA) at an excitation wavelength of 488 nm and emission filters of 525 and 625 nm. For MNs, the apoptotic assay was conducted on two different markers, FITC-conjugated Annexin V (ab201540) or CC3 (9669S). For Annexin V detection, MNs in 96-well plates were washed twice with PBS. Then, 100 μl of Annexin V binding buffer was added with 5 μl Annexin V-FITC for 5-min incubation at 37 °C, avoiding light. The stained plate was analyzed on a SpectraMax iD3 Multi‐Mode Microplate Reader (490/525 nm). For the CC3, the immunofluorescence protocol was used.
High-content analysis
For analyses of differentiation efficiency, neurite outgrowth, and apoptosis, the plates were imaged using the high-content imaging system, ImageXpress Micro (IXM, Molecular Devices); a set of 5 × 5 or 7 × 7 fields was collected from each well using the 10× or 20× objective. Data were further analyzed in MetaXpress 6 software (Molecular Devices).
Analysis of neurite length
During differentiation into MNs, cells were sampled at 5-day intervals between 20 and 60 days. MNs were cultured in 8-well chamber slides (LabTek II CC2-coated), stained with Tuj1 and imaged by IXM. MetaXpress 6 software was used to analyze the mean process length as a measure of total outgrowth divided by the number of processes of a cell. Analysis was performed on a total of 4–10 fields and at least 1000 cells per group from three independent experiments were analyzed. For neurite size comparison, the data were normalized to the values at day 20; shrinkage of neurites over time was determined as the net change from neurite sizes on days 20 and 25.
ELISA
ELISA was analyzed for IP-10 protein (DIP100, R&D System, Minneapolis, MN) and the sample dilution was performed according to the manufacturer’s protocol (1:20–1:50). The stained plate was analyzed on a SpectraMax iD3 Multi‐Mode Microplate Reader.
Antibody array
CMs from PSCs or differentiated cells were analyzed on a human L1000 antibody capture array (AAH-BLG-1000, Raybiotech, Norcross, CA), processed according to the manufacturer’s protocol. The array slides were imaged by a Molecular Devices 4000b scanner and data were calculated by Genepix. After normalization to dF-CM, proteins with levels over twofold up- or 0.5-fold down-regulated were selected.
Epigenetic inhibitor screen
Separate cultures of hiPSCs were incubated for 3 days with one of the following small molecules: 5 μM Nanomycin A (A8191, Apexbio, Walnut, CA), 10 μM LSD1 inhibitor (489476, Millipore-Sigma, Temecula, CA), 5 μM 5-Azacytidine (C832A53, Thomas Scientific, Sanjose, CA), and 10 μM GSK126 (67-905, Tocris, Tracy, CA). After 3 days of treatment, CM was collected (without the inhibitors) using the CM collection protocol as described above.
Measurement of mitochondrial stress
MNs were detached with Accutase, washed with PBS and incubated for 10 min with 5 μM Mitosox Red (M36008, ThermoFisher) at 37 °C in the dark. After three washes in warm PBS, the stained MNs were resuspended in warm FACS buffer (1% FCS in PBS) and analyzed by a Guava easyCyte flow cytometer (620/52 nm).
Bioinformatics analysis
The genes of differentially regulated proteins were analyzed using DAVID Bioinformatics Resources (version 6.8, https://david.ncifcrf.gov). The GO (Gene Ontology) functional annotation based on the biological process, molecular function and cellular component, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway were performed. Heat maps were generated with ClustVis [27]. Violin plots were constructed with the Python seaborn package and normalized with a standard min–max scaler.
CM treatment and behavioral test
The CM of ALS iPSCs (CS07 and CS053, Cedar-Sinai) or CM of fibroblasts was injected subcutaneously at the neck in a volume of 200 μl every other day as described [28].
For behavioral test, we monitored the weight, neurological scoring, and hanging time every other day. First, all mice were weighed. Then, we assayed them by neurological scoring, as published [29] with criteria as follows:
-
Score of 0 While mouse is suspended by the tail, the hindlimbs are fully extending away from the lateral midline and the status kept for 2 s.
-
Score of 1 Partial collapse of leg extension or trembling of hind legs during tail suspension. This stage is considered an onset of symptoms.
-
Score of 2 While mouse is suspended by the tail, the hindlimbs are not extending much or completely collapse. During walking of 90 cm, toes curling or dragging of leg parts is observed.
-
Score of 3 Rigid paralysis or minimal joint movement is observed. Hindlimbs not being used much for forwarding motion. We considered this stage the end-stage.
All mice were euthanized at a score of 3. All information was recorded for each mouse every other day until the end-stage (a score of 3).
For hanging test, mice were placed on a grid and then turned upside down. Then, the latency to fall was measured. The latency time measurement began from the point when the mouse was hanging free on the grid and ended with the animal falling to the cage underneath the grid.
Survival
For survival assessment, 11 female (8 ALS iPSC-CM group and 3 dF-CM group) and 11 male (8 ALS iPSC-CM group and 3 dF-CM group) SOD1G93A mice were used. Animals that showed signs of paralysis with a score of 3 were euthanized, e.g., the non-survival group.
Tissue collection
When mice showed the first signs of paralysis in hind limbs or at 120 days, euthanasia was proceeded with the guidelines of UC Berkeley’s OLAC administration. For histological comparison, some ALS mice were euthanized at 120 days. Spinal cords and muscles (gastrocnemius and tibialis anterior) were isolated. Then, spinal cords were fixed with 4% paraformaldehyde overnight at 4 °C and muscle tissues were fixed for 30 min at room temperature. Then, all tissues were transferred to 30% sucrose solution overnight and embedded in tissue-tek optimal cutting temperature (OCT, Sakura Finetek, The Netherlands) and snap frozen in isopentane cooled to − 70 °C with dry ice. Then, all samples were stored at − 80 °C before use.
For histological analysis, all OCT-embedded tissues were cryosectioned by a Cryostar NX50 (ThermoFisher). Muscle tissues were sectioned to 20-µm thickness while spinal cords were cut at 10 µm.
Muscle weight
To compare the wet muscle weight of the gastrocnemius and tibialis anterior in all groups, mice were euthanized and muscle tissues were isolated, and both left and right muscles were weighed on a scale. For comparison, the weight recorded was normalized to the body weight.
Cresyl violet staining and quantification
Mounted frozen sections were dried overnight at 37 °C and slides placed directly into 1:1 alcohol/chloroform overnight, then rehydrated through 100% alcohol to distilled water. Next, the sections were stained in 0.1% cresyl violet solution for 5–10 min at 37 °C, rinsed with distilled water, dehydrated with ethanol, cleared in xylenes, and mounted in Permount (ThermoFisher). Stained sections were imaged at 5× and 10× magnifications. For quantification, five sections were selected randomly and the total number of MNs of the ventral spinal cord was counted using unbiased methods, as described [30].
Neuromuscular junction (NMJ) staining and quantification
NMJ analysis was performed as previously described [31]. Briefly, dried sections were permeabilized with 1% Triton X-100 and blocked with 5% FCS in PBS for 1 h. The fixed cells were incubated for 2 h at room temperature in PBS + 1% FCS with Alexa 488-conjugated anti-neurofilament (1:500, 8024, Cell Signaling Technology), Alexa 488-conjugated anti-synaptophysin (1:500, MAB5258A4, Millipore-Sigma, Temecula, CA), or Alexa 555-conjugated anti-α-bungarotoxin (1:500, B35451, Life Technologies). The stained slides were covered with Slowfade antifade with DAPI (Life Technologies) for nuclear staining and covered with a glass coverslip. Images were captured with a fluorescence microscope (DM5000B, Leica). For quantification, five sections were selected randomly and counted. Intact NMJs (yellow) were identified as those that exhibited an overlay of α-bungarotoxin (red) and neurofilament (green). Denervated NMJs were defined by labeling with only α-bungarotoxin (red). Percent of intact NMJs was calculated as the number of intact NMJs (yellow)/total number of motor end plates (yellow + red) as described [31].
