
Abstract
Background
Plasmodium falciparum genetic diversity can add information on transmission intensity and can be used to track control and elimination interventions.
Methods
Dried blood spots (DBS) were collected from patients who were recruited for a P. falciparum malaria therapeutic efficacy trial in three malaria endemic sites in Ethiopia from October to December 2015, and November to December 2019. qPCR-confirmed infections were subject to amplicon sequencing of polymorphic markers ama1-D3, csp, cpp, cpmp, msp7. Genetic diversity, the proportion of multiclonal infections, multiplicity of infection, and population structure were analysed.
Results
Among 198 samples selected for sequencing, data was obtained for 181 samples. Mean MOI was 1.38 (95% CI 1.24–1.53) and 17% (31/181) of infections were polyclonal. Mean He across all markers was 0.730. Population structure was moderate; populations from Metema and Metehara 2015 were very similar to each other, but distinct from Wondogent 2015 and Metehara 2019.
Conclusion
The high level of parasite genetic diversity and moderate population structure in this study suggests frequent gene flow of parasites among sites. The results obtained can be used as a baseline for additional parasite genetic diversity and structure studies, aiding in the formulation of appropriate control strategies in Ethiopia.
Similar content being viewed by others


Background
Malaria caused an estimated 247 million cases and 619,000 deaths worldwide in 2022 [1]. The disease is a major public health and economic problem in Ethiopia. In 2020, the Federal Ministry of Health estimated that 75% of the land mass is malarious and over 50% of the population is at risk of malaria [2]. Malaria transmission generally occurs at elevations < 2000 m and peak transmission occurs from September to December and March to May [3].
Genetic diversity of Plasmodium falciparum populations and multiplicity of infection (MOI) in humans might vary according to transmission intensity in different geographical regions [4,5,6,7,8,9]. The study of genetic diversity helps researchers to understand the distribution, dynamics, and genetic structure of the parasite population [10,11,12,13,14].
Next-generation sequencing (NGS) tools are increasingly being applied to evaluate diverse markers in the parasite genome [4, 7,8,9,10]. Compared to other targets such as antigenic markers (merozoite surface proteins and Glutamic rich proteins), single nucleotide polymorphisms (SNPs) and microsatellites, highly diverse amplicon generate more sensitive detection of minority clones in polyclonal infections [4]. This method is becoming the new gold standard in treatment efficacy studies as it has a greater ability to distinguish between new and persisting infections [11].
Amplicon sequencing studies reveal population structure and genetic relatedness among parasite populations, which can be used to understand the spread of the parasite within a region and to evaluate the effectiveness of ongoing control efforts [7, 15,16,17,18,19,20,21,22].
In this study, P. falciparum from northern, southern and eastern part of Ethiopia were genotyped by amplicon sequencing at 5 highly diverse markers to assess parasite diversity and malaria transmission in the country.
Methods
Ethics statement
The study obtained ethical approval from the Institutional Review Board (IRB) of College of Natural and Computational Sciences, Addis Ababa University, certificate reference number IRB/033/2018. Written informed consent/assent was obtained from participants or parents/guardians for minors. Malaria positive cases were treated as per the national treatment guidelines for malaria [23].
Study area
Samples for this study were collected in three sites: Metema, Wondogent and Metehara. Metema is in northern Ethiopia, 925 km northwest of Addis Ababa, with altitudes 1608 m above sea level River include the Genda Wuha, is a notable mosquito habitat near the study area. Wondogent in southern Ethiopia is in the Rift Valley 261 km southern of Addis Ababa at an elevation of 1,723 m. It is surrounded by primary forests and water bodies which provide suitable habitats for mosquitos. Metehara is situated in eastern-central Ethiopia in the Rift Valley area 128 km southeast of Addis Ababa at an elevation of 947 m. Nearby rivers and an irrigated sugarcane farms provide breeding sites for malaria mosquitoes (Fig. 1).

Map of the study area shows sample collection site
Study population and sample collection
Study participants were febrile patients recruited from a health center in each site from October to December 2015 and again in Metehara from November to December 2019. Dried blood spots (DBS) were collected on Whatman 3 mm™ filter and stored in individual plastic bags containing desiccant.
