
Abstract
The merozoite surface protein-1 (MSP1) is a prime candidate for an asexual blood stage vaccine against malaria. However, polymorphism in this antigen could compromise the vaccine’s efficacy. Although the extent of sequence variation in MSP1 has been analyzed from various Plasmodium species, little is known about structural organization and diversity of this locus in Plasmodium malariae (PmMSP1). Herein, we have shown that PmMSP1 contained five conserved and four variable blocks based on analysis of the complete coding sequences. Variable blocks were characterized by short insertion and deletion variants (block II), polymorphic nonrepeat sequences (block IV), complex repeat structure with size variation (block VI) and degenerate octapeptide repeats (block VIII). Like other malarial MSP1s, evidences of intragenic recombination have been found in PmMSP1. The rate of nonsynonymous nucleotide substitutions significantly exceeded that of synonymous nucleotide substitutions in block IV, suggesting positive selection in this region. Codon-based analysis of deviation from neutrality has identified a codon under purifying selection located in close proximity to the homologous region of the 38 kDa/42 kDa cleavage site of P. falciparum MSP1. A number of predicted linear B-cell epitopes were identified across both conserved and variable blocks of the protein. However, polymorphism in repeat-containing blocks resulted in alteration of the predicted linear B-cell epitope scores across variants. Although a number of predicted HLA-class II-binding peptides were identified in PmMSP1, all variants of block IV seemed not to be recognized by common HLA-class II alleles among Thai population, suggesting that diversity in this positive selection region could probably affect host immune recognition. The data on structural diversity in PmMSP1 could be useful for further studies such as vaccine development and strain characterization of this neglected malaria parasite.
Similar content being viewed by others


Introduction
Despite annual declines in global malaria cases caused by the two major human malaria parasites Plasmodium falciparum and P. vivax during the past 2 decades due to integrative control measures, an increase in the number of infections by the low prevalent species including P. malariae and P. ovale spp. has been observed in some African endemic areas, such as Tanzania, Gabon, Democratic Republic of Congo and Uganda1,2,3,4. Although P. malariae infection usually does not result in acute severe symptoms, repeated and long-term exposures may be associated with chronic glomerulonephritis in children and adolescents in some endemic areas, especially Sub-Saharan Africa and Papua New Guinea5,6,7,8. While more compelling evidences are required to document chloroquine-resistance in P. malariae, the blood stage infection of this Plasmodium species may persist for an unusually long period and can recrudesce after many years of dormancy9,10,11. Like other human malaria parasites, P. malariae has been incriminated in transfusion-transmitted malaria in which the prevalence seems to vary across endemic areas12,13. Meanwhile, the low parasite density of P. malariae among infected individuals has hampered efficient detection by conventional microscopy, especially when it co-infects with other malaria species14,15,16. On the basis of microscopy diagnosis, P. malariae infection accounted for approximately 0.1% of all malaria cases in Thailand17 whereas PCR could diagnose about five times higher than microscopic examination15,18,19,20. To achieve malaria control and elimination, it may require effective interventions against the low prevalent Plasmodium species including P. malariae.
One of the leading vaccine candidates against asexual blood stages of malaria parasites is merozoite surface protein-1 (MSP1) which is believed to play a crucial role in invasion of host erythrocytes by the merozoites and during their egression from infected cells after asexual reproductive maturation21,22. The MSP1 of P. falciparum (PfMSP1) is synthesized as a precursor protein during schizogony and subsequently processed into 4 polypeptides of 83, 30, 38 and 42 kDa. Prior to erythrocyte entry of the merozoites, secondary processing of the C-terminal 42-kDa fragment ensues, yielding 33- and 19-kDa protein fragments23. On the basis of amino acid sequence identity, PfMSP1 have been divided in to 17 blocks, containing five conserved, five semi-conserved and seven variable blocks24. The 19-kDa fragment containing two epidermal growth factor (EGF)-like domains has been considered to be a potential vaccine candidate because it is a target for invasion inhibitory antibodies while naturally acquired antibodies against this fragment have been associated with protection against symptomatic malaria among individuals living in malaria endemic areas25,26. Likewise, the tripeptide repeats in PfMSP1 could elicit protective antibodies among children in Sub-Saharan Africa27. Furthermore, erythrocyte-binding domains have been identified in the 83-, 38- and 33-kDa fragments of PfMSP128,29,30,31. Meanwhile, the MSP1 gene of P. vivax (PvMSP1) displays mosaic organization of variable blocks whereas those of P. ovale spp. (PoMSP1) and P. knowlesi (PkMSP1) exhibit structural variation that are different from that of PfMSP132,33,34.