Statistical analysis
All statistical analyses were performed using GraphPad Prism software version 9 (GraphPad software Inc., San Diego, CA). All values are expressed as means ± SEM for independent experiments, or SD for replicates. To determine the significance of differences among groups, comparisons were made using Student’s t test. Survival and onset data were analyzed with Kaplan–Meier curves and log rank test. P < 0.05 was considered as statistically significant.
Results
Autologous ALS iPSC-CM is neuroprotective for MNs of ALS patients
hESC-CM was previously shown to enhance proliferation of myoblasts and rat neural precursor cells [19, 20], but the effects on cell viability were less studied. We decided to explore a protective role of hESC-CM when cells experience oxidative damage, which is physiologically and clinically relevant in a number of human diseases [32, 33]. hESC-CM protected human fibroblasts and human MNs that were differentiated from the hESCs from H2O2-caused cell death (Additional file 2: Figures S1 and S2). Additional antioxidant activity of hESC-CM was tested and not found (Additional file 2: Figure S3), suggesting that it did not simply neutralize ROS to preserve cell viability. The control dF-CM did not have such neuroprotective properties and was used as a negative control throughout this study.
Encouraged by the neuroprotective effects of hESC-CM on human MNs, we tested the hypothesis that iPSC-CM is also neuroprotective and will promote the viability of ALS patient-derived MNs. If true, this would suggest a novel completely autologous approach to treating neurodegenerative diseases. We studied two ALS patient-derived iPSC lines (CS53-male and CS07-female) with the most common mutation in the SOD1 gene, A4V (Additional file 1: Table S2). These ALS-iPSC lines were confirmed for their markers of pluripotency and then differentiated into MNPs and MNs, in parallel with normal, wild-type SOD1 human iPSCs (WTC11), as confirmed by the immuno-detection of OLIG2, Tuj1, and HB9 (Additional file 2: Figure S4a) and by qPCR on OCT4, NANOG, OLIG2, HB9, and CHAT (Additional file 2: Figure S4b). Interestingly, the WTC11 and ALS iPSC cells did not differ in the expression of pluripotency markers or morphology at iPSC stage, or in their MN markers when differentiated (Additional file 2: Figures S2 and S4). Additionally, there was no difference in the derivation efficiency of WTC11-MN and ALS-MN (Additional file 1: Tables S3 and S4).
When exposed to H2O2, MNs had diminished viability and increased apoptosis, and interestingly, all tested PSC CM, hESC-CM, WT iPSC-CM and most importantly the ALS patient-derived iPSC-CM, had neuroprotective effects in this experimental set-up (Fig. 1a). We further explored the timing of this neuroprotective effect of healthy human iPSC-CM and ALS patient iPSC-CM on MNs by either pre-treating the cells with these CMs at variable time points before H2O2 (Pre-treatment) or alternatively, by treating the cells with H2O2 first for variable times before adding the CMs (Post-treatment). The viability and apoptotic assays demonstrated that either WT iPSC-CM (Additional file 2: Figure S5) or ALS iPSC-CM (Fig. 1b) was critically needed to be present at least 10 min before H2O2 was added to protect the MNs from the cytotoxic effects, whereas the addition of iPSC-CMs after H2O2 (10 min to 24 h) did not confer this neuroprotection.

ALS iPSC conditioned medium (CM) is neuroprotective for ALS-MNs. a The effect of ALS iPSC-CM on viability and apoptosis in ALS-MNs. The viability of ALS-MNs exposed to H2O2 was measured by MTT assay. The ratio of apoptotic to live ALS-MNs was assayed by Annexin V fluorescence in a plate reader. n = 4, *P < 0.05. b The time course of viability (560/590 nm) and apoptosis (490/590 nm) of MNs that were pre-treated or post-treated (H2O2 first, then ALS iPSC-CM) with ALS iPSC-CM. c Representative images of MNs at 30 days and 60 days. WT, WT iPSC differentiated into MNs; ALS CS53, ALS iPSC differentiated into MNs; ALS CS53 + CM, iPSC differentiated into MNs in the presence of ALS iPSC CM. Scale bar, 200 μm. d Experimental schematic. The CMs from WT-iPSC, ALS-iPSC, and iPSCs-derived fibroblasts (dF-CM) were used to treat the differentiating MN cultures on days 30–60, after which MN assays were performed. e The neurite size was measured during MN differentiation from hESC, iPSC and ALS-iPSCs (± autologous ALS iPSC-CM on days 30, 35, 40, 45, and 50). ALS-MN neurites (CS53 and CS07) were maintained by ALS iPSC-CM when treatment started on day 30 or 35, but not on later days. For the negative control dF-CM, there were no cells that maintained neurites. Mean ± SEM, experiments were repeated with four independently derived MN cell lines from ALS hiPSCs (i.e., n = 4). *P < 0.05. f The net change of neurite size was examined during differentiation of ALS-MNs treated with autologous ALS iPSC-CM, as indicated. g Left, the percent of cleaved caspase-3 (CC3)-positive cells was quantified during this directed differentiation of MNs. Right, representative images of CC3 (green)-positive MNs with DAPI staining of cell nuclei (blue). Mean ± SEM, n = 5. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar, 100 μm
ALS-MNs are known to show a decrease in the size of their neurites as compared to wild-type MNs [14], and thus we examined this physiologically important parameter in the primary ALS-MN lines (CS53 and CS07) that were treated with their autologous ALS iPSC-CMs. The neurite size increased gradually for 40 days of differentiation of iPSCs into MNs with no difference between the wild-type and ALS cells (Fig. 1c, Additional file 2: Figure S6a and b). However, at 45 days of differentiation and all subsequent time points, the neurite size markedly shrunk in the ALS cohort as compared to the wild-type cells (Fig. 1c, Additional file 2: Figure S6b). The neurite length fluctuation suggested that the time from 40 to 45 days represents a key transition time-point of regulation of neurite outgrowth, where shrinkage becomes prominent in ALS cells.
Considering that pathological changes in the MNs started to manifest at ~ 40 days of their differentiation from the ALS hiPSCs (Additional file 2: Figure S6a, b), the CM was added to the culture medium on day 30, 35, 40 or 45 (Fig. 1d). The assays of neurite size showed that the maintenance of neurites was improved in the groups treated with ALS iPSC-CM starting on either day 30 or day 35 (Fig. 1e and Additional file 2: Figure S6c). Moreover, the rate of neurite size degeneration was significantly reduced by ALS iPSC-CM added on day 30 or 35 in both CS53 and CS07 ALS-MN lines (Fig. 1f). However, when added at later time point, e.g., on day 40, ALS iPSC-CM was not able to rescue the neurite size of ALS-MNs (Fig. 1e and Additional file 2: Figure S6c).
We also analyzed the relative numbers of apoptotic cells in CS53 and CS07 ALS-MNs. There was a slight upward trend in the number of CC3-positive cells between the wild-type and ALS-MN lines up to day 30, but from day 35 onward the number of CC3-positive cells started to dramatically increase in the ALS-MN cultures as compared to the healthy MN cultures (Fig. 1g and Additional file 2: Figure S6d). Notably, the rate of ALS-MN apoptosis was significantly attenuated by the autologous ALS iPSC-CM, for both ALS-MN cell lines (Fig. 1g and Additional file 2: Figure S6e).
These data reveal that ALS patient iPSC-CM has multi-functional neuro-protective effects on their ALS-MNs, improving cell viability and the maintenance of neurites.
Neuroprotection, improved neuro-muscular function, and delayed morbidity of ALS mice bearing the human disease SOD1G93A
To determine whether the CMs from ALS patient-iPSCs could have the same neuro-protective effects in vivo, we used transgenic mice engineered to carry the mutant human SOD1 (SOD1G93A) gene, which display progressive degeneration of MNs and the phenotypes of ALS and are used extensively to study human ALS [29, 34, 35], to assess the effects of ALS iPSC secretome in vivo. The first symptoms occurred in these mice at 80–90 days of age and morbidity at 128 ± 9 days [36]. So we studied from the asymptomatic stage (68 days) through the entire progression of the disease to the morbidity endpoint.