Laboratory methods
A piece roughly 5 mm2 was cut from each DBS and DNA was extracted using the NucleoMag Blood kit (Macherey–Nagel). All samples were screened by qPCR for P. falciparum using a varATS assay, targeting a multicopy gene with approximately 20 copies per genome [24]. The 3D7 P. falciparum culture strain was used as a positive control and to generate a standard curve for quantification. Primers and probes of qPCR are in Additional file 1: Table S1.
Parasite genotyping
198 Plasmodium falciparum-positive samples with Ct values < 30 based on the varATS assay were selected for parasite genotyping at 5 highly diverse markers: apical membrane antigen 1 (ama1-D3, PF3D7_1133400), circumsporozoite surface protein (csp, PF3D7_0304600), a conserved Plasmodium protein (cpp, PF3D7_1475800), a conserved Plasmodium membrane protein (cpmp, PF3D7_0104100), and merozoite surface protein 7 (msp7, PF3D7_1335100) [12]. Amplicons were generated by PCR and samples were pooled into a sequencing library as previously described (Additional file 2: Table S2) [25]. The library was sequenced in paired-end mode in one run using the Illumina MiSeq reagent kit v3 600 cycles (2 × 300 bp) with 15% Enterobacteria phage phiX control v3 (Illumina).
Data analysis
Haplotype calling and population analyses were conducted using R software version 4.2.2 [26] and Haplotype R package version 0.3.3 as previously described [25]. In short, low-quality reads and samples with < 10 reads were excluded. Single nucleotide polymorphisms with a mismatch rate ≥ 0.5 occurring in two or more samples were considered real. Sequences with coverage ≥ 3 and within-host frequency ≥ 1% in samples with ≥ 25 reads were considered haplotypes. A phylogenetic tree was constructed in R using ama1-D3 major clone sequences, the K80 nucleotide substitution model, and the neighbour-joining method [27]. Principal components analysis (PCA) was conducted using the adegenet package in R. Population structure analysis was done using STRUCTURE software version 2.3.4 [28] and Bayesian clustering analysis was done with Structure harvester v0.6.94 [29].
SPSS (IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp.) was used to conduct additional statistical analyses. MOI was determined by the maximum number of alleles observed at an individual locus and the average MOI was calculated for each sub-population. The Student’s t-test was used to assess the relationship between MOI, parasite density, and patient age, and the spearman’s rank correlation coefficient was used to see association between age and MOIs. A chi-squared test for independence was done to compare the proportion of polyclonal infections at each site. The expected heterozygosity (He) was calculated using the formula \(He = \left( {\frac{n}{n - 1}} \right)\left( {1 - \sum {p^{2} } } \right),\) where ‘n’ is the number of samples analysed and ‘p’ the frequency of each allele at a locus [30].
Results
qPCR screening
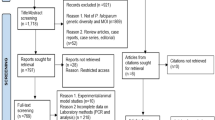
Of a total 661 samples that were collected, 248 (38%) were collected in 2015 and 413 (62%) were collected in 2019. qPCR screening identified 280 (42%) samples positive for P. falciparum, 198 of which were selected based on parasite density for amplicon sequencing. Sequencing and haplotyping at two or more markers was successful for 91% (181/198) of samples (Table 1). Demographic data for 181 samples successfully sequenced is given in (Table 2).
Parasite genotyping
Markers ama1-D3, csp, cpp, cpmp, and msp7 were successfully sequenced in 177 (89.3%), 179 (90.4%), 177 (89.3%), 176 (88.9%), and 158 (79.8%) of 198 samples, respectively. All five markers were sequenced in 150/198 (75.8%) samples. Between 11 (csp and msp7) and 23 (ama1-D3) different alleles per marker were observed (Table 3). The median coverage for each allele in each sample (including minority clones) was 2006 reads (range 21–13,682, 2.5 and 97.5 percentiles). Among the 150 samples with sequences for all markers, 58 major clones had unique haplotypes. The most common haplotype appeared as the major clone of 13 samples, all from Metehara (3 from 2015 and 10 from 2019).
