
Abstract
Background
Despite its immunogenicity, the polymorphic nature of merozoite surface protein 1, an important vaccine candidate for Plasmodium falciparum malaria, remains a concern. This study analyses the impact of genetic variability and parasite population structure on epitope organization of different MSP1 segments.
Methods
Altogether 98 blood samples collected from P. falciparum infected mild and severe malaria patients of Chhattisgarh and West Bengal were used to sequence regions encoding block 2 and MSP1-19 of msp1. Sequences were analysed using MEGA7, DnaSPv5, Arlequin3.5 and BepiPred.
Results
All three major MSP1 block 2 allele families namely K1, MAD20 and RO33 were detected in the samples and they together resulted in 41 indel variants. Chhattisgarh samples displayed an average MOI of 2.07 ± 1.59 which was higher in mild malaria and in age group < 18 years. Ultra-structure of block 2 alleles revealed that mutation and repeat expansion were two major mechanisms responsible for allelic variability of K1 and MAD20. Regions flanking block 2 were highly variable in Chhattisgarh with average mismatch differences (k) ranging from 1.198 to 5.156 for three families. In contrast, region encompassing MSP1-19 exhibited limited heterogeneity (kChhattisgarh = 1.45, kWest Bengal = 1.363). Of the 50 possible B cell linear epitopes predicted from block 2 variants, 94.9% (131 of 138) of the parasites could be represented by three conserved antigens.
Conclusions
Present data indicates that natural selection and transmission intensity jointly play a role in controlling allelic diversity of MSP1 in Indian parasite isolates. Despite remarkable genetic variability, a limited number of predominant and conserved epitopes are present in Indian parasite isolates reinstating the importance of MSP1 as a promising malaria vaccine candidate.
Similar content being viewed by others
Background
The estimated rate of malaria mortality has reduced by 47% worldwide between 2000 and 2013 [1]. This reduction of malaria burden has been achieved through coordinated control of parasites and vectors using a variety of interventions [2]. To sustain this encouraging statistics and prevent clinical disease in sub-Saharan Africa, Asia and Latin America which continue to share a disproportionately high global malaria load, development of vaccine against the most virulent species, Plasmodium falciparum, in particular, is urgently needed [3]. Till date RTS, S remains the most advanced malaria vaccine, although its mechanism action and factors responsible for inter-individual differences in vaccine efficacy are poorly characterized [4]. Of the different vaccine development strategies, those targeting pre-erythrocytic stage proteins and asexual blood stage antigens are primarily intended to prevent clinical disease. However, many blood stage merozoite proteins that elicit protective immunity against malaria use parallel redundant pathways and/or are extremely polymorphic [5, 6]. A polymorphic antigen with strong immunogenicity may still be considered as the component of a multistage polyvalent vaccine and protect the vulnerable populations in diverse transmission settings. As a proof of concept, a synthetic vaccine was constructed by fusing block 2 variants with conserved block 1 of P. falciparum merozoite surface protein 1. This hybrid vaccine produced high titre antibodies in experimental animals inhibiting parasite growth in vitro and showed strong reactivity against antibodies isolated from naturally exposed malaria patients in a Ghanaian cohort [7].
MSP1 is the most abundant surface antigen in the blood stage of P. falciparum. It plays a crucial role in the initial low affinity attachment of parasite to RBC membrane during erythrocyte invasion [8]. MSP1 contains 17 blocks of which block 2 shows extensive allelic polymorphism worldwide [9, 10]. Block 2 alleles are mainly represented by three families namely K1, MAD20 and RO33 in the field isolates based on their characteristic tri-peptide motifs. Different allelic sequences belonging to these families show highly skewed and continent specific geographical distribution [11]. Besides, the pattern and extent of fragment size polymorphism of block 2 alleles serve as molecular indicators host immunity and malaria transmission dynamics [12].
MSP1 is synthesized as a ~ 195 kDa precursor which is proteolytically cleaved into four major fragments prior to schizont rupture [13]. One of these fragments, MSP1-42 is further processed to produce MSP1-19 that enters with merozoite into RBCs whereas others are shed off [14]. MSP1-19 is immunogenic in both human and animal infections and is considered as an attractive vaccine candidate [15,16,17,18,19,20,21]. Studies evaluating the immunogenic potential of the rest of the MSP1 molecule identified block 2 region as a target of protective immunity and showed that antibodies to block 2 are also associated with reduced risk of clinical malaria [7, 22, 23].
Given this, the present study evaluates the genetic diversity of two most immunogenic segments of msp1 namely block 2 and MSP1-19 in parasite isolates from Chhattisgarh, in central India and West Bengal, in eastern India. In parallel, the question, how the observed allelic variation of these segments affects the distribution of B-cell epitopes, is also addressed. The results indicate that msp1 block 2 gene pool is shaped by a localized pattern of parasite transmission and its immunogenic repertoire is furnished with a limited number of conserved epitopes. The suitability of the MSP1 block 2 as a potential vaccine target, as revealed by the present report, may have significant implications in the global malaria eradication initiatives.
Methods
Sample collection and DNA analysis
Peripheral blood samples for parasite DNA analysis were collected from Ambikapur situated in Chhattisgarh, central India, during a period of 2010–2013. Owing to its distinct ecological and geographical conditions, malaria exhibits a discrete pattern in Chhattisgarh contributing to ~ 12% of total disease burden and the highest share of deaths (17%) in India [24]. Genomic DNA extracted from peripheral blood samples of P. falciparum malaria patients admitted in Calcutta National Medical College & Hospital, Kolkata, in the year 2010 were also included in the study [25]. Kolkata is the capital of West Bengal which accounts for about 10% of the total malaria cases in India [26]. The two study regions differ with regard to malaria transmission intensity and disease characteristics [27, 28].
Peripheral blood samples collected from P. falciparum malaria patients of Ambikapur were employed to isolate genomic DNA using QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) following manufacturer’s protocol. Overall, 98 P. falciparum infected blood samples, 41 from Chhattisgarh and 57 from West Bengal, detected through Giemsa-stained thick and thin smears, were selected for this study. Patients suffering from co-infection with Plasmodium vivax were excluded from the analysis. In addition, patients with acute lower respiratory tract infection, bacteraemia, measles, severe diarrhoea with dehydration and other chronic or severe conditions, such as cardiac, renal or hepatic diseases, AIDS, G6PD deficiency, sickle cell anaemia, typhoid and cancer were also excluded.