To date, mainly partial sequences of the MSP1 gene of P. malariae (PmMSP1) have been determined using isolates from French Guiana, Cameroon, Brazil and Thai-Myanmar border which reveals conserved and variable blocks35,36,37,38,39. However, the organization of these blocks based on the complete coding sequences remains to be elucidated. Herein, we analyzed the complete coding sequence of PmMSP1 among clinical isolates from diverse endemic areas of Thailand. Results have shown that PmMSP1 contained four variable blocks flanked by five conserved blocks. Like other human malarial MSP1s, intragenic recombination and natural selection have influenced diversity at this locus24,32,33,34. Furthermore, analysis of predicted linear B-cell and helper T-cell epitopes has suggested that polymorphism in this protein could affect host immune recognition.
Results
Amplification and sequencing of PmMSP1
The complete coding region of PmMSP1 was amplified from 35 Thai isolates (PM1–PM35). The origins and years of sample collections are shown in Fig. 1. Of these, 15 isolates contained single infections of P. malariae and the remaining samples were co-infected with P. falciparum (n = 2), P. vivax (n = 17) and both P. falciparum and P. vivax (n = 1). However, the PCR primers used in this study were specific for amplification of PmMSP1 because direct sequencing of the PCR-amplified products yielded clear electropherogram without superimposed signals of the sequences of this locus. Therefore, no cross amplification of PfMSP1 and PvMSP1 was observed in isolates containing P. falciparum or P. vivax. The complete coding sequences of PmMSP1 in this study varied from 5088 to 5493 bp. In total, 20 alleles were identified in which alleles III, VI, X, XIII and XV contained more than one isolate (Fig. 1). Interestingly, the same alleles could be found from different sampling periods and from diverse endemic areas of the country. For example, allele XIII consisting of 5418 bp, occurred in five isolates from Mae Hong Son, Tak, Trat, Ranong and Yala Provinces collected during 1994, 2004, 2007 and 2008 (Fig. 1).

Map of Thailand showing the provinces and period of sample collection. Distribution of the PmMSP1 alleles is shown as corresponding color circles. The map is modified from GADM maps and data (https://gadm.org/index.html) under the GADM license version 6.0.
Structural organization of PmMSP1
To determine the structural organization of PmMSP1, nucleotide diversity was determined across the aligned complete coding sequences of 35 Thai isolates and the sequence from a Cameroonian patient (GenBank accession no. FJ824669) whose nucleotide and amino acid positions of the gene/protein were used as reference. Results revealed that the extent of nucleotide diversity was variable across PmMSP1 with two regions containing nucleotide diversity > 0.04 (Fig. 2A), a comparable level for variable blocks of PvMSP132. One was from codons 210 to 241, designated block IV, and the other was in block VI spanning amino acids 682 and 832. Short insertions and deletions (indels) were found between codons 57 and 69 at the N-terminal part of PmMSP1, designated block II, (Fig. 3). Meanwhile, Tandem Repeats Finder algorithm has identified two repeat-containing regions in PmMSP1, one corresponding to block VI and the other from codons 989 to 1032 (block VIII). Therefore, the remaining nonrepeat regions encompassing approximately 86% of the entire coding region in PmMSP1 with nucleotide diversity < 0.02 were assigned to conserved blocks, consisting of blocks I, III, V, VII and IX (Fig. 2B).

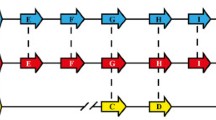
(A) Nucleotide diversity across the complete coding region of PmMSP1 using window length of 100 nucleotides and step size of 25 nucleotides. (B) Corresponding scheme of PmMSP1 showing organization of the protein regions. The potential cleavage sites for the 42-kDa and 19-kDa fragments are shown in open and filled downward arrow heads, respectively.