MOI ranged from 1 to 7 with a mean of 1.38. The percentage of polyclonal infections was 17.5% (7/40) in Metema, 24.4% (10/41) in Wondogent, 32% (8/25) in Metehara in 2015, and 13.6% (6/44) in Metehara in 2019 and 0.716 for ama1-D3, 0.870 for cpmp, 0.786 for cpp, 0.593 for csp, and 0.681 for msp7 (Table 3).
Mean MOI across all markers was 1.38 Detailed monoclonal and polyclonal infections by marker are given in Table 3. In 2015 18.9% (25/132) of infections were polyclonal compared to 12.2% (6/49) in 2019 (Table 3). The proportion of polyclonal infections decreased from 24 to 12% between 2015 and 2019 in Metehara; however, this trend did not reach significance (p = 0.48). No significant association between MOI and age or relative parasitaemia was observed (p > 0.05). Mean He across all sites was 0.730, indicating a moderate to high level of genetic diversity.
Population structure
Bayesian clustering analysis of admixed indicated that the data best fit three genetic clusters. STRUCTURE analysis revealed moderate population structure among parasites. Parasites from Metema-2015 and Metehara-2015 likely share ancestry from a single genetic group. Parasites from Wondogent-2015 largely belong to a second ancestral group. Roughly 25% of parasites from Metehara-2019 share ancestry with the parasites from Wondogent, and the remaining parasites belong to a third ancestral group (Fig. 2).

Structure plot (k = 3, admixture) showed moderate population structure. Parasites collected from Metema and Metehara-2015 shared ancestry from one group (red). Parasites from Wondogent and Metehara-2019 shared ancestry from another group (blue). Parasites with ancestry from the third group (green) were largely identified in Metehara-2019. Parasites from Metehara-2019 were dominated by two ancestral groups without much admixture (blue and green), suggesting possible clonal expansion of the two lineages
In a phylogenetic tree constructed based on the ama1-D3 sequences of major clones, samples from all sites were intermixed. No clades were unique to a single study site, suggesting a high level of genetic relatedness (Fig. 3). PCA considering alleles of all five markers showed that there is more diversity within parasite populations than between them as the samples are widely scattered yet overlap almost entirely (Fig. 4). This indicates gene flow among the three sites, and that populations did not change much from 2015 to 2019 in Metehara.

Phylogenetic tree based on ama1-D3 alleles from major clones using neighbor-joining method. Genetic distances between parasites are small and parasites from multiple sites appear in most clades, indicating that parasites are genetically similar at this locus

Principal components analysis considering alleles of all five markers. Parasite populations overlap, indicating gene flow and genetic similarity among groups
Discussion
Like many African countries, Ethiopia is working toward malaria elimination to alleviate the burden of the disease [1]. Molecular studies on P. falciparum genetic diversity and population structure can aid in monitoring the impact of different intervention strategies. This study employed amplicon sequencing to characterize parasite genetic diversity and structure across three malaria-endemic regions in Ethiopia. This study found moderate population structure and high genetic diversity among parasites with little distinction between parasites collected from the various study sites, likely due to gene flow among sites. High diversity of multi-locus haplotypes is typical in regions of high malaria transmission, as mosquitoes are more likely to take up multiple P. falciparum clones during a blood meal, thereby increasing the chance of recombination between unidentical parasites and generating a highly diverse parasite population [12, 14].
In this study, He among all samples was 0.73. This is similar to previous He measurements in Kenya, Nigeria, and Ghana [21, 25, 31], and lower than He values reported in Asia [25, 32]. The He reported here is substantially greater than those reported in other recent Ethiopian studies (0.17, 0.47, 0.54) [33,34,35]. This highlights the importance of noting methodological differences before comparing study results. Findings in this study of 17% polyclonal infections and mean MOI of 1.38 were in alignment with other recent findings in Ethiopia [33,34,35].