PCR amplification and cloning of PCR amplicons
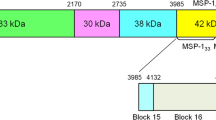
Oligonucleotide primers were designed for each target region using P. falciparum genomic DNA sequence (3D7 strain: GenBank accession number U65407.1) (Fig. 1). Primers were designed from the conserved sequences located on the both end of hypervariable block 2 (MSP1 block 2 forward: 5′-CACATGAAAGTTATCAAGAACTTGTC-3′, MSP1 block 2 reverse: 5′-TAAGTACGTCTAATTCATTTGCACG-3′) [29]. Region encoding the receptor binding site of MSP1 (MSP1-19) was also PCR amplified using MSP1-19 forward: 5′-CGTCACCAGCAAAAACAGACGAAC-3′ and MSP1-19 reverse: 5′-TGCTACCTGAATCTTCTTCGGTAC-3′ primers. Amplification of both target regions was performed in 15 μL reaction mixtures containing 0.2 mM dNTP, 1.5 mM MgCl2, 0.4 μM of each primer, and 1 U of GoTaq® Flexi DNA polymerase (Promega). The cycling conditions for PCR consisted of an initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 45 s, annealing at 58 °C for 45 s, extension at 72 °C for 45 s, and a final extension at 72° C for 5 min using a thermal cycler (Applied Biosystems® GeneAmp® PCR System 9700). The amplicons were visualized using UV transillumination on gel documentation system (Biostep) following electrophoresis on 2% agarose gel (Promega). PCR products showing single band were purified by Qiaquick gel extraction kit (QIAGEN India Pvt. Ltd, Hilden, Germany) and sequenced. PCR amplicons showing more than one band in gel electrophoresis images were suspected to represent multiclonal infections. To analyse those samples, each PCR product showing multiple bands was cloned in pTZ57R/T vector using InsTAclone PCR Cloning Kit (Fermentas) and transformed into DH5α Escherichia coli strain. Transformed E. coli were cultured on Luria–Bertani Agar containing 100 μg/μL Ampicillin. Ten colonies were chosen arbitrarily for each PCR amplicon to isolate the plasmids. Altogether, 16 samples showing multiple bands were analysed.

Schematic representation of P. falciparum merozoite surface protein 1 (MSP1). The regions subjected to sequence analysis were highlighted using broken lines
Sequencing
Purified PCR products showing single band and each of the isolated plasmids containing single genotype were sequenced using the same primers used in PCR. Sequencing PCR protocol was programmed with an initial denaturation at 94 °C for 30 s followed by 25 cycles (or 29 cycles, while plasmid DNA was used as templates) of denaturation at 94 °C for 10 s, holding for 10 s at 50 °C and extension for 4 min at 60 °C and finally stored at 4 °C. Sequencing was carried out in both directions, using the forward and reverse primers and Big Dye v3.1 dye terminator chemistry. The products were resolved on ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Sequence alignment and data analysis
Raw sequence data files from field isolates were manually revised to exclude signal noises. To compare the sequence identity, NCBI BLAST analysis was performed with all test sequences [30, 31]. Nucleotide sequences generated were submitted to the GenBank database under accession numbers MF772523–MF772713. MEGA7 tool was used to perform multiple sequence alignment and to translate DNA sequences into amino acid codes. Allele specific sequence motifs were used to search MSP1 block 2 sequences to assign family types. Sequences belonging to a given family were clustered to detect the pattern of fragment length polymorphism [32]. Based on nucleotide sequences pertaining to block 2, phylogenetic tree representing each allele family was constructed using maximum Parsimony method (MEGA7). This assisted further sub-classification of each allele types.
Association of allele frequency with transmission intensity, disease severity and multiclonality were examined using Chi square statistics while between group comparisons of multiplicity of infection (MOI) were conducted using Student’s t-test [33]. A p value < 0.05 was considered to be statistically significant. Single nucleotide variations (SNVs) were used to estimate several genetic diversity parameters using DnaSPv5 [34, 35]. These included (i) number of segregating sites (S), (ii) average number of pairwise nucleotide differences within population (k), (iii) average number of observed nucleotide differences per site between any two sequences (π), (iv) Watterson’s θ (θw). Estimation of Tajima’s D, Fu & Li’s statistics and the minimum number of recombination event (Rm) in regions corresponding to MSP1 block 2 and MSP1-19 was carried out using DnaSPv5. Tajima’s D and Fu & Li’s statistics were used to assess the neutral theory of evolution. The significance of Tajima’s D statistics was indicated by its confidence limits while that of Fu and Li’s D* and F* statistics were represented by its critical values [36,37,38]. The intra- and inter-population genetic differentiation were measured by the fixation index (FST) using the Arlequin software package version 3.5 [39, 40].
Prediction of B-cell linear epitopes for block 2 and MSP1-19 allelic variants
Linear B-cell epitopes were predicted from MSP1 block 2 and MSP1-19 amino acid sequences using BepiPred [41]. BepiPred combines predictions of a hidden Markov model and a propensity scale method developed by Parker et al. [42, 43]. It analyses each amino acid independently to assign a score between − 3 and 3. The strength of prediction by BepiPred is defined in terms of sensitivity and specificity. On the basis of a benchmark calculation containing 85 B-cell epitopes, dependence of sensitivity and specificity of BepiPred at different selected thresholds was estimated (Additional file 1: Table S1) [44]. In this study, analysis of epitopes was conducted using two different threshold scores namely 0.35 and 1.30. The threshold score of 0.35 was chosen since at this score the sensitivity and specificity estimates were optimum. A stringent threshold of 1.3 was chosen to improve the strength of prediction by maximizing the specificity feature. A minimum of 7 consecutive residues each displaying a score above the specified threshold was considered to be an epitope.