Variation in block VI of PmMSP1 spanning amino acid residues 683 and 827 (positions after GenBank accession no. FJ824669). Repeats are shown in brackets, bolds, italics or underlined residues. Numbers of repeat units are indicated by subscripts. Mutations in nonrepeat regions are highlighted. Representative isolates are shown in parentheses after the alleles. Alleles in bolds are found in P. brasiliamum. Allele A1.10 belongs to both P. malariae (KR072216) and P. brasilianum (JX045641). Asterisks denote alleles with two or more isolates: KR072218 and KR072215 in allele A1.11; PM17-PM19 in allele B1.1; PM3-PM7 in allele B1.2; KX672047 and KX672048 in allele B1.4; PM22-PM26 in allele B1.5; PM12 in allele B2.2; and PM29 and PM30 in allele B2.4.
Diversity of indels in PmMSP1
Previous reports have shown that P. malariae and P. brasilianum possessed similar or almost indistinguishable genetic background38,40,41. To gain further insight into sequence diversity in PmMSP1, the previously reported partial sequences of PmMSP1 and the MSP1 sequences of P. brasilianum (PbrMSP1) were included for comparison between the corresponding regions38,39. Despite short indels in block II, nine variants were identified. Of these, six variants occurred in Thai isolates whereas five variants were found in PbrMSP1 in which alleles V and VI were shared between PmMSP1 and PbrMSP1 (Table 1).
Diversity of block IV in PmMSP1
Of 32 codons in block IV of PmMSP1, amino acid substitutions were found in 20 residues, resulting in 16 haplotypes based on analysis of isolates from Thailand and elsewhere including those belonging to PbrMSP1 (Table 2). Of these, 10 haplotypes were identified among Thai isolates in which three haplotypes were shared across endemic countries. All haplotypes of available PbrMSP1 sequences (n = 4) were distinct from those of PmMSP1. However, allele VI from four Brazilian isolates (GenBank accession nos. KR072269, KR072272, KR072278 and KR072279) and allele XV from a Peruvian Saimiri monkey (KR072284) were closely related with a single amino acid difference (I233K)38. It is noteworthy that seven of nine amino acid substitutions in block IV of PbrMSP1 were shared with those of PmMSP1 (Table 2).
Diversity of repeats in PmMSP1
Block VI contained complex repeat motifs with multiple patterns of repeat arrays and arrangements. Together with previously reported sequences of PmMSP1 (n = 16) and PbrMSP1 (n = 5), 35 haplotypes have been identified in this block in which 19 haplotypes occurred among Thai isolates (Fig. 3). The N-terminal part of this block contains non-repetitive amino acid sequences with variable indels, resulting in eight to 40 residues in this region. On the basis of distinct repeats and arrangements, block VI could be classified into types A and B. Type A contained 17 alleles (A1.1–A1.17) whereas type B could be further subdivided into subtypes B1 and B2, containing eight and 10 alleles, respectively (Fig. 3). Interestingly, the amino acid sequence of allele A1.10 was shared between P. malariae from a Brazilian patient (KR072216) and P. brasilianum from a Peruvian Saimiri monkey (JX045641) whereas the remaining PbrMSP1 and most other PmMSP1 type A alleles seemed to be closely related. It is noteworthy that none of PbrMSP1 sequences belonged to type B. Meanwhile, the other repeat-containing region was located in block VIII spanning codons 989 and 1032 (residues after FJ824669), characterized by a degenerate octapeptide repeat motif, P(A)Q(T)P(S, T or Q)QA(S)A(S or T)L(S or V)P(V or -), with variation in the number of repeat units among isolates. Of 35 Thai isolates and 14 previously reported sequences, 13 haplotypes were identified in this block in which haplotype I was most common and occurred in isolates from Thailand, Myanmar and Brazil, followed by haplotype XIII which was shared between PmMSP1 and PbrMSP135,38,39 (Table 3).
