
Abstract
Background
Colorectal cancer (CRC) and inflammatory bowel disease (IBD) are the most prevalent diseases of the digestive system, and their association is unequivocal. A long-standing inflammatory process is one of the causes of sporadic as well as inherited cancers as it impacts on malignant transformation in a wide variety of neoplastic diseases, including colorectal cancer.
Methods
An extensive publication search was performed in Medline and PubMed database. The keywords: colorectal carcinoma, inflammation, Crohn disease, ulcerative colitis and inflammatory bowel disease were used.
Results
The nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and toll like receptor (TLR) signaling pathways are clearly involved in the inflammatory process and are therefore implicated in the transformation of normal colonic mucosa to premalignant and malignant disease. Focal sites of inflammation could significantly increase the risk of initiation and development of cancer. Altered inflammatory activity is likely to be a result of either a disturbance of intestinal bacterial flora or an inadequate cellular response to it. Additionally, increasing the level of inflammation-related factors may also interfere with the control of cellular proliferation.
Conclusions
This review shows an overview of the genetic and environmental factors that appear to influence both the occurrence of IBD and CRC with particular reference to NOD2 and TLRs as well as pro- and anti-inflammatory cytokines associated with tumor initiation and progression (encompassing both tumor invasion and metastases), as they constitute potential targets for therapeutic intervention.
Similar content being viewed by others
Background
Colorectal cancer (CRC) is the third most common malignancy and fourth leading cause of cancer-related mortality worldwide. There is a statistically significant association between countries affluence and the incidence of CRC. Developed countries account for greater than 55% of all CRC cases [1, 2]. Differences in the occurrence of CRC are over 10-fold: from the highest rates observed in Australia, New Zealand, Western Europe and the United States to the lowest in Africa and Central Asia [3, 4].
At present, over 75% of diagnosed CRC are classified as sporadic disease with no apparent first degree relatives affected with disease. Multiple environmental factors that include smoking, obesity and a diet rich in meat products and/or excessive alcohol consumption increases the risk of disease. CRC comprises a group of molecularly heterogeneous diseases, which are a result of different types of genomic, epigenomic, environmental and physiological factors that influence CRC development [4]. Most notably, an increased risk of developing CRC is associated with a variety of inherited disorders that include Familial adenomatous polyposis (FAP), Lynch syndrome, Peutz-Jegher’s syndrome and Juvenile Polyposis. In addition to these well-known inherited predispositions, chronic inflammation of the gastrointestinal tract is also tightly associated with a high risk of CRC [5,6,7,8]. We emphasize patients with inflammatory bowel diseases (IBD) and particularly those with ulcerative colitis (UC) represent a distinct entity burdened by an elevated risk of CRC. In this group of patients the incidence of CRC increases by 60% (i.e., colitis-associated colorectal cancer CAC) compared to that of the general population [9].
Inflammatory bowel disease-related colorectal cancer
IBD constitutes a group of chronic, recurrent and incurable diseases of unknown etiology affecting the gastrointestinal tract that can ultimately lead to the destruction of normal intestinal architecture. The etio-pathogenesis of IBD is complex and multifactorial with a significnat influence associated with genetic susceptibility, immunological and environmental factors, which altogether interact and result in the dysregulation of the mucosal immune system of the gut leading to uncontrolled inflammation. Clinically, two main subtypes of IBD are defined: Crohn’s disease (CD), where inflammation may develop anywhere in the gastrointestinal tract from the mouth to the anus, and UC, where inflammation is limited to the colonic mucosa.
Patients with long-standing UC and CD are at a higher risk of developing CRC compared to the general population [9]. Although IBD-associated CRC accounts for approximately 2% of all CRC, the rate of death resulting from CRC in IBD patients ranges from 10 to 15% [10]. Population-based studies reveal that IBD and CRC have a similar prevalence worldwide with an increasing incidence in northwestern Europe and the USA. Furthermore, both diseases are associated with westernized behaviors and lifestyle [2].
The first meta-analysis performed by Eaden et al. in 2001 on the risk of IBD-associated CRC was based on the data of 54 478 patients from 116 studies, which revealed an overall CRC prevalence of 3.7% in UC patients. The incidence rate corresponded to a cumulative probability of 2% by 10 years; 8% by 20 years; and 18% by 30 years. The mean age of UC-associated cancer diagnosis was 43.2 years [11]. A nationwide Korean study involving 7061 UC patients reported comparable results with an estimated UC-associated CRC cumulative risk of 0.7% at 10 years, 7.9% at 20 years, and 33.2% at 30 years [12]. In contrast, a few population studies from Hungary, Sweden, United Kingdom and Denmark reported contradictory results with a cumulative risk of CRC in UC between 0 and 1.5% at 10 years, 1.1–5.4% at 20 years, and 2.1–10.8% at 30 years [13,14,15,16]. The meta-analysis performed by Canavan et al. based on 12 studies revealed the cumulative risk for patients with CD was 2.9% at 10 years, 5.6% at 20 years, and 8.3% at 30 years after a diagnosis of CD [17]. These results indicate the risk of developing CRC is similar for both CD and UC. Several subsequent reports from Caucasian populations revealed that CRC in the course of CD was characterized by much younger ages of cancer onset with tumors distributed evenly along the entire colon [18, 19].