Results
Indel polymorphism of MSP1 block 2 and multiplicity of infection
A total of 98 malaria patients (41 from Chhattisgarh and 57 from West Bengal) were employed for the genetic analysis of msp1 block 2 of P. falciparum. All three major allelic families namely K1, MAD20 and RO33 were detected in Chhattisgarh and West Bengal with frequencies of K1 (χ2 = 14.7, p < 0.001) and MAD20 (χ2 = 16.1, p < 0.001) differing significantly between two study sites (Fig. 2a). Since the patients of West Bengal suffered from mild malaria, the correlation between MSP1 allelic varieties with severity of disease was examined in Chhattisgarh data only. Frequency of RO33 (χ2 = 9.83, p < 0.01) was significantly higher in mild infection (Fig. 2b). Keeping in line with the low transmission intensity of the region, multiclonal infections were not detected in West Bengal samples. On the other hand, 39.02% of Chhattisgarh patients suffered from multi-genotypic infections, resulting in a mean MOI of 2.07 ± 1.59. MOI was higher in patients with mild malaria (2.33 ± 1.78) than those with severe malaria (1.57 ± 1.02), although the difference was not statistically significant in two-tailed Student’s t test (Fig. 2c). To detect if there was any association of MOI with age, Chhattisgarh patients were classified into two age groups namely (i) ≤ 18 years (n = 7) and (ii) > 18 years (n = 34). MOI was higher in the patients below 18 years of age (≤ 18 years: 3 ± 2.24 and > 18 years: 1.88 ± 1.39 (Fig. 2d). A comparative analysis of distribution of the msp1 allelic families between single and multiple infections showed a statistically significant prevalence of MAD20 (χ2 = 18.1, p < 0.001) in patients suffering from multi-genotype infections whereas RO33 (χ2 = 29.1, p < 0.001) predominated in single infection (Fig. 2e). Taken together, MAD20 displayed an extensive within and between population variation whereas K1 exhibited polymorphism only within Chhattisgarh patients.

Analysis of frequencies of msp1 block 2 alleles and multiplicity of infections in different groups. a Frequencies of msp1 alleles in Chhattisgarh and West Bengal. b Distribution of msp1 alleles in Chhattisgarh patients with mild and severe malaria. c Comparison of MOI in the mild and severe malaria patients of Chhattisgarh. d Differences of MOI in two different age groups of Chhattisgarh patients. e Frequencies of K1, MAD20, RO33 alleles associated with single and multiple infections in Chhattisgarh. Asterisk indicates p < 0.05 in Chi square test
To refine the analysis further, K1 and MAD20 families were classified into multiple sub types according to the copy number and arrangement of tri-peptide motifs present. The parasite population of Chhattisgarh and West Bengal differed remarkably with respect to the distribution and frequency of sub-alleles (Fig. 3). Overall bin sizes of indel subtypes under K1 and MAD20 families were 16 (15 in Chhattisgarh and 1 in West Bengal) and 24 (17 in Chhattisgarh and 11 in West Bengal), respectively. Of the 6 distinct tri-peptide motifs observed in K1, four (SGT, SGP, SAQ and SGA, coded as 1–4) were previously reported, while two rare motifs namely STQ (conversion of GCT to ACT codon resulting in A to T substitution) and SAR (conversion of CAA to CGA codon resulting in Q to R substitution) were derived from SAQ to detected in two mild malaria patients having multiclonal infections. The members of MAD20 allele family were represented by four previously reported tri-peptide motifs such as SGG, SVA, SVT, and SKG (coded as 5–8) [45]. Three rare motifs including SGD (GGT > GAT), PGG (TCA > CCA), PVA (TCA > CCA), coded by 5*, 5#, 6*, respectively were also detected in Chhattisgarh population (Fig. 3 and Additional file 2: Table S2). Phylogenetic trees constructed based on tri-peptide copy number variation of K1 and MAD20 families in Chhattisgarh revealed a characteristic pattern of evolutionary relationship (Fig. 3). For instance, K1H15 (repeat motif = 34343434343431221) seemed to be originated from K1H14 (repeat motif = 343434343431221) by repeat expansion of SAQ-SGA tri-peptide (Figs. 3, 4). On the other hand, MH17 (repeat motif = 5755665) seemed to be derived from MH16 (repeat motif = 5757565) through deletion of one SVT and insertion of one SVA motif (Fig. 4).

Phylogenetic relationship and prevalence of different msp1 sub-alleles. a Organization of tri-peptide motifs in the alleles belonging to K1 family in Chhattisgarh parasite population and their respective proportions. b Organization and prevalence of tri-peptide motifs in the alleles belonging to MAD20 family in Chhattisgarh sample. c Organization and prevalence of tri-peptide motifs in the alleles belonging to MAD20 in West Bengal samples. Bootstrap values were shown for each branch of the Maximum Parsimony tree. SGT, SGP, SAQ, SGA, STQ and SAR repeats were present in K1 and denoted as 1, 2, 3, 4, 3* and 3#, respectively and SGG, SVA, SVT, SKG, SGD, PGG, PVA motifs were present in MAD20 and denoted as 5, 6, 7, 8, 5*, 5# and 6*, respectively. Each letter in the tri-peptide motifs represents an amino acid

Possible mechanisms leading to allelic variability of msp1 block 2. Repeat expansion and insertion/deletion are presumably responsible for generating K1H15 and MH17 from K1H14 to MH16, respectively
Genetic diversity of msp1 based on SNVs
To identify the footprints of genetic and population level forces shaping the msp1 genetic diversity, multiple sequence alignment was performed using reads covering the regions flanking repeat expanse of block 2. Genomic region encompassing 64–120 amino acid residues in K1 and 81–131 residues in MAD20 were excluded from this analysis (Fig. 1) [29]. Since RO33 family lacked any indel variations, the complete sequence reads representing RO33 allele was available for identification of single nucleotide changes. All three allele families from Chhattisgarh parasite sequences harboured extensive sequence variation in the non-repetitive part of block 2. In contrast, West Bengal parasite population harbored variations only in the sequences belonging to MAD20 family. This was reflected in the nucleotide diversity estimates. For example, mean pairwise mismatches (k) for alleles belonging to K1, MAD20 and R033 in Chhattisgarh were 1.198, 6.414, 5.156, respectively; while those estimated for West Bengal sequences were 0, 6.104 and 0, respectively (Table 1). In Chhattisgarh samples nucleotide substitutions were distributed to both upstream and downstream regions flanking the tri-peptide motifs of K1 and MAD20 whereas in West Bengal population SNVs were clustered only in the region downstream to repeat motifs of MAD20. Finally, most variants found in K1 and MAD20 allelic background in Chhattisgarh samples were rare in frequency as evidenced by the negative Tajima’s D statistic (K1: − 2.536, MAD20: − 1.360) and statistically significant Fu & Li’s D* and F* estimates (K1: − 4.523 and − 4.574, MAD20: − 3.804 and − 3.492, respectively). In contrast, all the segregating sites found in MAD20 group in West Bengal were of intermediate frequency resulting in a positive Tajima’s D (1.305) as well as positive Fu & Li’s D* and F* indices (1.657 and 1.820), suggesting a signature of diversifying selection (Table 1).