Twelve ALS mice were studied for the effects of ALS iPSC-CM (CS07 and CS53) and 6 ALS mice were studied in parallel, using the negative control, dF-CM. Age-matched males and females were identically treated with either ALS iPSC-CM or dF-CM. Throughout the study, there was no significant difference in body weight between ALS iPSC-CM-treated and the dF-CM-treated mice (Additional file 2: Figure S7).
The control dF-CM-treated mice showed rapid deterioration of neuro-muscular function, as measured through neurological scoring [29] and four-limb hanging tests [37]. Remarkably, the ALS iPSC-CM rescued the agility, coordination, and overall muscle function of the SOD1 mutant mice, delayed the onset of pathological symptoms (Fig. 2a, b), delayed morbidity (Fig. 2c), and improved the neurological scores of the ALS model animals (Fig. 2d). Consistent with the neuro-muscular improvements, survival was significantly prolonged in the ALS iPSC-CM group as compared to the dF-CM group (141.5 ± 3.26 days vs 120.3 ± 3.07 days, P < 0.001) (Fig. 2c).

In vivo neuroprotection of ALS iPSC secretome in the SOD1G93A mouse ALS model. a Hanging test of the transgenic mice (ALS iPSC-CM-treated mice n = 12 and dF-CM-treated mice n = 6). The decline in performance was significantly delayed in the mice treated with ALS iPSCs-CM, as compared to the dF-CM (114.8 ± 4.29 days vs 91.0 ± 4.31 days, P < 0.01). Values are mean ± SEM. b Kaplan–Meier curves of the disease onset showing that onset was delayed in the ALS iPSC-CM, as compared to the dF-CM group. Onset: (116.8 ± 3.64 days vs 92.3 ± 3.44 days, P < 0.001). c Kaplan–Meier curves of animal survival showing that the ALS iPSC-CM increased lifespan, as compared to dF-CM (141.5 ± 3.26 days vs 120.3 ± 3.07 days, P < 0.001). d Neurological scores were found to be improved by the ALS iPSC-CM, as compared to dF-CM. e Histological analysis (WT for SOD1 C57.B6 mice, n = 6, and ALS mice, n = 8) at 120 days of age, and at the end stage (morbidity time point). Representative images for Cresyl violet staining (and their quantification, bar graph) in the ventral lumbar spinal cord at 120 days showing that ALS iPSC-CM improved the number and the morphology of motor neurons of ALS mice, as compared to the dF-CM. Arrowheads, shrunken MNs. Scale bar, 200 μm. f Representative images of immunostaining of NMJ in gastrocnemius muscle at 120 days and quantification showing that the percent of colocalized NMJ decreased in the dF-CM group, as compared to the WT mice, whereas this percent was improved in the ALS iPSC-CM group. Arrows, colocalized endplates; arrowheads, mis-localized endplates. NF, Neurofilament, SYN, Synaptophysin, AchR, Acetylcholine receptors. Scale bar, 100 μm. g Mass of tibialis anterior (TA) and gastrocnemius (GA) muscles was also increased in the ALS mice treated with ALS iPSC-CM as compared to dF-CM
To investigate the effect of ALS iPSC-CM on the lumbar spinal cord and neuromuscular junction, these structures were analyzed postmortem in 120-day-old mice. Compared to the C57.B6 wild-type animals, the SOD1G93A mice with dF-CM treatment showed decreased total number of cresyl violet-stained MNs in the ventral spinal cord (58.5 ± 3.33 vs 13.5 ± 1.25, P < 0.001), indicating progression of the disease (Fig. 2e), and many cresyl violet-stained MNs showed shrunken morphology with no neurites (Fig. 2e). However, the number of MNs was significantly increased in the SOD1G93A mice treated with ALS iPSC-CM, as compared to the dF-CM group (41.25 ± 5.12 vs 13.5 ± 1.25, P < 0.001), which was also observed at the end-stage (at 150 days; 25.50 ± 5.32 vs 13.5 ± 1.25, P < 0.05; Fig. 2e). These results demonstrate that the treatment of ALS model mice with ALS iPSC-CM prevents deterioration of spinal MNs.
NMJ defects contribute to the decline in muscle mass and health [37], and NMJ alterations are a feature of neurological diseases [38,39,40]. Thus, we investigated NMJ integrity and muscle innervation, using the specific markers acetylcholine receptor, neurofilament and synaptophysin. Our results showed that the number of NMJs was significantly reduced in the dF-CM group, as compared to the C57.B6 group at day 120 (Fig. 2f), while the ALS iPSC-CM group had significantly more intact NMJs than the dF-CM group (48.46 ± 5.43 vs 22.87 ± 6.12, P < 0.001). As expected, at the end-stage, there was no difference between the dF-CM and the ALS iPSC-CM groups (Fig. 2f). These findings suggest that the treatment of SOD1G93A mice with ALS iPSC-CM preserves muscle innervation for a longer time, which is consistent with the improved performance in four-limb handing test and better neurological scores.
Improved NMJ integrity suggests a reduction in muscle atrophy, so we determined the weights of gastrocnemius anterior and tibialis anterior of the mice. The mass of gastrocnemius and tibialis anterior was normalized to the body weight. As expected, there was a significant decrease in the average weights of gastrocnemius and tibialis anterior in the dF-CM group, compared to the wild-type C57.B6 group. However, these weights were significantly increased in the ALS iPSC-CM group, as compared to the dF-CM group (Fig. 2g).
In summary, these data demonstrate that the ALS iPSC-CM significantly delays the onset of paralysis and functional decline in the SOD1G93A mice, extends their lifespan and protects MNs, NMJs and muscle mass. Such profound multiparametric positive effects in ALS, averting and delaying all key manifestations of this deadly disease, have not been reported previously with other tested methods.
Mechanisms, relation to pluripotency, sub-cellular details and improvement over CsA
To understand the mechanisms by which the iPSC secretome (CS07) is neuroprotective in ALS, we performed biochemical characterizations. First, we investigated whether the effect was produced by proteins. Heat inactivation and proteinase K treatments abrogated the positive effects of the iPSC secretome, demonstrating that the neuroprotective activity is contained in the protein fraction (Fig. 3a).