The isolates of P. falciparum analysed in this study were genetically diverse and generally did not cluster by collection site. This is indicative of gene flow among the geographic locations sampled, likely due to immense human traffic through the country as part of routine socio-economic activities. Additionally, this study revealed evidence of a shift in parasite populations between 2015 and 2019 in Metehara. Between the two time points MOI, heterozygosity, and the proportion of polyclonal infections decreased, although the differences were not statistically significant. Structure analysis showed a shift from a diverse parasite population arising from three ancestral groups in 2015 to a more structured population comprising just two ancestral groups in 2019. Additionally, this decrease in genetic diversity may represent a decrease in malaria transmission or a reduction in gene flow to and from Metehara by 2019. In this study, there is a reduction of MOI similar to the study done in Indonesia 1.6 [36] and a study done in Senegal 1.35 [37]. In this study, there is also a high level of heterozygosity, similar to the study done in Ghana [38].
The low to moderate level of genetic differentiation of parasites between the study sites is likely a consequence of immense human traffic as part of routine socio-economic activities. Further sampling of more populations along this North, South and East of Ethiopia including asymptomatic infections, will refine the boundaries of gene flow and inform the Ethiopia National Malaria Control Programme (NMCP) on local approaches to tackle malaria elimination.
Conclusion
This study employed amplicon sequencing of five polymorphic markers to characterize P. falciparum diversity and structure across malaria-endemic sites in Ethiopia. Overall, parasites were highly diverse, with little distinction by collection site. This is indicative of gene flow among parasite populations around the country. Further sampling of more populations along this North, South and East of Ethiopia including asymptomatic infections, will refine the boundaries of gene flow and inform the Ethiopia National Malaria Control Programme (NMCP) on local approaches to tackle malaria elimination. Therefore the study will help as baseline for further study.
Availability of data and materials
The dataset supporting the conclusion of this article is included within the article and the primers used to genotype the five genes are presented in attached Additional file 2: Table S2.
Abbreviations
- ama1-D3 :
-
Apical membrane protein1
- cpp :
-
Conserved Plasmodium protein
- csp :
-
Circumsporozoite surface protein
- cpmp :
-
Conserved Plasmodium membrane protein
- DBS:
-
Dry blood spot
- He :
-
Expected heterozygosity
- MOI:
-
Multiplicity of infection
- msp7 :
-
Merozoite surface protein 7
- NGS:
-
Next generation sequencing
- PCA:
-
Principal components analysis
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Quantitative polymerase chain reaction
- SNP:
-
Single nucleotide polymorphism
- WHO:
-
World Health Organization
References
WHO. World malaria report. Geneva: World Health Organization; 2022.
Federal Ministry of Health of Ethiopia. National malaria guidelines. 5th ed. Addis Ababa: Federal Ministry of Health of Ethiopia; 2020.
Federal Ministry of Health of Ethiopia. Ethiopia malaria elimination strategic plan: 2021–2025. Addis Ababa: Federal Ministry of Health of Ethiopia; 2020.
Zhong D, Koepfi C, Cui L, Yan G. Molecular approaches to determine the multiplicity of Plasmodium infections. Malar J. 2018;17:172.
Zhong D, Lo E, Wang X, Yewhalaw D, Zhou G, Atieli HE, et al. Multiplicity and molecular epidemiology of Plasmodium vivax and Plasmodium falciparum infections in East Africa. Malar J. 2018;17:185.
Koepfli C, Waltmann A, Ome-Kaius M, Robinson LJ, Mueller I. Multiplicity of infection is a poor predictor of village-level Plasmodium vivax and P. falciparum population prevalence in the Southwest Pacific. Open Forum Infect Dis. 2018;5: ofy240.
Chen JT, Li J, Zha G-C, Huang G, Huang Z-X, Xie D-D, et al. Genetic diversity and allele frequencies of Plasmodium falciparum msp1 and msp2 in parasite isolates from Bioko Island, Equatorial Guinea. Malar J. 2018;17:458.
Ndiaye T, Sy M, Gaye A, Siddle KJ, Park DJ, Bei AK, et al. Molecular epidemiology of Plasmodium falciparum by multiplexed amplicon deep sequencing in Senegal. Malar J. 2020;19:403.
Babiker H, Cartwright LC, Currie D, Charlwood J, Billingsley P, Teuscher T, Walliker D. Random mating in a natural population of the malaria parasite Plasmodium falciparum. J Parasitol. 1994;109:413–21.