A study by Watanabe et al. in Japan showed a decreased survival rate for patients with stage III UC-associated CRC than those with stage III sporadic CRC. In this study CRC associated with UC also tended to be multifocal and have a higher histological grade [20]. Jensen et al. reported similar findings from the Danish population, in which the overall mortality rate ratio of UC-associated CRC compared to sporadic CRC was 1.24 (95%CI: [1.02–1.51]) in the first year, and 1.17 (95%CI: [1.01–1.36]) after 5-years follow-up [21]. Furthermore, there are reports suggesting the risk of CRC is increased by at least 2-fold in IBD patients with a positive family history of CRC compared to patients without a family history of CRC [22, 23].
Inflammation in colorectal cancer
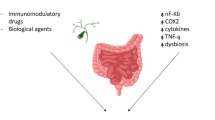
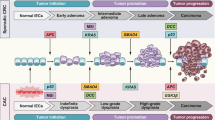
The relationship between cancer and chronic inflammation was first described over 150 years ago by Rudolf Virchow (reviewed in [24, 25]). Since this hypothesis was put forward it has been confirmed by many clinical studies, but the pathogenesis and molecular biology of cancers associated with chronic inflammation remains the subject of much research. Chronic inflammation is observed in approximately 20% of all human cancers. The strongest correlation between long-standing inflammation and tumorigenesis is most apparent in CRC. [26]. Inflammation is one of the physiological factors that occurs as a protective response elicited by injury or the destruction of tissue. In effect, inflammatory-driven genetic alteration and epigenetic change occur, both important aspects of tumor initiation in CRC and particularly in CAC [27].
Immune cells and cytokines in CRC
In IBD chronic inflammation is controlled by a complex interplay of innate and adaptive immune response mechanisms associated with disequilibrium in the production of Th1, Th2 and Th17 cytokines. This imbalance coupled with a defective immune response to pathogens promotes disease recurrence. The current concepts concerning circulating cytokines in IBD are still contradictory, but the cytokine profile in IBD tissue reveals that CD and UC are immunologically distinct [28]. Studies examining cytokines at the sites of inflammation in the colon has led to a paradigm in which UC is characterized by a predominance of cytokines related to T helper 2 (Th2) cells, whereas in the course of CD, Th1-associated cytokines prevail [29]. This is not reflected in the levels of systemic cytokines, many of which cannot be detected in the serum of IBD patients, mainly because they are restricted to the sites of inflammation or have relatively short half-lives. Further complicating this are significant issues in regards to the frequent extra-intestinal symptoms or comorbid diseases (such as stomatitis, skin inflammation, ocular manifestations, liver inflammation, rheumatoid arthritis and osteoporosis) that accompany IBD, reflecting the disruptive serum cytokine milieu [28, 30]. Cytokines secreted in response to infection and inflammation in the course of IBD may influence several stages of cancer initiation and progression. Tumour cells can react to host-derived cytokines that promote growth, attenuate apoptosis and facilitate invasion and metastasis. Furthermore, cytokine signaling is pleiotropic depending on the environment, one cytokine has the power to act on many different cell types to mediate diverse and sometimes opposing effects [31].
Cytokines such as TNF-α, IL-6 and IL-1 are involved in the promotion of CRC and CAC development whereas TGF-β and IL-10 are associated with an anti-inflammatory response. TNF-α is expressed in response to the initiation of inflammation and is secreted mainly by monocytes or macrophages. TNF-α maintains chronic inflammation and promotion of tumour progression and angiogenesis [32]. Activated TNF-α also triggers signaling pathways and transcription factors such as NF-κB. In turn, NF-κB plays a significant role in CRC and CAC tumorigenesis. Aberrant NF-κB activation occurs in 50% of CRC and CAC tumors [33]. Similarly to TNF-α, IL-1 acts as an initial alarm signal, which can activate a cascade of overlapping up-regulatory and pro-inflammatory responses. IL-1α and IL-1β are the most extensively studied cytokines within the IL-1 family.
Another pro-inflammatory cytokine is IL-6, which promotes tumor growth in human CRC cells. IL-6 appears critical for long-standing inflammation, where it is indispensable for the recruitment and activation of Th17 cells, and inhibits the function of regulatory T cells. IL-6 participates in the activation of signal transduction and activation of transcription 3 (STAT3). This trans-signaling pathway starts when IL-6 binds to the sIL-6R receptor on target cells, followed by dimerization of gp130 subunit and then activation of the JAK (Janus kinase). JAK induces recruitment and activation of STAT3, an oncogenic transcription factor involved in cell proliferation, survival, apoptosis and immune response [34].