Mechanisms of neuroprotection by ALS iPSC-CM. a MTT assay for viability of MNs cultured with proteinase K-treated or heat-inactivated ALS iPSC-CM. ***P < 0.001, n = 4 for each condition. b MTT assay for viability of MNs that were cultured with 200 μg/ml H2O2 alone or co-treated with complete unfractionated ALS iPSC-CM, heparin-binding protein fraction, exosome fraction or HPB flow-through and exosome-free CM. *P < 0.05, **P < 0.01, n = 4 for each condition. c MTT assay for viability of MNs that were cultured with secretome of ALS iPSC-derived embryoid bodies (EBs). The viability was significantly decreased in the EB-CM group compared to that in the ALS iPSC-CM group (0.66 ± 0.02 vs 0.44 ± 0.05, P < 0.001). ***P < 0.001, n = 4 for each condition. d The effect of epigenetic modifiers on the neuroprotective activity of the iPSC secretome: ALS iPSCs were treated with the indicated inhibitors of pluripotency that influence epigenetics, and the activity of their CM was subsequently tested with H2O2-exposed ALS-derived MNPs, in quadruplicate MTT and Annexin V assays. EZH2 inhibitor GSK126 diminished the neuroprotective activity of the iPSC-CM with high statistical significance (viability: 0.64 ± 0.04 vs 0.47 ± 0.02, P < 0.01; apoptosis: 1.71 ± 0.17 vs 2.65 ± 0.23, P < 0.01), *P < 0.05, **P < 0.01, ***P < 0.001, n = 4 for each condition. e Levels of mitochondrial ROS. All MNs were stained with mitoSOX red and then the mean fluorescence intensity (MFI) was measured by a Guava Easycyte Flowcytometer. Autologous ALS iPSC-CM normalized mitochondrial activity of ALS-MNs statistically same as the known modifier, Cyclosporin A (CsA). Graphs show Mean ± SEM, n = 4. **P < 0.01. f The effects on inflammation were assayed by qRT-PCR. The indicated markers of inflammation were diminished by ALS iPSC-CM similarly to CsA. n = 3, ***P < 0.001, ns not statistically significant. n = 4, **P < 0.01, ***P < 0.001, ns not statistically significant. g The effect of CsA on MN viability and apoptosis. The viability of ALS-MNs as compared to healthy WTC-11 MNs was measured by MTT assay. The ratios of apoptotic to live MNs were assayed by Annexin V fluorescence in a plate reader. ALS-MNs were prone to cell death, as compared to WTC-11 MNs; ALS iPSC-CM, but not dF-CM or CsA, protected ALS-MNs from the ROS (mutant SOD1)-induced cell death
Several reports have shown that the exosomes from stem cell-CM have a protective effect on cellular damage/stress [26, 41]. Our previous studies documented that heparin-binding proteins from hESC-CM promote proliferation of muscle and neural cells [19, 20]. Accordingly, we examined whether the neuroprotective activity is contained in the heparin-bound (HB) versus unbound soluble fractions and exosome fraction. First, we determined the maximum amount of HB fraction tolerated by cells, and the HB fraction became cytotoxic at 300 μg/ml (Additional file 2: Figure S8a). Exosomes, purified from hESC-CM by ultracentrifugation [26], negatively affected cell viability at 200 μg/ml (Additional file 2: Figure S8b). Based on these data, 200 μg/ml heparin-binding proteins and 100 μg/ml exosome preparations were used to treat MNs. There was an increase in the viability of human MNs that were co-treated with H2O2 and HB fraction, as compared to H2O2 alone (0.58 ± 0.02 vs 0.49 ± 0.01, P < 0.05); however, the efficiency of that neuroprotection was less than that displayed by the unfractionated ALS iPSC-CM (0.66 ± 0.04 vs 0.58 ± 0.02, P < 0.05) (Fig. 3b). There was no improvement in viability or decrease in apoptosis of MNs that were co-treated with H2O2 and the exosome fraction (Fig. 3b). The soluble fraction that did not contain exosomes or HB proteins, had a slight, not statistically significant neuroprotection (Fig. 3b). These data suggest that the neuro-protective activity is contained in multiple fractions of PSC-CM, and mostly in the soluble fractions (heparin-bound and-unbound).
Based on the absence of neuroprotective activity in dF-CM, we decided to confirm and mechanistically extrapolate the relationship between the protective effects and pluripotency. As shown in Fig. 3c, differentiation of PSCs into embryoid bodies negated the neuroprotective activity, expanding the data on PSC-derived fibroblasts and confirming that a state of pluripotency is required for the neuroprotective activity. To follow on the requirement for pluripotency, we employed a screen with inhibitors of pluripotency that modify the epigenetic states of iPSCs. We treated separate cultures of ALS iPSCs with the inhibitors: Nanomycin A, LSD1 inhibitor, 5 Azacytidine, or GSK126 (Fig. 3d and Additional file 2: Figure S9). After 3 days in culture with each of these small molecules, the expression of pluripotency markers decreased (Additional file 2: Figure S9a). However, pluripotent colony-like morphologies were maintained, and OCT4 and NANOG expression was higher in each single inhibitor culture, as compared to the embryoid bodies differentiated from iPSCs (Additional file 2: Figure S9a and b). To determine the effects of epigenetic modifiers on the neuroprotective properties, we studied the viability and apoptosis of MNPs exposed to H2O2 that were pre-treated with CM from ALS iPSC exposed to the epigenetic inhibitors. Interestingly, treatment of ALS iPSCs with EZH2 inhibitor, GSK126, significantly reduced the neuroprotective activity of the ALS iPSC-CM (Fig. 3d), while other inhibitors showed no difference or non-statistical trend (Additional file 2: Figure S9c). This suggests the possibility that an EZH2-dependent pathway regulated by GSK126 is important for the neuroprotective activity of the ALS iPSC secretome.
To understand the sub-cellular effects of the ALS iPCS secretome on MN, we assayed mitochondrial health, which is known to diminish in ALS-MNs, and generally in neurons of patients with neurological diseases [42, 43]. Specifically, we performed the mitoSox red mitochondrial ROS analysis on ALS and healthy MNs, and on the ALS-MNs that were cultured with the ALS iPSC-CM as compared to the dF-CM (Fig. 3e). As expected, ALS-MNs had higher mean fluorescence intensity (MFI) of mitoSox red as compared to the healthy MNs, and the positive control, treatment with immunosuppressive CsA that blocks mitochondrial pores, reduced the MFI (Fig. 3e). Notably, autologous ALS iPSC-CM significantly reduced the MFI of mitoSox red, as compared to the untreated or dF-CM-treated MN, to a degree that was statistically indistinguishable from CsA (Fig. 3e).
We also explored the relationship of ALS iPSC-CM to neuroinflammation, the shared phenotype of many neurological disorders, including ALS [44,45,46]. ALS iPSC-CM significantly diminished the expression of pro-inflammatory INFB1, TNF-α, and IP-10 in ALS-MNs, and these effects were similar in the CsA-treated ALS-MNs (Fig. 3f).
Since CsA and ALS iPSC-CM both improved mitochondria and reduced expression of INFB1, TNF, and IP-10, we compared them side-by-side for neuroprotection in diminishing apoptosis and improving the viability of human ALS-MNs. Interestingly, only ALS iPSC-CM, but not CsA, reduced apoptosis and increased viability of the SOD1 mutant ALS-MNs that were derived from patients with this disease (Fig. 3g).
These results establish that the ALS iPSC-CM activity is mediated by multiple soluble secreted proteins (not exosomes), which likely act together since fractionation results in diminished activity of every fraction, as compared to the unfractionated CM. The ALS iPSC-CM activity is dependent on the state of pluripotency and in an EZH2-dependent process; it improves mitochondrial health and reduces neuro-inflammatory gene expression. These data also show that while CsA blocks mitochondrial pores and both CsA and ALS iPSC-CM stabilize mitochondria, there are additional effects of ALS iPSC-CM on averting the death of ALS-MNs.
Identification of a neuro-protective protein interactome
To identify candidate neuroprotective proteins, we performed comparative proteomic antibody capture arrays that detect 1000 factors, comparing hESC-CM, WT iPSC-CM and ALS iPCS-CM (CS07 and CS53) that all had neuroprotective activity, with each other and with the dF-CM that lacked neuroprotective activity (Fig. 4a). This screen identified 106 candidates (Venn diagram, Fig. 4b) that were up-regulated in all PSC-CM groups, as compared to the dF-CM (> twofold change with P < 0.05). The violin plot showed that the proteome patterns of hESC-CM, WT iPSC-CM and ALS iPSC-CM were significantly different from those of dF-CM (Additional file 2: Figure S10a). The list of KEGG pathways was constructed through grouping the proteins by their key functions (Fig. 4c). PSC-CMs and dF-CMs significantly differed in the levels of regulatory proteins that participate in canonical morphogenic signaling pathways, including PI3K-Akt, Jak-STAT, BMP/TGF-β, and Ras, all of which were shown to be important for development, viability and/or maintenance of neurons (Fig. 4d) [47].

Proteomic profiling analysis. a Heat mapping of WTC11, H9, CS07, CS53, and dF-CM groups. The dF-CM is farthest from the other groups. b The Venn diagram of the proteins that were present at different levels in studied conditioned media. c The KEGG pathway list according to biological functions. d Heat mapping of representative KEGG pathways, iP3K-AKT signaling pathway (P = 5.00E−04) and TGF-β signaling pathway (P = 9.30E−03). e Heat mapping of negative regulators of apoptotic process (P = 3.00E−06) and inflammatory response (P = 2.60E−06). *Negative regulation of neuronal apoptosis (P = 8.00E−03). f Heat mapping of inflammation-related proteins. The levels of these proteins were lower in PSC-CM than in dF-CM. g Heat mapping of Heparin-bound proteins and neuroprotective proteins. h Volcano plot of the PSC-CM and dF-CM. The red dots represent differently expressed proteins (P < 0.05; FC > 1.75), while the grey dots represent proteins with P > 0.05
Additional protein characterizations according to the biological process, molecular function, and cellular component by DAVID (Additional file 2: Figure S10b) showed that consistent with the functional data, the biological process group had 170 GO terms, including positive regulation of cell proliferation, signal transduction, positive regulation of peptidyl-tyrosine phosphorylation, negative regulation of apoptosis, control of wound healing, and beta-amyloid clearance; the latter highly consistent with our previously reported [20] neuroprotection from ectopic beta-amyloid (Fig. 4e and Additional file 2: Figure S10b).