Tessema SK, Raman J, Duffy CW, Ishengoma DS, Amambua-Ngwa A, Greenhouse B. Applying next-generation sequencing to track falciparum malaria in sub-Saharan Africa. Malar J. 2019;18:268.
Jones S, Kay K, Hodel EM, Gruenberg M, Lerch A, Felger I, et al. Should deep-sequenced amplicons become the new gold standard for analyzing malaria drug clinical trials? Antimicrob Agents Chemother. 2021;65:e00437-e521.
Lerch A, Koepfi C, Hofmann NE, Messerli C, Wilcox S, Kattenberg JH, et al. Development of amplicon deep sequencing markers and data analysis pipeline for genotyping multi-clonal malaria infections. BMC Genom. 2017;18:864.
Miller RH, Hathaway NJ, Kharabora O, Mwandagalirwa K, Tshefu A, Meshnick SR, et al. A deep sequencing approach to estimate Plasmodium falciparum complexity of infection (COI) and explore apical membrane antigen 1 diversity. Malar J. 2017;16:490.
Gruenberg M, Lerch A, Beck HP, Felger I. Amplicon deep sequencing improves Plasmodium falciparum genotyping in clinical trials of antimalarial drugs. Sci Rep. 2019;9:17790.
Koepfli C, Mueller I. Malaria epidemiology at the clone level. Trends Parasitol. 2017;33:974–85.
Lerch A, Koepfli C, Hofman NE, Kattenberg JH, Rosanas-Urgell A, Betuela I, et al. Longitudinal tracking and quantification of individual Plasmodium falciparum clones in complex infections. Sci Rep. 2019;9:3333.
Atuh NI, Anong DN, Jerome F-C, Oriero E, Mohammed NI, D’Alessandro U, et al. High genetic complexity but low relatedness in Plasmodium falciparum infections from Western Savannah Highlands and coastal equatorial Lowlands of Cameroon. Pathog Glob Health. 2022;116:428–37.
Ralinoro F, Rakotomanga TA, Rakotosaona R, Rakoto DAD, Menard D, Jeannoda V, et al. Genetic diversity of Plasmodium falciparum populations in three malaria transmission settings in Madagascar. Malar J. 2021;20:239.
Pumpaibool T, Arnathau C, Durand P, Kanchanakhan N, Siripoon N, Suegorn A, et al. Genetic diversity and population structure of Plasmodium falciparum in Thailand, a low transmission country. Malar J. 2009;8:155.
Roh ME, Tessema SK, Murphy M, Nhlabathi N, Mkhonta N, Vilakati S, et al. High genetic diversity of Plasmodium falciparum in the low-transmission setting of the Kingdom of Eswatini. J Infect Dis. 2019;220:1346–54.
Ajogbasile FV, Kayode AT, Oluniyi PE, Akano KO, Uwanibe JN, Adegboyega BB, et al. Genetic diversity and population structure of Plasmodium falciparum in Nigeria insights from microsatellite loci analysis. Malar J. 2021;20:236.
Agaba BB, Anderson K, Gresty K, Prosser C, Smith D, Nankabirwa JI, et al. Genetic diversity and genetic relatedness in Plasmodium falciparum parasite population in individuals with uncomplicated malaria based on microsatellite typing in Eastern and Western regions of Uganda, 2019–2020. Malar J. 2021;20:242.
Federal Ministry of Health of Ethiopia. National malaria guidelines. 4th ed. Addis Ababa: Federal Ministry of Health of Ethiopia; 2017.
Hofmann N, Mwingira F, Shekalaghe S, Robinson LJ, Mueller I, Felger I. Ultra-sensitive detection of Plasmodium falciparum by amplifcation of multi-copy subtelomeric targets. PLoS Med. 2015;12: e1001788.
Huwe T, Kibria MG, Johora FT, Phru CS, Jahan N, Hossain MS, et al. Heterogeneity in prevalence of subclinical Plasmodium falciparum and Plasmodium vivax infections but no parasite genomic clustering in the Chittagong Hill Tracts, Bangladesh. Malar J. 2022;21:218.