Microheterogeneity in conserved blocks
The complete sequences of all 5 conserved blocks have been available from 35 Thai isolates and an isolate from Cameroon (FJ824669). All nucleotide substitutions in conserved blocks were dimorphic, i.e. either one or the other of any two bases occurred at given positions. In total, 37 mutations were observed in conserved blocks, resulting in three haplotypes in blocks I and III, nine in block V, four in block VII and 13 in block IX (Table 4). The levels of nucleotide diversity in conserved regions ranged from 0.00098 to 0.00232 in blocks I and VII, respectively, which was an order or two orders of magnitude less than those in variable blocks (blocks IV, VI and VIII). Since the 19-kDa fragment of PfMSP1 has been considered to be an asexual blood stage vaccine target26, microheterogeneity in this region is of concern for vaccine development. Analysis of the homologous region to the 19-kDa-fragment-encoding sequence in PmMSP1 has revealed 4 nucleotide substitutions: c.5045G>A (G1681E), c.5055A>C (E1684D), c.5060A>T (E1686V) and c.5074C>A (Q1691K) (positions after the FJ824669 sequence). In total, four haplotypes occurred in the putative 19-kDa fragment of PmMSP1, characterized by (1) G-E-E-Q, (2) E-E-E-K, (3) E-D-E-K and (4) E-E-V-K, in which haplotype I was found in the Cameroonian isolate (FJ824669) whereas the remaining haplotypes co-existed among P. malariae populations in Thailand. Of these four substituted residues, c.5044G>C+5045G>A (G1681Q) was found in three isolates from Brazil and constituted another haplotype characterized by Q-E-E-K although singletons were previously observed in other five positions of this region38.
Neutrality test
To test for departure from neutrality, nucleotide substitutions in nonrepeat regions were analyzed by comparing the rate of synonymous substitutions per synonymous site (dS) and that of nonsynonymous substitutions per nonsynonymous site (dN) for each block of PmMSP1. Results revealed that dN exceeded dS in conserved blocks II, V and IX, and variable block IV. However, significant difference between dN and dS was observed only in block IV (Z-test, p < 0.0005) (Table 4). Meanwhile, codon-based detection of deviation from neutrality by the FUBAR method has shown evidence of positive selection in blocks I (E26K), IV (P223L/S/H and K241E), V (P294R) and IX (P1045Q/R and E1684D). On the other hand, evidence of purifying selection was found at codon 1374 (GAT⟶GAC, p.D1374) in conserved block IX of PmMSP1, a homologous residue located in close proximity to the 38 kDa/42 kDa cleavage site in PfMSP1 (Supplemental Fig. S1).
Recombination
Evidence of intragenic recombination in the PmMSP1 gene was determined from 35 Thai isolates by using the RDP4 package which revealed 21 potential recombination sites across the coding region of this gene (Table 5). Recombination breakpoints were detected more commonly in repeats or variable blocks (28 of 42 sites, 66.7%) than in conserved blocks. On the other hand, no recombination event was detected in conserved blocks I, II and V. Recombination breakpoints spanned 41–3978 bp with an average length of 784 bp.
Phylogenetic analysis
Analysis of the complete coding sequences of PmMSP1 has revealed two distinct clades in the phylogenetic tree (Fig. 4). The maximum likelihood tree inferred from the sequences of block VI per se has revealed 2 clades corresponding to characteristic repeats assigned to types A and B. It is noteworthy that the bifurcating clusters of taxa in the clade belonging to type B were in line with the isolates bearing types B1 and B2 repeats (Figs. 3, 4). On the other hand, the tree inferred from the sequences excluding block VI showed a different topology.

Maximum likelihood trees inferred from 21 distinct complete coding sequences of PmMSP1 with PocMSP1 and PowMSP1 as outgroup sequences. Numbers on the branches are the percentage of 1000 bootstrap samples supporting the branch; only values greater than 50 are shown. Types are based on repeat classification in Fig. 3. Scale bar indicates nucleotide substitution per site.
Predicted linear B-cell epitopes
The graphical presentation from BepiPred 2.0 analysis has revealed a number of potential linear B-cell epitopes across PmMSP1, spanning both conserved and variable blocks (Fig. 5A). Short indels in block II did not affect predicted B-cell epitopes encompassing this region. Interestingly, amino acid substitutions in variable block IV seemed not to affect predicted linear B-cell epitopes in all variants (Fig. 5B). On the other hand, the predicted epitope scores were variable among different alleles of blocks VI and VIII (Fig. 5C,D). Variation in the predicted scores was more pronounced among variants in block VI in which some regions were below the cutoff threshold value for being linear B-cell epitopes.