One of the most important causes of MN degradation is the inflammatory response [44,45,46]. The proteomic screening showed that several proteins and cytokines known to induce inflammation, including IL-1, IL-6, IL-12, and TNF-α, were reduced in PSC-CMs compared to the dF-CM (Fig. 4f).
Interestingly, in concert with the above fractionation experiments and our previous findings on heparin-binding activity of pro-regenerative hESC-CM [19, 20], we identified several heparin-binding proteins that were elevated in the PSC-CM cohorts as compared to the dF-CM, including angiogenin (ANG), Angiopoietin-1 (ANG-1), angiopoietin like 3, apolipoprotein E (ApoE), secreted frizzled related protein 1, serpin family A member 5, thrombospondin 4, vascular endothelial growth factor B, fibroblast growth factor 11, and fibroblast growth factor 19 (Fig. 4g). Most of these heparin-affinity factors promote cell proliferation and attenuate cell death [48,49,50,51].
Among proteins that do not bind to heparin, but were identified in the neuroprotective PSC secretome (Fig. 4g), TIMP1 has shown protective effects against not only traumatic and ischemic brain injury but also HIV-1-induced neuronal apoptosis [51, 52]. IGFBP-2 is involved in regulation of cell proliferation and has an anti-apoptotic effect via modulation of CC3 [53, 54]. IGFBP-2 may have a role in neuroprotection from the hypoxic–ischemic injury and protects neurons from beta-amyloid-induced toxicity [55, 56]. HSP27 has an intrinsic responsibility in neuroprotection by increasing cell viability and decreasing apoptotic signaling [57,58,59]. Comparing PSC-CM and dF-CM through volcano plots, the identity of proteins which may play important roles in various aspects of neuroprotection was determined (Fig. 4h).
The comprehensive comparative proteomics suggests that activities of multiple ALS iPSC-secreted proteins have the capacity, in concert, to avert the degeneration and death of ALS-MNs: some proteins inhibit apoptosis by mechanisms independent from each other, while others reduce protein aggregation, reduce ROS damage, improve post-ROS repair, and/or support neuronal cell fate.
Discussion
This work demonstrates that proteins secreted by PSCs are neuroprotective and importantly, that the ALS patient-derived iPSCs can be used to combat their disease. Induced oxidative stress (such as by H2O2) is known to induce the death of many types of neuronal and non-neuronal cells. In sporadic and familial ALS, mutations in genes of different functions, promote oxidative stress, which is thought to contribute to neurodegeneration [4, 5, 60, 61]. Thus, we first established that hESC-CM was able to protect fibroblasts and MNs from H2O2-induced apoptosis. This neuroprotective activity was then confirmed with ALS patient hiPSC-CM. H2O2 is an extreme neurotoxin, hence it is important that in various screens and all independent experiments, PSC-CMs consistently enhanced the viability of MNs and diminished their apoptosis by ~ 20% in the presence of H2O2. Next, we established that proteins secreted by the WT and ALS hiPSCs promote cell viability, formation and stability of neurites, and mitochondrial health of SOD1 mutant human ALS-MNs. In vivo, human ALS iPSC-CM delayed the onset of symptom and extended the lifespan of SOD1 mutant ALS mice, through improved maintenance of MNs and NMJs. ALS iPSC-CM could slow down, but not overcome the progression of ALS, which might be improved when the defined proteins are determined and tested in future work.
The autologous patient’s iPSC secretome avoids the problems of cell transplantation: incomplete differentiation, danger of transplanting cancer-causing cells and difficulty of iPSC maturation [62,63,64]. Moreover, the autologous approach using the patient’s own hiPSCs will minimize the variability of heterologous hiPSCs and the immune response to heterologous proteins. Reprogramming of disease-specific differentiated cells into iPSCs often results in normal pluripotency, even when there are disease-associated mutations, and in our work, ALS-iPSCs were typical pluripotent stem cells, based on their markers and the efficiency of MN differentiation [65,66,67].
Mechanistically, our results suggest that the activity is protease-sensitive and that a combination of proteins from different biochemical fractions are needed for the neuroprotection (Fig. 3a). Based on our comparative proteomics, the candidate proteins that were enriched in the PSC-CM as compared to the inactive dF-CM, are either secreted or can be secreted when cells are damaged, and/or via exosomes or microvesicles [68, 69] (Additional file 1: Table S5). Because the neuroprotective activity of the hiPSC secretome is diminished when either exosomal or soluble fractions are removed, both fractions likely have proteins that confer neuroprotection through their interactome.
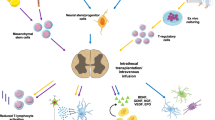
Uncovering the specific interactions of specific proteins that are necessary and sufficient is clearly a subject of future work. At the same time, several neuroprotective candidates seem to be particularly promising, and Fig. 5 summarizes their literature-predicted activities. TIMP1, ANG, ANG1, HSP27, and APOE were enriched in PSC-CM as compared to the negative control dF-CM, and each of these proteins has known neuroprotective effects [50,51,52, 70,71,72,73,74,75,76,77].

Mechanisms of multifunctional neuroprotection through PSC-CM. Oxidative stress alters SOD1 activity, and dysfunctional or mutant SOD1 causes further oxidative damage; this induces apoptosis, destabilizes mitochondria and promotes hyperinflammation. Distinct PSC-CM proteins through different mechanisms have the capacity to act together to attenuate ROS, improve proteostasis, inhibit Cas3 activation and apoptosis, and promote neuronal repair. The key candidates in PSC-CM as neuroprotective factors are TIMP1, ApoE, ANG, ANG1, and HSP27, whose neuroprotective effects have been confirmed in literature. Ultimately, PSC-CM delays or inhibits death and denervation of MNs, promoting muscle innervation and preventing degeneration. Figure created using BioRender (https://biorender.com/)
TIMP1 protects neurons against not only traumatic and ischemic brain injury but also HIV-1-induced neuronal apoptosis [51, 52]. Angiogenic ANG [78, 79] is necessary for success in maturation of neurons, including MNs in the spinal cord and brain [80, 81], protects MNs from hypoxia, and contributes to enhanced viability of MNs and longer lifespan of ALS mice [72, 73]. ANG-1 is another angiogenic factor in the secretomes of stem cells [79, 82, 83] that has anti-inflammatory [84] and neuroprotective anti-apoptotic effects [74,75,76]. HSP27 is an antioxidant and inhibitor of procaspase-3 activation [85,86,87], and these activities are being explored in Alzheimer’s disease [59, 88] and ALS models [70, 77]. ApoE reduces oxidative stress and contributes to beta-amyloid disposal [50, 71]. Moreover, all these proteins interact at the levels of apoptotic, TGF-β, AKT, and ERK signaling pathways [72,73,74,75,76, 87], suggesting a possible functional synergy.
Several clinical trials have suggested positive effects of mesenchymal and adipose stem cell secretomes [21, 89,90,91,92], but interestingly, here we showed that pluripotency was required for the positive effects and that differentiation of PSCs into fibroblasts or even into embryoid bodies abrogated the neuroprotection. Epigenetic modification is one of the major regulators of pluripotency [93]. Our study demonstrated an EZH2-mediated mechanism of the neuroprotective effect. EZH2 is a key part of the Polycomb repressive complex, histone methyltransferase [94]. The expression of EZH2 is not only required for pluripotency [95, 96], but influences differentiation into various cell fates, including neurons [97,98,99]. And EZH2 depletion has been shown to induce cell cycle arrest, decrease cell proliferation and induce apoptosis [100, 101]. Consistently, we found that GSK126 treatment of iPSCs reduced the neuroprotective anti-apoptotic activity of their secretome. Interestingly, some of the proteins identified in our comparative proteomics crosstalk with EZH2. For example, HSP90 is involved in EZH2 stability, level and function [102, 103]; ANG-1 and HSP27 are regulated by EZH2 and their expression is decreased upon EZH2 inhibition [104, 105]. The conclusion that the state of pluripotency is needed for the neuroprotective effects of PSC secretome fits well with the notion that pluripotent stem cells maintain low levels of oxidative stress via oxidation–reduction (redox) signaling, as compared to their differentiated progeny [106,107,108,109].