Unlike pro-inflammatory cytokines, TGF-β together with IL-10 acts as an anti-inflammatory factor important in maintaining intestinal homeostasis and suppression of CRC tumorigenesis in early-stage cancer. However, these cytokines have a dual role during cancer development and may transform from an inhibitor to a promoter of disease [29]. Recently research has focussed on the use of manipulating cytokine signaling to create a treatment strategy that can protect or treat patients with chronic inflammation or CRC. The characteristic of cytokines acting on IBD and CRC is shown in Table 1 [35,36,37,38,39,40,41,42,43].
Pathogenesis of IBD associated CRC
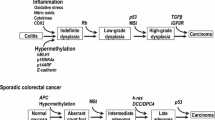
Carcinogenesis proceeds via a sequence of molecular alterations that result in a premalignant state culminating in genomic instability and the appearance of carcinoma. IBD-associated CRC appears quite similar to sporadic CRC with disease development comprising almost the same alterations as found in sporadic CRC. CAC is manifested in epithelial dysplasia and resulting from two types of instability: CIN (chromosomal instability) and MSI (microsatellite instability) [44, 45]. These modifications are observed in both pathways of carcinogenesis, but the timing and frequency of these alterations in epithelial cells are different that allow the distinction between IBD-CRC and sporadic CRC [46, 47]. The sequence of events in the development of sporadic cancer consists of a molecular cascade that includes the loss of adenomatous polyposis coli (APC) function, aneuploidy, methylation changes, microsatellite instability, activation of the KRAS gene and COX-2 enzyme, changes in DCC/DPC4 genes and loss of p53 function [48]. The differences in IBD-CRC formation concern the loss of APC and p53 function. Loss of APC function typically occurs earlier in patients with sporadic CRC, whereas in patients with IBD it is less frequent and takes place later in the development of malignancy. By contrast, loss of p53 function occurs much earlier in IBD-associated carcinogenesis, compared to patients with sporadic CRC [45, 46, 49]. Mutations in TP53 seem crucial for neoplasia initiation in IBD patients and are observed in 50–85% of CACs [48].
NOD2 and TLRs signaling pathways in IBD and CRC
Kurzawski et al. first described the link between NOD2 polymorphisms and the risk of CRC in 2004 [50]. Initially, studies confirmed DNA lesions constituting an base insertion at nucleotide position 3020, which was linked to CRC developing at older ages. Suchy et al. who analyzed sequence variants in genes engaged in inflammatory response, confirmed this finding and concluded that polymorphisms in genes involved in the inflammatory response might be genetic modifiers of the disease and influence CRC risk in older persons [51]. Interestingly, Freire et al. found a significantly higher incidence of NOD2 mutations in patients with CRC diagnosed under 60 years of age (28.6% vs. 10.4%, p = 0.015) and in female patients (24.4% vs. 10.4%, p = 0.048) [52]. Askling et al. in a population-based cohort study of 19,876 individuals with UC or CD revealed that familial CRC was associated with a more than 2-fold risk of CRC and an increase in the absolute risk of CRC at 54 years of age from 3.8 to 6.9%. Patients with a first-degree relative diagnosed with CRC before 50 years of age had a higher relative risk (9.2, C.I. = 3.7–23.0) and the highest absolute risk (29%) of disease. However, they did not observe any association with familial IBD [53].
A large meta-analysis performed by Tian et al. summarized earlier reports and proved that R702W, G908R, and 3020insC variants in NOD2 irrefutably contribute to CRC susceptibility in Caucasians [54]. The latest report by Samadder et al., on a 16-year long study following a group of 9505 patients with IBD, 101 of whom developed CRC, revealed that patients with IBD have a 3- to 5-fold increased risk of CRC, but more specifically, those with CRC in a first-degree relative had an almost 8-fold increase in disease risk. They emphasized that family history may act as a simple measure to identify individuals with IBD at highest risk for CRC and suggested the need for enhanced surveillance in this particular group of patients [55]. Recent in vitro studies performed by Udden et al. have shown that NOD2 activates the NF-κB and MAPK (mitogen-activated protein kinase) signaling axis in response to bacterial muramyl dipeptide (MDP) and inhibits TLR-mediated activation of these pathways and production of inflammatory mediators [56].
Toll-like receptors (TLRs) are mediators of inflammation in the gastrointestinal tract and may act as modulators of CRC risk. Activation of TLR signal transduction pathways induces numerous genes, including inflammatory cytokines, chemokines, and antigen presenting molecules. There are TLR variants that are associated with deregulation of their signaling function that are major contributing factors to the predisposition and susceptibility to IBD. [57].