Predicted linear B-cell epitopes in PmMSP1 based on the BepiPred 2.0 method. (A) Epitope scores across the entire protein (GenBank accession no. FJ824669). Variable blocks are shown as broken boxes. (B–D) represent epitope scores for representative alleles of blocks IV, VI and VIII, respectively. The cutoff value is indicated by a broken line.
Predicted helper T-cell epitopes
Analysis of HLA-class II-binding peptides in PmMSP1 based on common HLA-DR alleles in Thai population (allele frequencies > 10%) including HLA-DRB1*12:02, -DRB1*15:02, -DQB1*05:01, -DQB1*05:02, -DQB1*03:01, -DQB1*03:03, -DQA1*01:01, -DQA1*01:02, -DQA1*03:02 and -DQA1*06:01 has predicted a number of potential binding peptides predominantly outside blocks VI and VIII which contained repeats (Supplemental Fig. S2). Block IV did not receive adequate scores for being HLA-class II-binding peptides (percentile rank < 10 and MHC binding affinity IC50 < 1000 nM) for these common HLA class II alleles42,43. However, searching for potential HLA-class II-binding peptides among alleles spanning block IV from residues 207 to 221 has shown that alleles II, IV and V had percentile ranks less than 10 and MHC binding affinity IC50 < 1000 nM for some uncommon HLA class II alleles in Thailand44. On the other hand, a potential HLA-class II-binding peptide was identified in one of nine alleles (allele V) of block IV encompassing residues 211–225 (Table 6). Taken together, these peptide variants could be potential helper T-cell epitopes in this molecule albeit being recognized by some uncommon HLA class II alleles among Thai population44.
Discussion
In this study, we have shown that the complete coding sequence of PmMSP1could be partitioned into five conserved and four variable blocks. Like other malarial MSP1s, conserved blocks of PmMSP1 exhibited microheterogeneity of sequences with dimorphic nucleotide substitutions24,32,33,34,45,46. Comparative analysis has revealed that short indels in block II of PmMSP1 seemed to be homologous to a short indel region at the 5′ portion of PvMSP1. Likewise, variable nonrepeat block IV of PmMSP1 were found to be homologous to variable nonrepeat blocks of PoMSP1, and repeat domains of PkMSP1 and PvMSP1 (Supplemental Fig. 3). Likewise, repeat blocks VI and VIII of PmMSP1 were homologous to blocks VIII and X of PocMSP1 and PowMSP1, blocks IV and VI of PkMSP1, and blocks VI and VIII of PvMSP1 (Supplemental Fig. 3). Meanwhile, the distantly related PfMSP1 also contained repeats in blocks VIII homologous to block VI of PmMSP1. Although variable and semi-conserved blocks of PfMSP1 consisted of two distinct parental alleles (MAD20 and K1), sequences of these regions were highly conserved within each allelic family24,45. Therefore, intraspecific conserved blocks of these malarial MSP1 genes seemed to be largely found in corresponding locations. Taken together, the similarity in primary structural organization of MSP1s across Plasmodium species may suggest that this locus has evolved from a common ancestral sequence whereas the lack of homologous regions in some domains of the genes among species could imply post-speciation evolution of individual MSP1 lineages. Consistently, it has been suggested that positive selection could influence lineage-specific evolutionary history of some human and simian malarial MSP1s47.