At the cellular level, ALS iPSC-CM improved the mitochondrial stability of MNs and reduced the expression of inflammatory genes. Recently suggested as an ALS treatment, CsA [46] had similar effects on mitochondrial health and MN-expressed inflammatory genes, but in contrast to ALS iPSC-CM, CsA failed to prevent the death of ALS SOD1 mutant MNs (Fig. 3e–g). CsA is also an immunosuppressant. The fact that ALS iPSC-CM stabilizes mitochondria and the hyperinflammatory response in ALS-MN without immunosuppression potentially makes it a better therapeutic.
In addition, the lack of neuroprotective effect of late application of ALS iPSC-CM suggests that an early diagnosis and immediate treatment are needed for the optimal efficacy of PSC-CM and the autologous ALS iPSC-CM in attenuating the pathologies, which is the case with therapies for most neurological diseases.
Conclusion
Neuronal cell death is causal in many neurodegenerative diseases, including age-related loss of memory and dementias (such as Alzheimer’s disease), Parkinson’s disease, strokes, as well as diseases that afflict broad ages, i.e., traumatic brain injury, spinal cord injury, ALS, and spinal muscle atrophy. These diseases are characterized by neuroinflammation and oxidative cell damage, many involve perturbed proteostasis and all are devastating and without a cure. Our work describes a feasible meaningful disease-minimizing treatment for ALS and suggests a clinical capacity for treating a broad class of diseases of neurodegeneration, and excessive cell apoptosis [14, 110,111,112,113].
Availability of data and materials
All data required in the paper, including the primary raw data are provided in the original manuscript and supplementary material file.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- PSC:
-
Pluripotent stem cell
- MN:
-
Motor neuron
- SOD1:
-
Superoxide dismutase 1
- ESC:
-
Embryonic stem cell
- iPSC:
-
Induced pluripotent stem cell
- CM:
-
Conditioned medium
- CsA:
-
Cyclosporin A
- MNP:
-
Motor neuron precursor
- NBM:
-
Neurobasal medium
- PCR:
-
Polymerase chain reaction
- NMJ:
-
Neuromuscular junction
- dF-CM:
-
Conditioned medium from differentiated fibroblast cells
- CC3:
-
Cleaved caspase3
- MFI:
-
Mean fluorescence intensity
References
Check WA. Amyotrophic lateral sclerosis. JAMA. 1979;242(4):319.
Aoyama K, Nakaki T. Impaired glutathione synthesis in neurodegeneration. Int J Mol Sci. 2013;14:21021–44.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–5.
Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, et al. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–6.
Ezzi SA, Urushitani M, Julien JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem. 2007;102:170–8.
Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. 2006;1762(11–12):1051–67.
Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253.
Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–6.
Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–19.
Yamashita T, Kushida Y, Wakao S, Tadokoro K, Nomura E, Omote Y, et al. Therapeutic benefit of Muse cells in a mouse model of amyotrophic lateral sclerosis. Sci Rep. 2020;10:1–11.
Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, et al. Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther. 2014;22:498–510.
Zakrzewski W, Dobrzyński M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. 2019;10(1):68.
Comley L, Allodi I, Nichterwitz S, Nizzardo M, Simone C, Corti S, et al. Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience. 2015;291:216–29.
Fujimori K, Ishikawa M, Otomo A, Atsuta N, Nakamura R, Akiyama T, et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat Med. 2018;24:1579–89.
Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell. 2014;14:796–809.
Li M, Ona VO, Guégan C, Chen M, Jackson-Lewis V, Andrews LJ, et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–9.
Kostic V, Jackson-Lewis V, De Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–62.
Gutierrez-Aranda I, Ramos-Mejia V, Bueno C, Munoz-Lopez M, Real PJ, Mácia A, et al. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells. 2010;28(9):1568–70.
Yousef H, Conboy MJ, Mamiya H, Zeiderman M, Schlesinger C, Schaffer DV, et al. Mechanisms of action of hESC-secreted proteins that enhance human and mouse myogenesis. Aging (Albany NY). 2014;6:602–20.
Yousef H, Conboy MJ, Li J, Zeiderman M, Vazin T, Schlesinger C, et al. hESC-secreted proteins can be enriched for multiple regenerative therapies by heparin-binding. Aging (Albany NY). 2013;5:357–72.
Fukuoka H, Suga H, Narita K, Watanabe R, Shintani S. The latest advance in hair regeneration therapy using proteins secreted by adipose-derived stem cells. Am J Cosmet Surg. 2012;29:273–82.
Sagaradze G, Grigorieva O, Nimiritsky P, Basalova N, Kalinina N, Akopyan Z, et al. Conditioned medium from human mesenchymal stromal cells: towards the clinical translation. Int J Mol Sci. 2019;20:1656.
Yang D, Wang W, Li L, Peng Y, Chen P, Huang H, et al. The relative contribution of paracine effect versus direct differentiation on adipose-derived stem cell transplantation mediated cardiac repair. PLoS One. 2013;8:e59020.
Takahashi M, Li TS, Suzuki R, Kobayashi T, Ito H, Ikeda Y, et al. Cytokines produced by bone marrow cells can contribute to functional improvement of the infarcted heart by protecting cardiomyocytes from ischemic injury. Am J Physiol. 2006;291:886–93.
Qu Q, Li D, Louis KR, Li X, Yang H, Sun Q, et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat Commun. 2014;5:3449.
Wang Y, Zhang L, Li Y, Chen L, Wang X, Guo W, et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int J Cardiol. 2015;192:61–9.
Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015;43:W566–70.
Fontanilla CV, Gu H, Liu Q, Zhu TZ, Zhou C, Johnstone BH, et al. Adipose-derived stem cell conditioned media extends survival time of a mouse model of amyotrophic lateral sclerosis. Sci Rep. 2015;5:1–11.
Hatzipetros T, Kidd JD, Moreno AJ, Thompson K, Gill A, Vieira FG. A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J Vis Exp. 2015;2015:53257.
Cai MD, Choi SM, Yang EJ. The effects of bee venom acupuncture on the central nervous system and muscle in an animal hsOD1G93A mutant. Toxins (Basel). 2015;7:846–58.
Wang J, Zuzzio K, Walker CL. Systemic dental pulp stem cell secretome therapy in a mouse model of amyotrophic lateral sclerosis. Brain Sci. 2019;9:165.
Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141:4206–18.
Tarng DC, Tseng WC, Lee PY, Chiou SH, Hsieh SL. Induced pluripotent stem cell-derived conditioned medium attenuates acute kidney injury by downregulating the oxidative stress-related pathway in ischemia–reperfusion rats. Cell Transplant. 2016;25:517–30.
Alves CJ, De Santana LP, Dos Santos AJD, De Oliveira GP, Duobles T, Scorisa JM, et al. Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 2011;1394:90–104.
Rabinovich-Toidman P, Rabinovich-Nikitin I, Ezra A, Barbiro B, Fogel H, Slutsky I, et al. Mutant SOD1 increases APP expression and phosphorylation in cellular and animal models of ALS. PLoS One. 2015;10:e0143420.
Haenggeli C, Julien JP, Lee Mosley R, Perez N, Dhar A, Gendelman HE, et al. Therapeutic immunization with a glatiramer acetate derivative does not alter survival in G93A and G37R SOD1 mouse models of familial ALS. Neurobiol Dis. 2007;26:146–52.
Ari C, Poff AM, Held HE, Landon CS, Goldhagen CR, Mavromates N, et al. Metabolic therapy with deanna protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS One. 2014;9(7):e103526.