Aberrant TLR signaling causes changes in commensal bacterial composition, intolerance and disturbance of mucosal homeostasis. Additionally, as sensors of cell death and tissue remodeling, TLRs have also been shown to be key receptors involved in inflammation-driven carcinogenesis. Several TLRs have been examined in the intestine, but the most important with respect to inflammation appear to be TLR2 and TLR4, the expression of which has been shown to be increased in macrophages of IBD patients [58]. Slattery et al. examined 1555 individuals with CRC and 1956 controls for TLR2, TLR3, and TLR4 with the risk of developing colon or rectal cancer. They showed that TLR3 (rs3775292) and TLR4 (rs11536898) polymorphisms were significantly associated with rectal (although only weakly) and colon cancer, respectively. Although, they studied a group consisting of individuals with no previous history of CRC or any known (as indicated on the pathology report) diagnosis of FAP, UC or CD [59]. One of latest reports regarding TLRs variants in a group of patients with advanced and metastatic CRC indicated that TLR4 Asp299Gly, TLR4 Thr399Ile, TLR9 T1237C and T1486C and TLR2–196 to − 174 del/del homozygous genotypes were significantly associated with CRC. Moreover, TLR4 Asp299Gly and Thr399Ile polymorphisms were significantly associated with concomitant KRAS gene mutations [60]. Both of these SNPs are located in the coding region of the TLR4 ectodomain, which results in an amino acid substitution known to reduce cytokine expression, contributing to an increased susceptibility to CRC [61, 62].
Taken together, NOD2 plays a critical role in the suppression of inflammation and tumorigenesis in the colon via down-regulation of the TLR signaling pathways. There remains a need for further research into families where IBD patients originate, especially those who carry polymorphic variants in genes involved in the inflammatory response and in particular, NOD2 and the TLRs. A comprehensive study of NOD2 and TLRs that include a thorough examination of genetic variance in these genes, differential expression levels and epigenetic modifications are crucial to understand normal and pathological function of these genes such that appropriate design and development of novel therapeutic approaches for CRC treatment can be undertaken.
Epigenetic modifications in CRC
The CpG island hypermethylation in several gene promoters contributes to the pathogenesis of CRC resulting in gene silencing. Hypermethylation of the promoter region of a gene results in reduced gene expression and is associated with a failure to appropriately respond to the cellular environment. Two genes are most frequently affected by aberrant promoter methylation are hMLH1 and CDKN2A. A loss of MLH1 expression via epigenetic silencing is associated with microsatellite instability (MSI), observed in a proportion of both sporadic and IBD-associated CRCs [44, 63]. Silencing of CDKN2Aresults in aberrant signaling culminating in cellular proliferation [64].
Activation of cyclooxygenase enzyme COX-2 is also related to the pathogenesis of advanced CRC. Studies of COX-2 expression have shown it has elevated levels in various cancers and links the inflammatory response with CRC. Over-expression of COX-2 is induced by inflammation suggesting this is an early stage in tumour development. COX-2 contributes to increased proliferation and angiogenesis, both of which are correlated with tumour initiation and progression [44, 65]. Another key constituent of the pathogenic process is oxidative stress. Elevated levels of reactive oxygen and nitrogen species are associated with DNA damage thereby contributing to mutational load [44]. Active inflammation in IBD patients results in increased levels of inflammatory cells that include neutrophils and macrophages. These cells produce free radicals and pro-oxidant molecules called RONs (reactive oxygen and nitrogen forms) [48], which are linked to DNA and protein damage. Alterations in DNA or protein brought about by free radical attack can result in affecting essential growth-regulatory genes and may be implicated in IBD and CRC. DNA damage product concentration, namely the mutagen 8-hydroxydeoxyguanosine (8-OHdG) was reported to be increased in patients with IBD [66, 67], suggesting an etiological role in the development of malignancy in this disorder.
Microbiota in CRC
It is well established that the intestinal microbiota plays a crucial role in the pathogenesis of CRC by altering intestinal bacterial biofilms, micro-environmental homeostasis, and immune reaction. Bacterial biofilms act as a first line of defense against invading microbes capable of inducing an inflammatory response and the generation of genotoxic compounds derived from bacteria. There is evidence to indicate that microbiotic imbalance contributes to the development of IBD-associated CRC by initiating and maintaining colonic mucosal inflammation [68, 69]. Abnormal bacterial microflora or disruption of homeostasis may promote inflammation in the large intestine, resulting in damage to the mucous membrane and subsequently, the development of neoplastic lesions. Pathogenic microorganisms contribute to intestinal inflammation through the activation of pattern recognition receptors (TLRs) or by endocytotic processes, toxin secretion, invasion or adherence [70].
TLRs are specific detectors of microbes and signal the immune system of any disequilibrium in microbial homeostasis, via the activation of transcription factors (i.e. NF-κB) and the secretion of cytokines. Research using mice that over-expressed TLR-4 showed increased inflammatory mediator levels: TNF-a, COX-2, and IL-12 and accordingly higher severity of both DSS (dextran sulfate sodium) induced colitis and CAC [71].