Unlike msp1 block 2, sequences encoding MSP1-19 showed relatively conserved genetic configuration as reflected by the low nucleotide diversity estimates namely θ, π and k (Table 1). Four non-synonymous substitutions at amino acid positions 1691 (T > K), 1700 (S > N), 1701 (R > G) and 1716 (L > F) were shared between two study sites whereas three additional rare variants were recorded only in Chhattisgarh samples. Interestingly, one of these rare mutations at position 4998 bp (C > T) altered glutamine (CAA) at 1666 to a stop codon (TAA) in one Chhattisgarh isolate [46]. This was presumably tolerated by the presence of another rare mutation (5000A > T) in the same patient. The remaining rare variant corresponded to a synonymous change.
Comparison of sequence diversity among geographically diverse P. falciparum populations
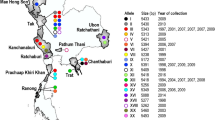
To understand the pattern of genetic differentiation with respect to geographical distance among Indian parasite sub-populations and those present in other malaria endemic countries, msp1 sequence data were retrieved from public databases [GenBank accession numbers: JF460898–JF460938, AB502443–AB502513, AB502514–AB502545, AB502546–AB502586, AB502587–AB502628, AB715434, AB502629–AB502704, AB502705–AB502745, AB715435–AB715519] [14, 47,48,49,50,51,52,53,54,55,56,57]. Except for the sub-populations from Assam and Orissa, all other pairwise comparisons in Indian isolates displayed statistically significant (p < 0.05) fixation indices (Table 2). Comparison of average allele frequencies of K1, MAD20 and RO33 in Indian sub-populations with that observed in other countries resulted in significant FST estimates for all pairwise tests (Additional file 3: Table S3). An analysis of frequency spectra of allele families indicated an overall prevalence of K1 and MAD20 in South East Asia, excepting Myanmar and Vanuatu. Abundance of RO33 was comparatively higher in African P. falciparum populations while it was absent in Peruvian Amazon of South America (Fig. 5). In summary, all inter population assessments indicated the existence of a strong local structure in the P. falciparum populations.

Worldwide distribution of P. falciparum msp1 block 2 alleles. Frequencies of K1, MAD20 and RO33 in different geographical regions. Proportion of each allele in a certain parasite population was shown using pi diagram
Assessment of antigenic organization of observed MSP1 block 2 and MSP1-19 alleles
The next section examined how this extreme genetic variability of MSP1 block 2 may influence its antigenic potential. Numbers of variants subjected to linear epitope mapping were 16, 24 and 3 for K1, MAD20 and RO33, respectively (Additional file 4: Table S4). Epitope evaluation was initially conducted using the threshold score of 0.35. This revealed that every residue of K1 and RO33 and those located in an internal stretch of MAD20 could potentially be incorporated as an epitope. Prediction of epitopes was then repeated using the stringent threshold score of 1.30. Two independent stretches of amino acids with variable lengths and sequences emerged as potential epitopes for each of K1 (13–66 residues) and MAD20 (8–57 residues) variants (Additional files 5 and 6: Figures S1, S2). Numbers of unique epitope predicted for K1, MAD20 and R033 were 18, 31 and 1, respectively (Table 3). Of the 18 different K1 epitopes, SNTSSGASPPADA was present in 84% (31 out of 37) parasite field isolates. Among MAD20 epitopes, GGSGNSRRTNPSDNSSDSDAK was present in 98.8% (89 out of 90) of parasites either as an independent motif (epitope #3) or as the part of a larger epitope (epitope #4, 5, 6, 7, 8, 11, 13, 16, 31) (Table 3). A single epitope, QSAKNPPGATVPSGTAS, with slightly variable scores represented all 3 RO33 alleles observed among 11 isolates. In summary, each block 2 family could be represented by a unique antigenic determinant and 94.9% (131 of 138) of parasites was represented by 3 predominant epitopes. Average epitope score of block 2 peptides was the highest for K1 (2.076 ± 0.145) followed by R033 (1.872 ± 0.007) and MAD20 (1.749 ± 0.129).
Similar analysis was conducted on 4 haplotypes of MSP1-19 and it revealed that average epitope score for this relatively conserved segment of MSP1 was significantly (< 0.05) lower (1.608 ± 0.091) than any of the probable block 2 antigens as per Student’s t test (Fig. 6).

Comparison of average epitope scores of MSP1 block 2 and MSP1-19 peptides in India. Epitopes were predicted based on a threshold score of 1.3. Asterisk indicates p < 0.05 in two-tailed Student’s t test
Discussion
The significant decline of global malaria burden achieved in the last 15 years is mainly attributed to the use of insecticide-treated nets and implementation of artemisinin-based combination therapy (ACT) [58]. Two factors that still hinder the progress of malaria control include the emergence of drug-resistant parasite strains and development of vectors resistant to insecticide [59]. Development of a malaria vaccine would be an additional arsenal to the existing tools for malaria control. One of the challenges in developing malaria vaccines is the extensive genetic diversity of parasite antigens that are vaccine targets. Individuals living in areas of high transmission intensity are often simultaneously infected by multiple parasite genotypes [60]. It is, therefore, important to characterize the level of parasite genetic variation in diverse geographical locations to identify the prevailing parasite strains. To this end, this article provides a comprehensive description of P. falciparum diversity for two most important immunogenic segments of MSP1 in disparate malaria affected regions of India. In addition it makes an attempt to correlate the variability of the protein sequences with its antigenic properties.