Cappello V, Francolini M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int J Mol Sci. 2017;18(10):2092.
Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Chem Neuroanat. 2016;76:35–47.
Bruneteau G, Bauché S, Gonzalez de Aguilar JL, Brochier G, Mandjee N, et al. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol. 2015;2:362–72.
Basu J, Ludlow JW. Exosomes for repair, regeneration and rejuvenation. Expert Opin Biol Ther. 2016;16:489–506.
Duffy LM, Chapman AL, Shaw PJ, Grierson AJ. Review: the role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2011;37(4):336–52.
Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019;710:132933.
Sultani M, Stringer AM, Bowen JM, Gibson RJ. Anti-inflammatory cytokines: important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemother Res Pract. 2012;2012:1–11.
Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7.
Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636-649.e18.
Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2011;116:1–9.
Huang JY, Hong YT, Chuang JI. Fibroblast growth factor 9 prevents MPP+-induced death of dopaminergic neurons and is involved in melatonin neuroprotection in vivo and in vitro. J Neurochem. 2009;109:1400–12.
Subramanian V, Crabtree B, Acharya KR. Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum Mol Genet. 2008;17:130–49.
Zhou S, Wu H, Zeng C, Xiong X, Tang S, Tang Z, et al. Apolipoprotein E protects astrocytes from hypoxia and glutamate-induced apoptosis. FEBS Lett. 2013;587:254–8.
Tejima E, Guo S, Murata Y, Arai K, Lok J, Van Leyen K, et al. Neuroprotective effects of overexpressing tissue inhibitor of metalloproteinase TIMP-1. J Neurotrauma. 2009;26:1935–41.
Chao C, Borgmann K, Brew K, Ghorpade A. Tissue inhibitor of metalloproteinases-1 protects human neurons from staurosporine and HIV-1-induced apoptosis: Mechanisms and relevance to HIV-1-associated dementia. Cell Death Dis. 2012;3:1–9.
Huynh HD, Zheng J, Umikawa M, Zhang C, Silvany R, Iizuka S, et al. IGF binding protein 2 supports the survival and cycling of hematopoietic stem cells. Blood. 2011;118:3236–43.
Migita T, Narita T, Asaka R, Miyagi E, Nagano H, Nomura K, et al. Role of insulin-like growth factor binding protein 2 in lung adenocarcinoma: IGF-independent antiapoptotic effect via caspase-3. Am J Pathol. 2010;176:1756–66.
Fletcher L, Isgor E, Sprague S, Williams LH, Alajajian BB, Jimenez DF, et al. Spatial distribution of insulin-like growth factor binding protein-2 following hypoxic-ischemic injury. BMC Neurosci. 2013;14:1–11.
Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSw mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–17.
Akbar MT, Lundberg AMC, Liu K, Vidyadaran S, Wells KE, Dolatshad H, et al. The neuroprotective effects of heat shock protein 27 overexpression in transgenic animals against kainate-induced seizures and hippocampal cell death. J Biol Chem. 2003;278:19956–65.
Toth ME, Gonda S, Vigh L, Santha M. Neuroprotective effect of small heat shock protein, Hsp27, after acute and chronic alcohol administration. Cell Stress Chaperones. 2010;15:807–17.
Venugopal A, Sundaramoorthy K, Vellingiri B. Therapeutic potential of Hsp27 in neurological diseases. Egypt J Med Hum Genet. 2019;20:1–8.
Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013;3:461.
Huang WJ, Zhang X, Chen WW. Role of oxidative stress in Alzheimer’s disease. Biomed Rep. 2016;4:519.
Jang SE, Qiu L, Chan LL, Tan EK, Zeng L. Current status of stem cell-derived therapies for Parkinson’s disease: from cell assessment and imaging modalities to clinical trials. Front Neurosci. 2020;14:558532.
Song CG, Zhang YZ, Wu HN, Cao XL, Guo CJ, Li YQ, et al. Stem cells: a promising candidate to treat neurological disorders. Neural Regen Res. 2018;13:1294–304.
Gorecka J, Kostiuk V, Fereydooni A, Gonzalez L, Luo J, Dash B, et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res Ther. 2019;10(1):87.
Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell. 2011;146:318–31.
Tucker BA, Mullins RF, Streb LM, Anfnson K, Eyestone ME, Kaalberg E, et al. Patient-specifc iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife. 2013;2013(2):e00824.
Douvaras P, Wang J, Zimmer M, Hanchuk S, O’Bara MA, Sadiq S, et al. Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Rep. 2014;3:250–9.
Gonzalez-Begne M, Lu B, Han X, Hagen FK, Hand AR, Melvin JE, et al. Proteomic analysis of human parotid gland exosomes by multidimensional protein identification technology (MudPIT). J Proteome Res. 2009;8:1304–14.
Kim HS, Choi DY, Yun SJ, Choi SM, Kang JW, Jung JW, et al. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J Proteome Res. 2012;11:839–49.
Pires AO, Mendes-Pinheiro B, Teixeira FG, Anjo SI, Ribeiro-Samy S, Gomes ED, et al. Unveiling the differences of secretome of human bone marrow mesenchymal stem cells, adipose tissue-derived stem cells, and human umbilical cord perivascular cells: a proteomic analysis. Stem Cells Dev. 2016;25:1073–83.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–16.
Sebastià J, Kieran D, Breen B, King MA, Netteland DF, Joyce D, et al. Angiogenin protects motoneurons against hypoxic injury. Cell Death Differ. 2009;16:1238–47.
Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG, et al. Control of motoneuron survival by angiogenin. J Neurosci. 2008;28:14056–61.
Jiang H, Zhang F, Yang J, Han S. Angiopoietin-1 ameliorates inflammation-induced vascular leakage and improves functional impairment in a rat model of acute experimental autoimmune encephalomyelitis. Exp Neurol. 2014;261:245–57.
Kosacka J, Figiel M, Engele J, Hilbig H, Majewski M, Spanel-Borowski K. Angiopoietin-1 promotes neurite outgrowth from dorsal root ganglion cells positive for Tie-2 receptor. Cell Tissue Res. 2005;320:11–9.
Yin J, Gong G, Liu X. Angiopoietin: a novel neuroprotective/neurotrophic agent. Neuroscience. 2019;411:177–84.
Patel YJK, Payne Smith MD, De Belleroche J, Latchman DS. Hsp27 and Hsp70 administered in combination have a potent protective effect against FALS-associated SOD1-mutant-induced cell death in mammalian neuronal cells. Mol Brain Res. 2005;134:256–74.
Chen YT, Tsai MJ, Hsieh N, Lo MJ, Lee MJ, Cheng H, et al. The superiority of conditioned medium derived from rapidly expanded mesenchymal stem cells for neural repair. Stem Cell Res Ther. 2019;10:1–15.
Hilkens P, Fanton Y, Martens W, Gervois P, Struys T, Politis C, et al. Pro-angiogenic impact of dental stem cells in vitro and in vivo. Stem Cell Res. 2014;12:778–90.
Aparicio-Erriu IM, Prehn JHM. Molecular mechanisms in amyotrophic lateral sclerosis: the role of angiogenin, a secreted RNase. Front Neurosci. 2012;6:167.
Subramanian V, Feng Y. A new role for angiogenin in neurite growth and pathfinding: implications for amyotrophic lateral sclerosis. Hum Mol Genet. 2007;16:1445–53.
Xiang J, Hu J, Shen T, Liu B, Hua F, Zan K, et al. Bone marrow mesenchymal stem cells-conditioned medium enhances vascular remodeling after stroke in type 2 diabetic rats. Neurosci Lett. 2017;644:62–6.
Zhao Q, Hu J, Xiang J, Gu Y, Jin P, Hua F, et al. Intranasal administration of human umbilical cord mesenchymal stem cells-conditioned medium enhances vascular remodeling after stroke. Brain Res. 2015;1624:489–96.
Pizurki L, Zhou Z, Glynos K, Roussos C, Papapetropoulos A. Angiopoietin-1 inhibits endothelial permeability, neutrophil adherence and IL-8 production. Br J Pharmacol. 2003;139:329–36.