R Development Core Team. R: a language and environment for statistical computing. 4.0.0. Vienna: R Foundation for Statistical Computing; 2021.
Toparslan E, Karabag K, Bilge U. A workflow with R: phylogenetic analyses and visualizations using mitochondrial cytochrome b gene sequences. PLoS ONE. 2020;15: e0243927.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Earl DA, Vonholt BM. Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–61.
Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89:583–90.
Nderu D, Kimani F, Karanja E, Thiong’o K, Akinyi M, Too E, et al. Genetic diversity and population structure of Plasmodium falciparum in Kenyan-Ugandan border areas. Trop Med Int Health. 2019;24:647–56.
Tessema S, Wesolowski A, Chen A, Murphy M, Wilheim J, Mupiri A-R, et al. Using parasite genetic and human mobility data to infer local and cross-border malaria connectivity in Southern Africa. Elife. 2019;8: e43510.
Tadele G, Jatieh FK, Oboh M, Oriero E, Dugassa S, Amambua-Ngwa A, et al. Low genetic diversity of Plasmodium falciparum merozoite surface protein 1 and 2 and multiplicity of infections in western Ethiopia following effective malaria interventions. Malar J. 2022;21:383.
Tessema SK, Belachew M, Koepfli C, Lanke K, Huwe T, Chale W, et al. Spatial and genetic clustering of Plasmodium falciparum and Plasmodium vivax infections in a low-transmission area of Ethiopia. Sci Rep. 2020;10:19975.
Holzschuh A, Ewnetu Y, Carlier L, Lerch A, Gerlovina I, Baker SC, et al. Plasmodium falciparum transmission in the highlands of Ethiopia is driven by closely related and clonal parasites. BioRxiv. 2023. https://doi.org/10.1101/2023.06.09.544365.
Pava IZ, Puspitasari AM, Rumaseb A, Handayuni I, Trianty L, Utami RAS, et al. Molecular surveillance over 14 years confirms reduction of Plasmodium vivax and falciparum transmission after implementation of artemisinin-based combination therapy in Papua, Indonesia. PLoS Negl Trop Dis. 2020;14: e0008295.
Bei AK, Niang M, Deme AB, Daniels RF, Sarr FD, Sokhna C, et al. Dramatic changes in malaria population genetic complexity in Dielmo and Ndiop, Senegal, revealed using genomic surveillance. J Infect Dis. 2018;217:622–7.
Amoah LE, Abukari Z, Amoah MED, Dieng CC, Lo E, Afrane YA. Population structure and diversity of Plasmodium falciparum in children with asymptomatic malaria living in different ecological zones of Ghana. BMC Infect Dis. 2021;21:439.
Acknowledgements
The authors would like to thank the study participants.
Authors would like to thank Addis Ababa University, Microbial Cellular Molecular Biology department for partial funding.
Funding
Funding partially was obtained from Addis Ababa University. Funding agency did not play a role in the design of the study, collection of sample, analysis, and interpretation of the data and writing of manuscript.
Author information
Authors and Affiliations
Contributions
AGR and TH contributed equally to this work. AG, involved in data collection, and drafting manuscript. TH laboratory analysis and result interpretation. CK, conceived the research idea, supervised AGR, TH and critically revised the manuscript and fully involved in all phases of the study. AS critically revised the manuscript. SKT critically revised the manuscript. AM involved in data analysis of the study. LG and HM critically revised the manuscript, supervised AGR. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was ethically approved by the Institutional Review Board (IRB) of College of Natural and Computational Sciences, Addis Ababa University certificate reference number IRB/033/2018. Written informed consent/assent was obtained from participants or parents/guardians for minors. Malaria positive cases were treated as per the national treatment guideline.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers and probe used for qPCR assay.
Additional file 2: Table S2.
Detailed procedure used for deep amplicon sequencing of five highly polymorphic markers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Reda, A.G., Huwe, T., Koepfli, C. et al. Amplicon deep sequencing of five highly polymorphic markers of Plasmodium falciparum reveals high parasite genetic diversity and moderate population structure in Ethiopia. Malar J 22, 376 (2023). https://doi.org/10.1186/s12936-023-04814-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-023-04814-w