MSP1 is one of the prime candidates for the development of malaria blood stage vaccine and it serves as a suitable marker for the identification of genetically distinct P. falciparum populations [50]. Analysis of msp1 block 2 reveals predominance of MAD20 in both geographical regions, studied. Similar prevalence of MAD20 was observed in studies conducted in Baikunthpur and Madhya Pradesh, two neighboring regions and those from Philippines, Papua New Guinea and Myanmar [28, 48, 49, 54]. In contrast, a higher frequency of K1 has been reported from Orissa, Madhya Pradesh, Assam in India and Mauritania and Uganda [47, 48, 56, 61]. It is important to note in this context that several of these studies including those conducted on Indian sub-populations used PCR followed by hybridization with allele-specific probes to capture the allelic diversity of block 2. Since this technique relies on size discrimination of products ranging from 400 to 600 bp, it is possible that some unique msp1 alleles remain indistinguishable because of their proximity of sizes [62].
The present study recovers a total number of 33 different indel parasite alleles based on the sequence diversity of block 2 in Chhattisgarh whereas the parasite sub-population from West Bengal harbours 13 indel sub-alleles. Only 10.42% of allele pools are shared between the hyper- and hypoendemic states of Chhattisgarh and West Bengal, respectively. This high level of genotypic diversification and low level of gene migration among Indian parasite sub-populations have been supported by statistically significant FST estimates. As expected, Ambikapur parasite population shares 34% and 28% of indel variants with those from neighbouring regions of Baikunthpur and Madhya Pradesh, respectively [28, 48].
Point mutation and repeat instability due to recombination are the major factors responsible for variability of K1 and MAD20. For instance, K1H7 (repeat motif = 3111111221) seems to be originated from K1H6 (repeat motif = 31111221) by repeat expansion of STG tri-peptide in Chhattisgarh. Such extensive variability has presumably been evolved as an immune evasion mechanism by the parasite in which protective immune response mounted by the host has favoured diversifying selection of block 2 [22]. A finer evaluation of msp1 repeat organization in Chhattisgarh data suggests that K1 alleles may be broadly classified in two sub-families (starting with code: 3/3#11… or 3/3*434343…) while MAD20 family exhibits three sub-groups (starting with code: 875…, or 8565…, or 575…). The complexity of Chhattisgarh parasite population is exemplified by the observation that 39.02% patients suffered from multi-genotypic infections (ranging from 2 to 7). This statistics is comparable with that of Baikunthpur where 37% of the samples carried polyclonal infections with a MOI of 1.67 [28]. This data and that of others indicate a possible positive association between MOI and endemicity of P. falciparum [63,64,65,66]. Nevertheless, this correlation may not be an absolute one as MOI of P. falciparum ranges from 1.00 to 2.70 in few hypoendemic regions of Southeast Asia [67, 68]. A very high MOI of 3 ± 2.24 detected in the age group ≤ 18, is suggestive of a weaker immunity of younger people. A negative correlation between MOI and disease severity in Chhattisgarh (mild malaria: 2.33 ± 1.78; severe malaria: 1.57 ± 1.02) is another notable observation.
To identify footprints of genetic and population level forces, sequences adjacent to the repeat expanse of block 2 and genomic region covering MSP1-19 are scanned for SNVs. MSP1-19 displays limited sequence heterogeneity. Of the ten MSP1-19 allelic forms reported globally, Indian field isolates harbor 4 non-synonymous substitutions suggesting the probable influence of purifying selection shaping the diversity of this functionally important portion of msp1 gene [46]. Thus, the present study demonstrates that different kinds of selection forces shape the complex genetic landscape of MSP1.
Of the different MSP1 segments, most vaccine studies focus on the conserved C-terminal region of MSP1-19, although the block 2 region also elicits functionally protective immune responses and is associated with reduced risk of malaria [7, 22, 23, 69,70,71]. The immune responses to MSP1-19 and block 2 mediated predominantly by IgG1 and IgG3 subclasses, respectively [72, 73]. In vitro assays with purified IgG3 from malaria immune individuals have established the functional superiority of IgG3 as an inhibitor of parasite growth [7, 73, 74]. However, the extensive polymorphic nature of block 2 is a potential challenge.
To this end, the present study elaborates the antigenic properties of MSP1 block 2 and MSP1-19 by evaluating their probable antigen conformations and potencies using BepiPred. Forty-three MSP1 block 2 variants observed in 138 P. falciparum field isolates generate 50 unique linear B-cell epitopes. However, 94.9% (131 of 138) of parasites may be represented by only 3 conserved block 2 epitopes. In addition, the average epitope score for each of these three representative block 2 antigens are noticeably higher compared to that of MSP1-19. A polyvalent recombinant protein incorporating these three block 2 epitopes together with a sequence from MSP1 block 1, has been shown to induce high titre antibodies against a wide range of allelic types of P. falciparum field isolates [75]. On the contrary, a recent comparative analysis suggests that the global MSP1-42 population is not as tightly conserved as it has been thought previously [76]. This reinforces the importance of MSP1 block 2 modules as effective blood-stage malaria vaccine.
One drawback of the current study is that due to lack of required crystallographic structure of block 2, the analysis remains limited to evaluation of linear B-cell epitopes instead of conformational epitopes which are believed to be better suited for most biomedical applications. However, this may also be borne in mind that predicted conformation-based antigenic determinants may not always be immunologically functional and biochemically verifiable. Prediction of B-cell linear epitopes has often been served as an alternative procedure for proteins that are not structurally well characterized [77].
Conclusion
Taken together, the present study identifies a high level of genetic differentiation between the parasite populations of Chhattisgarh and West Bengal which arises presumably due to lack of gene flow and difference in malaria transmission intensities. It also indicates that an opposing pattern of natural selection may operate on msp1 block 2 and MSP1-19. The most remarkable finding of the current study, nevertheless, is the presence of a limited number of conserved epitopes representing the MSP1 block 2 despite its extensive genetic diversity. This kindles the possibility of vaccine development based on this immunologically active merozoite segment.
Abbreviations
- MOI:
-
multiplicity of infection
- MSP1:
-
merozoite surface protein 1
- PCR:
-
polymerase chain reaction
- SNV:
-
single nucleotide variation
- WHO:
-
World Health Organization
References
WHO. World malaria report 2014. Geneva: World Health Organization; 2014.
Birkett AJ. Building an effective malaria vaccine pipeline to address global needs. Vaccine. 2015;33:7538–43.
Malaria Basics: Global Impact and Actions. 2002.