Bakthisaran R, Tangirala R, Rao CM. Small heat shock proteins: role in cellular functions and pathology. Biochim Biophys Acta. 2015;1854:291–319.
Weeks SD, Muranova LK, Heirbaut M, Beelen S, Strelkov SV, Gusev NB. Characterization of human small heat shock protein HSPB1 α-crystallin domain localized mutants associated with hereditary motor neuron diseases. Sci Rep. 2018;8:1–15.
Yu AL, Fuchshofer R, Birke M, Kampik A, Bloemendal H, Welge-Lüssen U. Oxidative stress and TGF-β2 increase heat shock protein 27 expression in human optic nerve head astrocytes. Investig Ophthalmol Vis Sci. 2008;49:5403–11.
Tóth ME, Szegedi V, Varga E, Juhász G, Horváth J, Borbély E, et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones. 2013;18:759–71.
Zhou BR, Xu Y, Xu Y, Guo SL, Wang Y, Zhu F, et al. The effect of conditioned media of Adipose-derived stem cells on wound healing after ablative fractional carbon dioxide laser resurfacing. Biomed Res Int. 2013;2013.
Kay AG, Long G, Tyler G, Stefan A, Broadfoot SJ, Piccinini AM, et al. Mesenchymal stem cell-conditioned medium reduces disease severity and immune responses in inflammatory arthritis. Sci Rep. 2017;7:1–11.
Dahbour S, Jamali F, Alhattab D, Al-Radaideh A, Ababneh O, Al-Ryalat N, et al. Mesenchymal stem cells and conditioned media in the treatment of multiple sclerosis patients: clinical, ophthalmological and radiological assessments of safety and efficacy. CNS Neurosci Ther. 2017;23:866–74.
Pawitan JA. Prospect of stem cell conditioned medium in regenerative medicine. Biomed Res Int. 2014;2014.
Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. 2014;115:311.
Sparmann A, Van Lohuizen M. Polycomb silencers control cell fate, development and cancer [Internet]. Nat Rev Cancer. 2006;6:846–56.
O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb -group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330–6.
Collinson A, Collier AJ, Morgan NP, Sienerth AR, Chandra T, Andrews S, et al. Deletion of the polycomb-group protein EZH2 leads to compromised self-renewal and differentiation defects in human embryonic stem cells. Cell Rep. 2016;17:2700–14.
Sher F, Rößler R, Brouwer N, Balasubramaniyan V, Boddeke E, Copray S. Differentiation of neural stem cells into oligodendrocytes: involvement of the polycomb group protein Ezh2. Stem Cells. 2008;26:2875–83.
Aoki R, Chiba T, Miyagi S, Negishi M, Konuma T, Taniguchi H, et al. The polycomb group gene product Ezh2 regulates proliferation and differentiation of murine hepatic stem/progenitor cells. J Hepatol. 2010;52:854–63.
Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell. 2009;36:61–74.
Yao Y, Hu H, Yang Y, Zhou G, Shang Z, Yang X, et al. Downregulation of enhancer of zeste homolog 2 (EZH2) is essential for the induction of autophagy and apoptosis in colorectal cancer cells. Genes (Basel). 2016;7:83.
Qi W, Chan HM, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–5.
Lee YC, Chang WW, Chen YY, Tsai YH, Chou YH, Tseng HC, et al. Hsp90α mediates BMI1 expression in breast cancer stem/progenitor cells through facilitating nuclear translocation of c-Myc and EZH2. Int J Mol Sci. 2017;18:1986.
Huang Q, He S, Tian Y, Gu Y, Chen P, Li C, et al. Hsp90 inhibition destabilizes Ezh2 protein in alloreactive T cells and reduces graft-versus-host disease in mice. Blood. 2017;129:2737–48.
Moore HM, Gonzalez ME, Toy KA, Cimino-Mathews A, Argani P, Kleer CG. EZH2 inhibition decreases p38 signaling and suppresses breast cancer motility and metastasis. Breast Cancer Res Treat. 2013;138:741–52.
Herrera-Merchan A, Arranz L, Ligos JM, De Molina A, Dominguez O, Gonzalez S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun. 2012;3:1–12.
Guo YL, Chakraborty S, Rajan SS, Wang R, Huang F. Effects of oxidative stress on mouse embryonic stem cell proliferation, apoptosis, senescence, and self-renewal. Stem Cells Dev. 2010;19:1321–31.
Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860–73.
Mandal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells. 2011;29:486–95.
Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–16.
Inoue H, Tsukita K, Iwasato T, Suzuki Y, Tomioka M, Tateno M, et al. The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 2003;22:6665–74.
Pedersen WA, Luo H, Kruman I, Kasarskis E, Mattson MP. The prostate apoptosis response-4 protein participates in motor neuron degeneration in amyotrophic lateral sclerosis. FASEB J. 2000;14:913–24.
Guerrero EN, Mitra J, Wang H, Rangaswamy S, Hegde PM, Basu P, et al. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum Mol Genet. 2019;28:2459–76.
Sathasivam S, Shaw PJ. Apoptosis in amyotrophic lateral sclerosis—what is the evidence? Lancet Neurol. 2005;4:500–9.
Acknowledgements
We thank Catherine Lomen-Hoerth, Director of ALS Center, UCSF, for helpful discussion and review of our manuscript, William How for the helpful discussion on antibody array analysis, and Mary West and the QB3 Cell and Tissue Analysis Facility for use of cryotome, microscopy, IXM and MetaXpress, and qPCR instruments.
Funding
This work was supported by NIBIB (R01 EB023776), NIA (R01AG071787), NHLBI (R01 HL139605), Open Philanthropy, Silicon Valley Community Foundation, Foster Foundation, San Francisco Foundation, Georges’ Harik Foundation, and Donors’ Trust grants to IC.
Author information
Authors and Affiliations
Contributions
D.K. designed the experiments, provided Figs. 1, 2, 3, 4 and 5 and Supplementary Figures and Tables, and co-wrote the manuscript. S.K., A.S., and N.P. contributed to Figs. 1, 2, 3 and 4. N.W. contributed to Fig. 3. M.J.C. designed the study and edited the manuscript. I.M.C. designed and directed the study, interpreted results, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experimental procedures were performed in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Office of Laboratory Animal Care (OLAC), UC Berkeley.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest in this manuscript.
Supplementary Information
Additional file 1
. Table S1. Primer sequences for real-time polymerase chain reaction. Table S2. The list of amyotrophic lateral sclerosis cell lines. Table S3. The efficiency of motor neuron differentiation from normal pluripotent stem cells. Table S4. The efficiency of motor neuron differentiation from ALS patient hiPSCs. Table S5. The 106 PSC-CM protein candidates and their secretory capacity.
Additional file 2
. Figure S1. Human embryonic stem cell-derived conditioned medium (hESC-CM) protects human fibroblast from H2O2 cytotoxicity. Figure S2. Human embryonic stem cell-derived conditioned medium (hESC-CM) protects motor neurons (MNs) from H2O2 cytotoxicity. Figure S3. Antioxidant activity is not different between human embryonic stem cell-derived conditioned medium (hESC-CM) and differentiated fibroblast-derived conditioned medium (dF-CM). Figure S4. Differentiation of ALS iPSCs into motor neurons (ALS-MNs). Figure S5. Profiling the time-point of the neuroprotective effect from iPSC-CM. Figure S6. The comparative effects of autologous ALS-derived hiPSC-conditioned media (ALS-CM) on neurites and apoptosis of ALS-motor neurons (ALS-MNs) in ALS patient-derived cell lines. Figure S7. The effect of CMs on the weight of SOD1G93A transgenic mice. Figure S8. The optimization of concentrations of heparin-binding protein (HBP) and exosome. Figure S9. The effect of epigenetic modifiers on the conditioned medium (CM). Figure S10. Differentially present proteins in each of the listed categories.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, D., Kim, S., Sung, A. et al. Autologous treatment for ALS with implication for broad neuroprotection. Transl Neurodegener 11, 16 (2022). https://doi.org/10.1186/s40035-022-00290-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-022-00290-5