Gastro-intestinal inflammation may be the result of a disturbances in the ratio between tolerogenic and pro-inflammatory microflora that results in a breakdown of gut homeostasis and consequent mucosal inflammation. Changes in the number, diversity and stability of bacteria, in particular belonging to the Clostridia group, may lead to the development of intestinal diseases including CRC. The change of normal physiological into pathological processes, referred to as dysbiosis, may also affect CAC development. There is increasing evidence to suggest that the enterotoxic bacteria, Bacteroides fragilis and Escherichia coli promote the development of CRC. The presence of these bacteria is associated with tissue damage, STAT3 activation and IL-17 production resulting in colitis. Other pathogens that may contribute to the tumor progression are Enterococcus faecalis that activate the immune system via the production of reactive oxygen species (ROS). In turn Streptococcus bovis or Bacteroides fragilis are associated with the release of pro-survival and pro-angiogenic cytokines (i.e. IL-6, IL-8, IL-17) [27]. Furthermore, several studies carried out on IBD rodent models have revealed that commensal or specific bacteria are implicated in the development of IBD-associated CRC. IL-10 knockout mice develop IBD as well as rectal dysplasia and adenocarcinoma induced by Enterococcus faecalis [72]. IL-10 knock-out mice, under pathogen-free conditions, also developed colitis after the age of 3 months. CRC was observed in 25 and 60% of these mice after the ages of 3 and 6 months, respectively [73]. Moreover, modification of the enteric flora in IL-10 knockout mice by probiotic lactobacilli was associated with a reduced prevalence of CRC and lower mucosal inflammatory activity [74]. There remains much still to be learned from studying gut microbiota and at present little is known about commensal bacteria and what role they play in IBD. At present, it appears that the key to a healthy gut is bacterial diversity rather than the presence of dominant bacterial strains.
Conclusions
The association between inflammation and the initiation and development of cancer is unquestionable. Both processes are considered intertwined, as is observed in the context of cancer risk in patients with IBD. Chronic inflammation is not always recognizable at a clinical level and alone it may not be sufficient to trigger disease development. Notwithstanding, focal sites of inflammation could significantly increase the risk of initiation and development of cancer. Altered inflammatory activity is likely to be a result of either a disturbance of intestinal bacterial flora or an inadequate cellular response to it. Additionally, increasing the level of inflammation-related factors may also interfere with the control of cellular proliferation. A better understanding of the patho-mechanisms that drive inflammation in IBD patients is essential, as it is highly likely to result in the identification of additional therapeutic possibilities and reduce the risk of CRC in this population of patients.
Availability of data and materials
Not applicable.
Abbreviations
- 8-OHdG:
-
8-hydroxydeoxyguanosine
- APC:
-
Adenomatous polyposis coli
- CAC:
-
Colitis-associated colorectal cancer
- CD:
-
Crohn’s disease
- CIN:
-
Chromosomal instability
- COX-2:
-
Cyclooxygenase 2
- CRC:
-
Colorectal cancer
- DCC:
-
Deleted in colon cancer gene
- DPC4:
-
Deleted pancreatic cancer gene
- DSS:
-
Dextran sulfate sodium
- FAP:
-
Familial adenomatous polyposis
- hMLH1:
-
human mutL homolog 1
- IBD:
-
Inflammatory bowel diseases
- IL-1, IL-6 e.t.c.:
-
Interleukin 1,6 e.t.c.
- JAK:
-
Janus kinase
- MAPK:
-
Mitogen-activated protein kinase
- MDP:
-
Muramyl dipeptide
- MSI:
-
Microsatellite instability
- NF-κB:
-
Nuclear Factor κB
- NOD2:
-
Nucleotide-binding oligomerization domain containing 2
- p53:
-
tumor protein p53
- RONs:
-
Reactive oxygen and nitrogen forms
- ROS:
-
Reactive oxygen species
- STAT3:
-
Signal transducer and activator of transcription 3
- TGF-β:
-
Transforming growth factor β
- Th1, Th2,Th17:
-
T helper (Th) cytokines
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor alpha
- UC:
-
Ulcerative colitis
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015. https://doi.org/10.1002/ijc.29210.
Favoriti P, Carbone G, Greco M, Pirozzi F, Pirozzi RE, Corcione F. Worldwide burden of colorectal cancer: a review. Updat Surg. 2016. https://doi.org/10.1007/s13304-016-0359-y.
Marley AR, Nan H. Epidemiology of colorectal cancer. Int J Mol Epidemiol Genet. 2016;7:105–14.
Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017. https://doi.org/10.1136/gutjnl-2015-310912.
Plawski A, Banasiewicz T, Borun P, Kubaszewski L, Krokowicz P, Skrzypczak-Zielinska M, Lubinski J. Familial adenomatous polyposis of the colon. Hered Cancer Clin Pract. 2013. https://doi.org/10.1186/1897-4287-11-15.
Stojcev Z, Borun P, Hermann J, Krokowicz P, Cichy W, Kubaszewski L, Banasiewicz T, Plawski A. Hamartomatous polyposis syndromes. Hered Cancer Clin Pract. 2013. https://doi.org/10.1186/1897-4287-11-4.
Banasiewicz T, Marciniak R, Kaczmarek E, Krokowicz P, Paszkowski J, Lozynska-Nelke A, Gronek P, Plawski A, Drews M. The prognosis of clinical course and the analysis of the frequency of the inflammation and dysplasia in the intestinal J-pouch at the patients after restorative proctocolectomy due to FAP. Int J Color Dis. 2011. https://doi.org/10.1007/s00384-011-1241-5.
Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009. https://doi.org/10.1016/j.coph.2009.06.006.