Nielsen CM, Vekemans J, Lievens M, Kester KE, Regules JA, Ockenhouse CF. RTS, S malaria vaccine efficacy and immunogenicity during Plasmodium falciparum challenge is associated with HLA genotype. Vaccine. 2018;36:1637–42.
Berzins K. Merozoite antigens involved in invasion. Chem Immunol. 2002;80:125–43.
Gaur D, Mayer DC, Miller LH. Parasite ligand-host receptor interactions during invasion of erythrocytes by Plasmodium merozoites. Int J Parasitol. 2004;34:1413–29.
Cowan GJ, Creasey AM, Dhanasarnsombut K, Thomas AW, Remarque EJ, Cavanagh DR. A malaria vaccine based on the polymorphic block 2 region of MSP-1 that elicits a broad serotype-spanning immune response. PLoS ONE. 2011;6:e26616.
Lin CS, Uboldi AD, Epp C, Bujard H, Tsuboi T, Czabotar PE, et al. Multiple Plasmodium falciparum merozoite surface protein 1 complexes mediate merozoite binding to human erythrocytes. J Biol Chem. 2016;291:7703–15.
Miller LH, Roberts T, Shahabuddin M, McCutchan TF. Analysis of sequence diversity in the Plasmodium falciparum merozoite surface protein-1 (MSP-1). Mol Biochem Parasitol. 1993;59:1–14.
Tanabe K, Mackay M, Goman M, Scaife JG. Allelic dimorphism in a surface antigen gene of the malaria parasite Plasmodium falciparum. J Mol Biol. 1987;195:273–87.
Aspeling-Jones H, Conway DJ. An expanded global inventory of allelic variation in the most extremely polymorphic region of Plasmodium falciparum merozoite surface protein 1 provided by short read sequence data. Malar J. 2018;17:345.
Branch OH, Takala S, Kariuki S, Nahlen BL, Kolczak M, Hawley W, Lal AA. Plasmodium falciparum genotypes, low complexity of infection, and resistance to subsequent malaria in participants in the Asembo Bay cohort project. Infect Immun. 2001;69:7783–92.
Woehlbier U, Epp C, Hackett F, Blackman MJ, Bujard H. Antibodies against multiple merozoite surface antigens of the human malaria parasite Plasmodium falciparum inhibit parasite maturation and red blood cell invasion. Malar J. 2010;9:77.
Chenet SM, Branch OH, Escalante AA, Lucas CM, Bacon DJ. Genetic diversity of vaccine candidate antigens in Plasmodium falciparum isolates from the Amazon basin of Peru. Malar J. 2007;7:93.
Wang Q, Zhao Z, Zhang X, Li X, Zhu M, Li P, et al. Naturally acquired antibody responses to Plasmodium vivax and Plasmodium falciparum merozoite surface protein 1 (MSP1) C-Terminal 19 kDa domains in an area of unstable malaria transmission in Southeast Asia. PLoS ONE. 2016;11:e0151900.
Blackman MJ, Heidrich HG, Donachie S, McBride JS, Holder AA. A single fragment of a malaria merozoite surface protein remains on the parasite during red cell invasion and is the target of invasion inhibiting antibodies. J Exp Med. 1990;172:379–82.
Egan AF, Morris J, Barnish G, Allen S, Greenwood BM, Kaslow DC, et al. Clinical immunity to Plasmodium falciparum malaria is associated with serum antibodies to the 19-kDa C-terminal fragment of the merozoite surface antigen, PfMSP-1. J Infect Dis. 1996;173:765–9.
Egan AF, Burghaus P, Druilhe P, Holder AA, Riley EM. Human antibodies to the 19 kDa C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 inhibit parasite growth in vitro. Parasite Immunol. 1999;21:133–9.
John CC, O’Donnell RA, Sumba PO, Moormann AM, de Koning-Ward TF, King CL, et al. Evidence that invasion-inhibitory antibodies specific for the 19-kDa fragment of merozoite surface protein-1 (MSP-1 19) can play a protective role against blood-stage Plasmodium falciparum infection in individuals in a malaria endemic area of Africa. J Immunol. 2004;173:666–72.
Okech BA, Corran PH, Todd J, Joynson-Hicks A, Uthaipibull C, Egwang TG, et al. Fine specificity of serum antibodies to Plasmodium falciparum merozoite surface protein, PfMSP-1(19), predicts protection from malaria infection and high-density parasitemia. Infect Immun. 2004;72:1557–67.
Stanisic DI, Richards JS, McCallum FJ, Michon P, King CL, Schoepflin S, et al. Immunoglobulin G subclasss specific responses against Plasmodium falciparum merozoite antigens are associated with control of parasitemia and protection from symptomatic illness. Infect Immun. 2009;77:1165–74.
Polley SD, Tetteh KK, Cavanagh DR, Pearce RJ, Lloyd JM, Bojang KA, et al. Repeat sequences in block 2 of Plasmodium falciparum merozoite surface protein 1 are targets of antibodies associated with protection from malaria. Infect Immun. 2003;71:1833–42.
Cavanagh DR, Dodoo D, Hviid L, Kurtzhals JA, Theander TG, Akanmori BD, et al. Antibodies to the N-terminal block 2 of Plasmodium falciparum merozoite surface protein 1 are associated with protection against clinical malaria. Infect Immun. 2004;72:6492–502.
Jain V, Basak S, Bhandari S, Bharti PK, Thomas T, Singh MP, Singh N. Burden of complicated malaria in a densely forested Bastar region of Chhattisgarh State (Central India). PLoS ONE. 2014;9:e115266.
Basu M, Das T, Ghosh A, Majumder S, Maji AK, Kanjilal SD, et al. Gene-gene interaction and functional impact of polymorphisms on innate immune genes in controlling Plasmodium falciparum blood infection level. PLoS ONE. 2012;7:e46441.
Joshi H, Valecha N, Verma A, Kaul A, Mallick PK, Shalini S, et al. Genetic structure of Plasmodium falciparum field isolates in eastern and north-eastern India. Malar J. 2007;6:60.
Saha P, Ganguly S, Maji AK. Genetic diversity and multiplicity of infection of Plasmodium falciparum isolates from Kolkata, West Bengal. India. Infect Genet Evol. 2016;43:239–44.
Patel P, Bharti PK, Bansal D, Raman RK, Mohapatra PK, Sehgal R, et al. Genetic diversity and antibody responses against Plasmodium falciparum vaccine candidate genes from Chhattisgarh, Central India: implication for vaccine development. PLoS ONE. 2017;12:e0182674.