It is noteworthy that the levels of nucleotide diversity of PkMSP1, PvMSP1 and PfMSP1 were comparable among Thai isolates. On the other hand, the level of nucleotide diversity of PmMSP1 was significantly less than those of PkMSP1, PvMSP1 and PfMSP1 but remarkably greater than those of PoMSP119,32,33,48,49,50. Consistent findings were observed when analysis was performed separately for synonymous (πS) and nonsynonymous sites (πN) (Supplemental Table 2). The extent of nucleotide diversity among MSP1s of different Plasmodium species in Thailand could be due to evolutionary and population genetic forces on parasite populations such as mutation, recombination and population processes. Meanwhile, the neutral theory of molecular evolution predicts that the level of nucleotide diversity is proportional to the mutation rate (μ) and effective population size (Ne) under mutation-drift equilibrium51. Since the mutation rates of malarial MSP1 genes seemed to be similar across species52,53,54, variation in the levels of nucleotide diversity of these loci could be due to the difference in effective population sizes among Plasmodium species in Thailand. Although some regions or residues in malarial MSP1s were deviated from selective neutrality, the remaining majority of sequences seemed to be under neutral evolution. Therefore, the level of nucleotide diversity may roughly reflect the number of breeding individuals in the population. On the other hand, the low level of nucleotide diversity in PmMSP1 could represent the low transmission rate and probably from bottleneck effects due to malaria control measures as previously noted38,46. Our previous surveys of malaria in Thailand have shown that the prevalence of P. malariae and P. knowlesi in Thailand was comparable15,18,19,20. Therefore, it is likely that the higher level of nucleotide diversity of PkMSP1 than that of PmMSP1 could stem from a hidden large reservoir of P. knowlesi in its macaque natural hosts in this country33,55. On the one hand, the haplotype diversity of most malarial MSP1 genes in Thailand was relatively high (> 0.9), implying that distinct or rare haplotypes were abundant in the populations (Supplemental Table S2). On the other hand, we observed some predominant PmMSP1 haplotypes in this country, i.e. haplotypes III, XIII and X, which occurred across endemic provinces and between long time intervals of sample collections (Fig. 1), suggesting that the parasites bearing these haplotypes could probably have reproductive advantage.
Conserved blocks in PmMSP1 displayed microheterogeneity of sequences in which nucleotide substitutions seems to have evolved neutrally because block-wise analysis revealed that dS was not significantly different from dN (Table 4). However, codon-based analysis has identified four positively selected codons in conserved blocks, suggesting that natural selection has influenced evolution of particular codons. Interestingly, one of these codons (residue E1684D) was located between the two EGF-like domains at the C-terminal part of PmMSP1 in which the homologous region in PfMSP1 has been a target for naturally acquired antibodies associated with clinical protection against falciparum malaria26. Intriguingly, positive selection in the EGF-like domain of PmMSP1 could probably be driven by host immune pressure. On the other hand, evidence for purifying selection was detected at codon 1374 (GAT ⟶ GAC) that was located in close proximity to the canonical 38 kDa/42 kDa cleavage site in PfMSP156. Importantly, cleavage at this site has been shown to be a rate-limiting processing step, suggesting its pivotal role for MSP1 proteolytic maturation57,58. Therefore, deviation from selective neutrality occurred at particular residues in conserved regions of PmMSP1.
The variable nonrepeat block IV of PmMSP1 spanned 32 codons with 21 amino acid substitutions, resulting in 16 alleles among Thai and global isolates (Table 2). The significant difference in dN exceeding dS in this block implies that positive selection could influence diversity in this region (Table 4). On the basis of amino acid alignment, block IV of PmMSP1 was homologous to block III of PfMSP1, a portion of the 83-kDa fragment which forms a flexible wing domain of the protein as demonstrated by single-particle cryo-electron microscopy31. Several lines of evidence have suggested that MSP1 could be detected as monomeric and dimeric forms58,59,60. It has been shown that dimerization of PfMSP1 involves the interaction between the 83-kDa and 42-kDa fragments31. Although the significance of dimerization of PfMSP1 remains unknown, the protective capability against falciparum malaria conferred by natural antibodies to the 83-kDa fragment could suggest the functional importance of this region61. Importantly, in silico analysis has shown that block IV of PmMSP1 contained both B-cell and helper T-cell epitopes. Consistently, recombinant proteins derived from various regions of PmMSP1 including the N-terminal fragment elicited strong immunogenicity in mice62 and were highly recognized in serum samples of primates and non-human primates from malaria endemic areas63,64. Although allelic variation in block IV of this protein seemed not to drastically change the propensity of being B-cell epitopes as predicted by the IEDB analysis resource (Fig. 5B), amino acid substitutions in this region were unlikely recognized by common HLA class II alleles among Thai population (Fig. 4B, Table 6, Supplemental Fig. S2). Importantly, mutations in block IV may reduce or totally abolish predicted binding capability of the peptides to some uncommon HLA class II alleles in Thai population (Table 6; Supplemental Fig. S2). Undoubtedly, further studies are required to address the immunological significance of helper T-cell epitopes in block IV of PmMSP1. Therefore, it seemed that positive selection in block IV could probably be driven by host immune pressure.