Long AG, Lundsmith ET, Hamilton KE. Inflammation and colorectal cancer. Curr Colorectal Cancer Rep. 2017. https://doi.org/10.1007/s11888-017-0373-6.
Eluri S, Parian AM, Limketkai BN, Ha CY, Brant SR, Dudley-Brown S, Efron JE, Fang SG, Gearhart SL, Marohn MR, Meltzer SJ, Bashar S, Truta B, Montgomery EA, Lazarev MG. Nearly a third of high-grade dysplasia and colorectal Cancer is undetected in patients with inflammatory bowel disease. Dig Dis Sci. 2017. https://doi.org/10.1007/s10620-017-4652-5.
Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001. https://doi.org/10.1136/gut.48.4.526.
Kim BJ, Yang SK, Kim JS, Jeen YT, Choi H, Han DS, Kim HJ, Kim WH, Kim JY, Chang DK. Trends of ulcerative colitis-associated colorectal cancer in Korea: a KASID study. J Gastroenterol Hepatol. 2009;24:667–71.
Winther KV, Jess T, Langholz E, Munkholm P, Binder V. Long-term risk of cancer in ulcerative colitis: a population-based cohort study from Copenhagen County. Clin Gastroenterol Hepatol. 2004. https://doi.org/10.1016/S1542-3565(04)00543-9.
Rutter MD, Saunders BP, Wilkinson KH, Rumbles S, Schofield G, Kamm MA, Williams CB, Price AB, Talbot IC, Forbes A. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology. 2006. https://doi.org/10.1053/j.gastro.2005.12.035.
Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol. 2008. https://doi.org/10.3748/wjg.14.3937.
Söderlund S, Brandt L, Lapidus A, Karlén P, Broström O, Löfberg R, Ekbom A, Askling J. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology. 2009. https://doi.org/10.1053/j.gastro.2009.01.064.
Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment Pharmacol Ther. 2006. https://doi.org/10.1111/j.1365-2036.2006.02854.x.
Kiran RP, Khoury W, Church JM, Lavery IC, Fazio VW, Remzi FH. Colorectal cancer complicating inflammatory bowel disease: similarities and differences between Crohn's and ulcerative colitis based on three decades of experience. Ann Surg. 2010. https://doi.org/10.1097/SLA.0b013e3181e61e69.
Ouaïssi M, Maggiori L, Alves A, Giger U, Sielezneff I, Valleur P, Sastre B, Panis Y. Colorectal cancer complicating inflammatory bowel disease: a comparative study of Crohn's disease vs ulcerative colitis in 34 patients. Color Dis. 2011. https://doi.org/10.1111/j.1463-1318.2010.02241.x.
Watanabe T, Konishi T, Kishimoto J, Kotake K, Muto T, Sugihara K. Japanese Society for Cancer of the Colon and Rectum. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: a nationwide Japanese study. Inflamm Bowel Dis. 2011. https://doi.org/10.1002/ibd.21365.
Jensen AB, Larsen M, Gislum M, Skriver MV, Jepsen P, Nørgaard B, Sørensen HT. Survival after colorectal cancer in patients with ulcerative colitis: a nationwide population-based Danish study. Am J Gastroenterol. 2006;101:1283–7.
Nuako KW, Ahlquist DA, Mahoney DW, Schaid DJ, Siems DM, Lindor NM. Familial predisposition for colorectal cancer in chronic ulcerative colitis: a case-control study. Gastroenterology. 1998;115:1079–83.
Cremer A, Demetter P, De Vos M, Rahier JF, Baert F, Moreels T, Macken E, Louis E, Ferdinande L, Fervaille C, Dedeurwaerdere F, Bletard N, Driessen A, De Hertogh G, Vermeire S, Franchimont D. Belgian Inflammatory Bowel Disease Research and Development (BIRD) Group. Risk of Development of More-advanced Lesions in Patients With Inflammatory Bowel Diseases and Dysplasia. Clin Gastroenterol Hepatol. 2019. https://doi.org/10.1016/j.cgh.2019.05.062.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001. https://doi.org/10.1016/S0140-6736(00)04046-0.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002. https://doi.org/10.1038/nature01322.
De Marzo AM, Platz EA, Sutcliffe S, Xu J, Gronberg H, Drake CG, Nakai Y, Isaacs WB, Nelson WG. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007. https://doi.org/10.1038/nrc2090.
Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010. https://doi.org/10.1053/j.gastro.2010.01.058.
Korolkova OY, Myers JN, Pellom ST, Wang L, M'Koma AE. Characterization of serum cytokine profile in predominantly colonic inflammatory bowel disease to delineate ulcerative and Crohn's Colitides. Clin Med Insights Gastroenterol. 2015. https://doi.org/10.4137/CGast.S20612.
Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008. https://doi.org/10.3748/wjg.14.4280.
Krela-Kaźmierczak I, Kaczmarek-Ryś M, Szymczak A, et al. Bone metabolism and the c.-223C>T polymorphism in the 5'UTR region of the Osteoprotegerin gene in patients with inflammatory bowel disease. Calcif Tissue Int. 2016. https://doi.org/10.1007/s00223-016-0192-9.
Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel). 2011. https://doi.org/10.3390/cancers3043856.
Klampfer L. Cytokines, inflamation and colon cancer. Curr Cancer Drug Targets. 2011;11:451–64.
Kojima M, Morisaki T, Sasaki N, Nakano K, Mibu R, Tanaka M, Katano M. Increased nuclear factor-kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res. 2004;24:675–81.
Waldner MJ, Foersch S, Neurath MF. Interleukin-6 - a key regulator of colorectal Cancer development. Int J Biol Sci. 2012. https://doi.org/10.7150/ijbs.4614.
Mager LF, Wasmer MH, Rau TT, Krebs P. Cytokine-induced modulation of colorectal Cancer. Front Oncol. 2016. https://doi.org/10.3389/fonc.2016.00096.
Vitale S, Strisciuglio C, Pisapia L, Miele E, Barba P, Vitale A, Cenni S, Bassi V, Maglio M, Del Pozzo G, Troncone R, Staiano A, Gianfrani C. Cytokine production profile in intestinal mucosa of paediatric inflammatory bowel disease. PLoS One. 2017. https://doi.org/10.1371/journal.pone.0182313.
Chen ML, Sundrud MS. Cytokine networks and T-cell subsets in inflammatory bowel diseases. Inflamm Bowel Dis. 2016. https://doi.org/10.1097/MIB.0000000000000714.
Cui G, Yuan A, Sun Z, Zheng W, Pang Z. IL-1β/IL-6 network in the tumor microenvironment of human colorectal cancer. Pathol Res Pract. 2018. https://doi.org/10.1016/j.prp.2018.05.011.
Lippitz BE, Harris RA. Cytokine patterns in cancer patients: a review of the correlation between interleukin 6 and prognosis. Oncoimmunology. 2016. https://doi.org/10.1080/2162402X.2015.1093722.
Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, Vives F, Moon C, Penaflor E, Turner S, Ayanoglu G, Chang C, Basham B, Mumm JB, Pierce RH, Yearley JH, McClanahan TK, Phillips JH, Cua DJ, Bowman EP, Kastelein RA, LaFace D. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol. 2014. https://doi.org/10.1038/mi.2013.101.
Wang Y, Wang K, Han GC, Wang RX, Xiao H, Hou CM, Guo RF, Dou Y, Shen BF, Li Y, Chen GJ. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014. https://doi.org/10.1038/mi.2013.126.
Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013. https://doi.org/10.1084/jem.20122308.
Jana A, Krett NL, Guzman G, Khalid A, Ozden O, Staudacher JJ, Bauer J, Baik SH, Carroll T, Yazici C, Jung B. NFkB is essential for activin-induced colorectal cancer migration via upregulation of PI3K-MDM2 pathway. Oncotarget. 2017. https://doi.org/10.18632/oncotarget.16343.
Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J Gastroenterol. 2014;20(29):9872–81. https://doi.org/10.3748/wjg.v20.i29.9872.
Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal Cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009;29:2727–38.
Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J Gastroenterol. 2012. https://doi.org/10.3748/wjg.v18.i29.3839.
Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011. https://doi.org/10.1053/j.gastro.2011.01.057.
Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004. https://doi.org/10.1152/ajpgi.00079.2004.
Mattar MC, Lough D, Pishvaian MJ, Charabaty A. Current management of inflammatory bowel disease and colorectal cancer. Gastrointest Cancer Res. 2011;4:53–61.
Kurzawski G, Suchy J, Kładny J, Grabowska E, Mierzejewski M, Jakubowska A, Debniak T, Cybulski C, Kowalska E, Szych Z, Domagała W, Scott RJ, Lubiński J. The NOD2 3020insC mutation and the risk of colorectal cancer. Cancer Res. 2004;64:1604–6.
Suchy J, Kłujszo-Grabowska E, Kładny J, Cybulski C, Wokołorczyk D, Szymańska-Pasternak J, Kurzawski G, Scott RJ, Lubiński J. Inflammatory response gene polymorphisms and their relationship with colorectal cancer risk. BMC Cancer. 2008. https://doi.org/10.1186/1471-2407-8-112.
Freire P, Portela F, Donato MM, Figueiredo P, Ferreira M, Amaro P, Sá A, Andrade P, Gouveia H, Sofia C. CARD15 mutations and colorectal cancer in a south European country. Int J Color Dis. 2010. https://doi.org/10.1007/s00384-010-1028-0.
Askling J, Dickman PW, Karlén P, Broström O, Lapidus A, Löfberg R, Ekbom A. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. Gastroenterology. 2001. https://doi.org/10.1053/gast.2001.24052.
Tian Y, Li Y, Hu Z, Wang D, Sun X, Ren C. Differential effects of NOD2 polymorphisms on colorectal cancer risk: a meta-analysis. Int J Color Dis. 2010. https://doi.org/10.1007/s00384-009-0809-9.
Samadder NJ, Valentine JF, Guthery S, Singh H, Bernstein CN, Leighton JA, Wan Y, Wong J, Boucher K, Pappas L, Rowe K, Burt RW, Curtin K, Smith KR. Family history associates with increased risk of colorectal Cancer in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2018. https://doi.org/10.1016/j.cgh.2018.09.038.