Jiang G, Daubenberger C, Huber W, Matile H, Tanner M, Pluschke G. Sequence diversity of the merozoite surface protein 1 of Plasmodium falciparum in clinical isolates from the Kilombero District, Tanzania. Acta Trop. 2000;74:51–61.
BLAST. https://blast.ncbi.nlm.nih.gov/. Accessed 29 May 2017.
GenBank. https://www.ncbi.nlm.nih.gov/genbank/. Accessed 20 May 2017.
Snounou G, Zhu X, Siripoon N, Jarra W, Thaithong S, Brown KN, et al. Biased distribution of MSP 1 and MSP 2 allelic variants in Plasmodium falciparum populations in Thailand. Trans R Soc Trop Med Hyg. 1999;93:369–374.
GraphPad QuickCalcs. https://www.graphpad.com/quickcalcs/ttest1.cfm. Accessed 29 June 2017.
Rozas J, Rozas R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–5.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–7.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Tajima F. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics. 1996;143:1457–65.
Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2007;1:47–50.
Arlequin ver 3.5.2.2. http://cmpg.unibe.ch/software/arlequin35/Arl35Downloads.html. Accessed 16 June 2017.
B Cell Help - IEDB Analysis Resource. http://tools.iedb.org/bcell/help/. Accessed 30 June 2017.
Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25:5425–32.
Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2.
BepiPred 1.0 Output format. http://www.cbs.dtu.dk/services/BepiPred-1.0/output.php. Accessed 23 Aug 2017.
Noranate N, Prugnolle F, Jouin H, Tall A, Marrama L, Sokhna C, et al. Population diversity and antibody selective pressure to Plasmodium falciparum MSP1 block 2 locus in an African malaria-endemic setting. BMC Microbiol. 2009;9:219.
Mamillapalli A, Sunil S, Diwan SS, Sharma SK, Tyagi PK, Adak T, et al. Polymorphism and epitope sharing between the alleles of merozoite surface protein-1 of Plasmodium falciparum among Indian isolates. Malar J. 2007;6:95.
Raj DK, Das BR, Dash AP, Supakar PC. Genetic diversity in the msp1 gene of Plasmodium falciparum in different malaria endemic localities. Am J Trop Med Hyg. 2004;71:285–9.
Bharti PK, Shukla MM, Sharma YD, Singh N. Genetic diversity in the block 2 region of the merozoite surface protein-1 of Plasmodium falciparum in central India. Malar J. 2012;11:78.
Tanabe K, Mita T, Palacpa NM, Arisue N, Tougan T, Kawai S, et al. Within-population genetic diversity of Plasmodium falciparum vaccine candidate antigens reveals geographic distance from a Central sub-Saharan African origin. Vaccine. 2013;31:1334–9.
Oyebola MK, Idowu ET, Olukosi YA, Iwalokun BA, Agomo CO, Ajibaye OO, et al. Genetic diversity and complexity of Plasmodium falciparum infections in Lagos, Nigeria. Asian Pac J Trop Biomed. 2014;4(suppl 1):S87–91.
Bouyou-Akotet MK, M’Bondoukwe NP, Mawili-Mboumba DP. Genetic polymorphism of merozoite surface protein-1 in Plasmodium falciparum isolates from patients with mild to severe malaria in Libreville, Gabon. Parasite. 2015;22:12.
Ahmedou Salem MS, Ndiaye M, OuldAbdallahi M, Lekweiry KM, Bogreau H, Konate L, et al. Polymorphism of the merozoite surface protein-1 block 2 region in Plasmodium falciparum isolates from Mauritania. Malar J. 2014;13:26.
Mohammed H, Mindaye T, Belayneh M, Kassa M, Assefa A, Tadesse M, et al. Genetic diversity of Plasmodium falciparum isolates based on MSP-1 and MSP-2 genes from Kolla-Shele area, Arbaminch Zuria District, southwest Ethiopia. Malar J. 2015;14:73.
Soe TN, Wu Y, Tun MW, Xu X, Hu Y, Ruan Y, et al. Genetic diversity of Plasmodium falciparum populations in southeast and western Myanmar. Parasit Vectors. 2017;10:322.
Khaminsou N, Kritpetcharat O, Daduang J, Charerntanyarak L, Kritpetcharat P. Genetic analysis of the merozoite surface protein-1 block 2 allelic types in Plasmodium falciparum clinical isolates from Lao PDR. Malar J. 2011;10:371.
Baruah S, Lourembam SD, Sawian CE, Baruah I, Goswami D. Temporal and spatial variation in MSP1 clonal composition of Plasmodium falciparum in districts of Assam, Northeast India. Infect Genet Evol. 2009;9:853–9.
Hussain MM, Sohail M, Kumar R, Branch OH, Adak T, Raziuddin M. Genetic diversity in merozoite surface protein-1 and 2 among Plasmodium falciparum isolates from malarious districts of tribal dominant state of Jharkhand, India. Ann Trop Med Parasitol. 2011;105:579–92.
Cibulskis RE, Alonso P, Aponte J, Aregawi M, Barrette A, Bergeron L, et al. Malaria: global progress 2000–2015 and future challenges. Infect Dis Poverty. 2016;5:61.
Phillips RS. Current Status of Malaria and Potential for Control. Clin Microbiol Rev. 2001;14:208–26.
Ulrich Y, Schmid-Hempel P. Host modulation of parasite competition in multiple infections. Proc Biol Sci. 2012;279:2982–9.
Kiwuwa MS, Ribacke U, Moll K, Byarugaba J, Lundblom K, Färnert A, et al. Genetic diversity of Plasmodium falciparum infections in mild and severe malaria of children from Kampala, Uganda. Parasitol Res. 2013;112:1691–700.
Takala SL, Escalante AA, Branch OH, Kariuki S, Biswas S, Chaiyaroj SC, et al. Genetic diversity in the Block 2 region of the merozoite surface protein 1 (MSP-1) of Plasmodium falciparum: additional complexity and selection and convergence in fragment size polymorphism. Infect Genet Evol. 2006;6:417–24.