Repetitive amino acid sequences have been observed in several malarial antigens including MSP1s. Our analysis has revealed two repeat-containing regions in blocks VI and VIII of PmMSP1. Unlike block VIII that contained degenerate octapeptide motifs, repeats in block VI were more complex, characterized by a repertoire of different repeat arrays and arrangements. Meanwhile, the RDP4 package has identified 21 recombination breakpoints in PmMSP1. Interestingly, about half of recombination events involved block VI whereas about one-third of the breakpoints occurred within this block. Besides slip-strand mispairing mechanism that could generate sequence and size variation in repeat sequences, recombination may contribute to shuffle of repeat units in block VI. Although a number of linear B-cell epitopes were predicted in this block (Fig. 5A), in silico analysis has suggested that variation in repeat sequences could affect antibody recognition (Fig. 5C). Meanwhile, phylogenetic tree inferred from the block VI sequences of PmMSP1 has revealed two distinct clades, corresponding to repeat sequence types A and B of this block. Importantly, variation in the number of repeat units could affect intensity of antibody reactivity whereas distinct variants of repetitive antigens may abolish specific antibody response as shown by antibody recognition of repeat antigens in block II of PfMSP165,66. Therefore, sequence divergence of repetitive regions in PmMSP1 could probably enhance host immune evasion by the parasites.
One of the shared features of PmMSP1 and PvMSP1 was the presence of indels near the N-terminus of the proteins. Although indels in block II spanned 17 codons, 6 alleles have been identified among Thai isolates (Table 1). Indels are commonly found in both coding and noncoding regions of prokaryotes and eukaryotes genomes while they may occur within repeats and nonrepeat regions67,68. The generation of indels related with repeats could be due to polymerase slippage69,70. On the other hand, the formation of indels in nonrepeat regions required pre-existing palindromic or quasi-palindromic sequences, provoking a double-stranded break intermediate during DNA replication while the ensuing repair process was imperfect71,72,73,74. It is noteworthy that quasi-palindromic repeats were identified around indels of PmMSP1 and PvMSP1, supporting the mechanisms for indel formation in nonrepeats of these genes (Supplemental Fig. S4). Although analysis of natural selection on these indels was not possible due to unknown ancestral state of this region, the lack of frame-shift mutation following indels in both PmMSP1 and PvMSP1 could imply selective constraint on the protein structure and/or function.
Several lines of evidence have suggested that P. malariae and P. brasilianum were de facto either con-species or the same parasites38,40,41. A repertoire of alleles in block VI constituting the most polymorphic region of the gene has been identified among PmMSP1 and PbrMSP1 (Fig. 2; Table 4). Importantly, allele A1.10 of block VI was shared between the MSP1 genes of P. malariae and P. brasilianum whereas allele XIII of block VIII has been previously reported to occur in both species38. Like other genes or non-coding loci containing repeats in malarial genomes, variation in repeat sequences and the number of repeat units could be generated by the process of slip-strand mispairing mechanism75,76. Therefore, it is unlikely that identical complex repeats could have arisen from homoplasy. Furthermore, shared alleles between PmMSP1 and PbrMSP1 have been observed in variable blocks II (alleles V and VI) and VIII (allele XIII) whereas a single codon difference was observed between alleles VI and XV in variable block IV (alleles VI and XV) (Tables 1, 2, 3). Taken together, it is likely that P. malariae and P. brasilianum could be identical species or at least con-species as previously noted38,41.
In conclusion, analysis of the complete coding sequences of PmMSP1 from clinical isolates has revealed structural organization of this locus. Besides structural similarity across human malarial MSP1s, evidences of intragenic recombination and natural selection have been identified in PmMSP1. The information from this study could be useful for further studies such as vaccine development and strain characterization of P. malariae based on this molecule.