Udden SMN, Peng L, Gan JL, Shelton JM, Malter JS, Hooper LV, Zaki MH. NOD2 suppresses colorectal tumorigenesis via Downregulation of the TLR pathways. Cell Rep. 2017. https://doi.org/10.1016/j.celrep.2017.05.084.
Kordjazy N, Haj-Mirzaian A, Haj-Mirzaian A, Rohani MM, Gelfand EW, Rezaei N, Abdolghaffari AH. Role of toll-like receptors in inflammatory bowel disease. Pharmacol Res. 2018. https://doi.org/10.1016/j.phrs.2017.11.017.
Li TT, Ogino S, Qian ZR. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World J Gastroenterol. 2014. https://doi.org/10.3748/wjg.v20.i47.17699.
Slattery ML, Herrick JS, Bondurant KL, Wolff RK. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int J Cancer. 2011. https://doi.org/10.1002/ijc.26314.
Messaritakis I, Stogiannitsi M, Koulouridi A, Sfakianaki M, Voutsina A, Sotiriou A, Athanasakis E, Xynos E, Mavroudis D, Tzardi M, Souglakos J. Evaluation of the detection of toll-like receptors (TLRs) in cancer development and progression in patients with colorectal cancer. PLoS One. 2018. https://doi.org/10.1371/journal.pone.0197327.
Omrane I, Baroudi O, Kourda N, Bignon YJ, Uhrhammer N, Desrichard A, Medimegh I, Ayari H, Stambouli N, Mezlini A, Bouzayenne H, Marrakchi R, Benammar-Elgaaid A, Bougatef K. Positive link between variant toll-like receptor 4 (Asp299Gly and Thr399Ile) and colorectal cancer patients with advanced stage and lymph node metastasis. Tumour Biol. 2014. https://doi.org/10.1007/s13277-013-1075-6.
Santini D, Angeletti S, Ruzzo A, Dicuonzo G, Galluzzo S, Vincenzi B, Calvieri A, Pizzagalli F, Graziano N, Ferraro E, Lorino G, Altomare A, Magnani M, Graziano F, Tonini G. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol. 2008. https://doi.org/10.1111/j.1365-2249.2008.03776.x.
Li X, Yao X, Wang Y, Hu F, Wang F, Jiang L, Liu Y, Wang D, Sun G, Zhao Y. MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PLoS One. 2013. https://doi.org/10.1371/journal.pone.0059064.
Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011. https://doi.org/10.1021/bi200642e.
Azer SA. Overview of molecular pathways in inflammatory bowel disease associated with colorectal cancer development. Eur J Gastroenterol Hepatol. 2013. https://doi.org/10.1097/MEG.0b013e32835b5803.
Polanczyk-Piechota A, Fichna J. Review article: the role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn Schmiedeberg's Arch Pharmacol. 2014. https://doi.org/10.1007/s00210-014-0985-1.
Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006. https://doi.org/10.1515/BC.2006.049.
Keku TO, Dulal S, Deveaux A, Jovov B, Han X. The gastrointestinal microbiota and colorectal cancer. Am J Physiol Gastrointest Liver Physiol. 2014;308(5):G351–63. https://doi.org/10.1152/ajpgi.00360.2012.
Flemer B, Lynch DB, Brown JM, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2016;66(4):633–43. https://doi.org/10.1136/gutjnl-2015-309595.
Sansonetti PJ, Medzhitov R. Learning tolerance while fighting ignorance. Cell. 2009. https://doi.org/10.1016/j.cell.2009.07.024.
Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, España C, Ungaro R, Harpaz N, Cooper HS, Elson G, Kosco-Vilbois M, Zaias J, Perez MT, Mayer L, Vamadevan AS, Lira SA, Abreu MT. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis. 2010. https://doi.org/10.1002/ibd.21527.
Balish E, Warner T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol. 2002. https://doi.org/10.1016/S0002-9440(10)61172-8.
Berg DJ, Davidson N, Kühn R, Müller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996. https://doi.org/10.1172/JCI118861.
O'Mahony L, Feeney M, O'Halloran S, Murphy L, Kiely B, Fitzgibbon J, Lee G, O'Sullivan G, Shanahan F, Collins JK. Probiotic impact on microbial flora, inflammation and tumour development in IL-10 knockout mice. Aliment Pharmacol Ther. 2001. https://doi.org/10.1046/j.1365-2036.2001.01027.x.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AH, SH, MKR, EL wrote the manuscript. AH, RS and AP was responsible for conception, design and consultations, AP was responsible for critical revision for important intellectual content AP and RJS were responsible for final revision. All authors have given final approval of the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hnatyszyn, A., Hryhorowicz, S., Kaczmarek-Ryś, M. et al. Colorectal carcinoma in the course of inflammatory bowel diseases. Hered Cancer Clin Pract 17, 18 (2019). https://doi.org/10.1186/s13053-019-0118-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13053-019-0118-4