Atroosh WM, Al-Mekhlafi HM, Mahdy MA, Saif-Ali R, Al-Mekhlafi AM, Surin J. Genetic diversity of Plasmodium falciparum isolates from Pahang, Malaysia based on MSP-1 and MSP-2 genes. Parasit Vectors. 2011;4:233.
Gupta P, Singh R, Khan H, Raza A, Yadavendu V, Bhatt RM, Singh V. Genetic profiling of the Plasmodium falciparum population using antigenic molecular markers. Sci World J. 2014;2014:140867.
Mwingira F, Nkwengulila G, Schoepflin S, Sumari D, Beck HP, Snounou G, et al. Plasmodium falciparum msp1, msp2 and glurp allele frequency and diversity in sub-Saharan Africa. Malar J. 2011;10:79.
Schoepflin S, Valsangiacomo F, Lin E, Kiniboro B, Mueller I, Felger I. Comparison of Plasmodium falciparum allelic frequency distribution in different endemic settings by high resolution genotyping. Malar J. 2009;8:250.
Alam MS, Elahi R, Mohon AN, Al-Amin HM, Kibria MG, Khan WA. Plasmodium falciparum genetic diversity in Bangladesh does not suggest a hypoendemic population structure. Am J Trop Med Hyg. 2016;94:1245–50.
Ghanchi NK, Martensson A, Ursing J, Jafri S, Bereczky S, Hussain R, et al. Genetic diversity among Plasmodium falciparum field isolates in Pakistan measured with PCR genotyping of the merozoite surface protein 1 and 2. Malar J. 2010;9:1.
Singh S, Kennedy MC, Long CA, Saul AJ, Miller LH, Stowers AW. Biochemical and immunological characterization of bacterially expressed and refolded Plasmodium falciparum 42-kilodalton C-terminal merozoite surface protein 1. Infect Immun. 2003;71:6766–74.
Stoute JA, Gombe J, Withers MR, Siangla J, McKinney D, Onyango M, et al. Phase 1 randomized double-blind safety and immunogenicity trial of Plasmodium falciparum malaria merozoite surface protein FMP1 vaccine, adjuvanted with AS02A, in adults in western Kenya. Vaccine. 2007;25:176–84.
Cavanagh DR, Kocken CH, White JH, Cowan GJ, Samuel K, Dubbeld MA, et al. Antibody responses to a novel Plasmodium falciparum merozoite surface protein vaccine correlate with protection against experimental malaria infection in Aotus monkeys. PLoS ONE. 2014;9:e83704.
Cavanagh DR, Dobaño C, Elhassan IM, Marsh K, Elhassan A, Hviid L, et al. Differential patterns of human immunoglobulin G subclass responses to distinct regions of a single protein, the merozoite surface protein 1 of Plasmodium falciparum. Infect Immun. 2001;69:1207–11.
Tebo AE, Kremsner PG, Luty AJ. Plasmodium falciparum: a major role for IgG3 in antibody-dependent monocyte-mediated cellular inhibition of parasite growth in vitro. Exp Parasitol. 2001;98:20–8.
Galamo CD, Jafarshad A, Blanc C, Druilhe P. Anti-MSP1 block 2 antibodies are effective at parasite killing in an allele-specific manner by monocyte-mediated antibody-dependent cellular inhibition. J Infect Dis. 2009;199:1151–4.
Tetteh KK, Conway DJ. A polyvalent hybrid protein elicits antibodies against the diverse allelic types of block 2 in Plasmodium falciparum merozoite surface protein 1. Vaccine. 2011;29:7811–7.
Thái TL, Jun H, Lee J, Kang JM, Lê HG, Lin K, et al. Genetic diversity of merozoite surface protein-1 C-terminal 42 kDa of Plasmodium falciparum (PfMSP-142) may be greater than previously known in global isolates. Parasit Vectors. 2018;11:455.
Wang HW, Lin YC, Pai TW, Chang HT. Prediction of B-cell linear epitopes with a combination of support vector machine classification and amino acid propensity identification. J Biomed Biotechnol. 2011;2011:432830.
Authors’ contributions
SS conceived and designed the study. PG, MM and SDK helped in collecting blood samples from malaria patients. SG performed the experiments. SG and SS carried out the statistical and bioinformatic analysis. SG and SS wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We are grateful Dr. Soumalee Basu, Department of Microbiology, University of Calcutta, for her valuable suggestion and Ms. Pramita Chowdhury, Department of Biochemistry, University of Calcutta, India for her assistance in artwork. Authors also thank Dr. Srikanta Sen for his kind help and guidance in bioinformatic analysis. We are thankful to CAS (UGC), for providing sequencing facility at the Department of Biochemistry, University of Calcutta.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All sequence data are publicly available at NCBI (www.ncbi.nlm.nih.gov).
Consent for publication
Not applicable.
Ethics approval and consent to participate
Ethical clearance to conduct the study was obtained from the Calcutta National Medical College & Hospital and Institutional Ethics committee for human research, Pt. Ravishankar Shukla University, Raipur. Written informed consent, prior to sample collection, was collected from each study participant or guardian, in case where the participant was a child.
Funding
This work has been supported by the funding from Department of Biotechnology, Government of West Bengal (Project No: 215-BT(Estt)/RD-08/2015 dated 16. 03.2015) and fellowship from Council of Scientific & Industrial Research (CSIR), India (Sanction No.: 09/028(0957)/2015-EMR-1).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Selected thresholds and corresponding sensitivity & specificity estimates of BepiPred.
Additional file 2: Table S2.
Tripeptide motifs and their corresponding nucleotide sequences for K1 and MAD20.
Additional file 3: Table S3.
Pairwise FST estimated using msp1 block 2 allele frequencies in different P. falciparum populations worldwide.
Additional file 4: Table S4.
MSP1 block 2 and -19 alleles and NCBI accession numbers.
Additional file 5: Figure S1.
Distribution of epitope scores in different K1 block 2 sub-alleles. N, N-terminal; C, C-terminal.
Additional file 6: Figure S2.
Distribution of epitope scores in different MAD20 block 2 sub-alleles. N, N-terminal; C, C-terminal.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ghoshal, S., Gajendra, P., Datta Kanjilal, S. et al. Diversity analysis of MSP1 identifies conserved epitope organization in block 2 amidst high sequence variability in Indian Plasmodium falciparum isolates. Malar J 17, 447 (2018). https://doi.org/10.1186/s12936-018-2592-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2592-y