Materials and methods
Parasite isolates
Thirty-five Plasmodium malariae isolates were obtained from symptomatic malaria patients during surveys of Plasmodium species distribution in Thailand during 1994 and 2016 (Fig. 1). Either finger-pricked or venous blood samples were taken from each subject and spotted onto filter papers or preserved in EDTA, respectively. Both thin and thick blood films were prepared from fresh blood and stained with Giemsa solution for microscopic examination of malaria parasites. DNA was extracted from each blood sample using Qiagen DNA mini kit (Qiagen, Hilden, Germany) per the manufacturer’s recommendation and kept at − 40 °C until use. Definite species identification was performed by species-specific nested PCR targeting 18S rRNA, mitochondrial cytochrome b or cytochrome oxidase I as previously described15,19,20.
PCR amplification and sequencing of the PmMSP1 gene
The complete coding sequence of PmMSP1 was amplified by nested PCR using outer primers: Pmmsp1F0 (5′-TACTCTATATTATCAAGTTTAATTC-3′) and Pmmsp1R0 (5′-CATTCGTATCCTTCTTTTCTGT-3′), and inner primers: Pmmsp1F01 (5′-GTTTAATTCAAAAATGAAAGCAC-3′) and Pmmsp1R01 (5′-TCTTTTTTTCTTAAAGTAAGTTAAAC-3′). Amplification reaction and condition were as previously described34. All amplification reactions were done in an Applied Biosystem GeneAmpH PCR System 9700 thermocycler (PE Biosystems, Foster City, CA). PCR products were analyzed by 1% agarose gel electrophoresis. The PCR products were purified by using a QIAamp PCR purification kit (Qiagen) and used as templates for sequencing. Sequencing primers were deployed to obtain overlapping sequences of the gene in which both directions were determined directly from the PCR-purified templates (Supplemental Table S1). Validation of singletons and indels in the sequences was performed by sequencing of the PCR products from independent amplification reactions using the same genomic DNA as templates.
Data analysis
Alignment of the PmMSP1 nucleotide sequences was performed by using the default option of the MUSCLE program and manually edited77. Indels in coding regions were determined from multiple alignments of amino acid sequences to maintain the reading frame. The sequence of the first complete PmMSP1 gene from a Cameroonian patient was used as reference (GenBank accession number FJ824669)36. Tandem repeats were analyzed by scanning each sequence using window sizes per the default option of the Tandem Repeats Finder version 4.0 algorithm78. Nucleotide diversity was computed from the average number of nucleotide differences per site between two sequences in the sample and the standard errors were estimated by 1000 boostrap pseudoreplicates79. A sliding window analysis of nucleotide diversity was performed by using window length of 100 nucleotides and step size of 15 sites. Haplotype diversity and its sampling variance were determined by using the DnaSP program80. The number of synonymous substitutions per synonymous site and the number of nonsynonymous substitutions per nonsynonymous site was computed using Nei and Gojobori’s method79 with Juke and Cantor correction81. Standard errors of these parameters were estimated by the bootstrap method with 1000 pseudoreplicates using the MEGA 6.0 program82. Differences between the nucleotide diversity values were determined by a two-tailed Z-test. Deviation from selective neutrality at individual codons was identified using fast unconstrained Bayesian approximation (FUBAR) method implemented in the Datamonkey Web-Server83,84. To minimize the interfering signals from recombination on selection of individual codons, the data generated by elimination of recombination segments was deployed for analysis85. Determination of intragenic recombination was performed by using the Recombination Detection Program version 4 (RDP4)86. Phylogenetic trees were constructed by using the Maximum Likelihood method based on the General Time Reversible model with a discrete Gamma distribution to model evolutionary rate differences among sites84. The final tree is the one with the highest log likelihood value. Bootstrap supports for the branching patterns were estimated from 1000 pseudoreplicates of the sample data. Prediction of linear B-cell epitopes was done by using the BepiPred linear epitope prediction 2.0 implemented in the Immune Epitope Database (IEDB) And Analysis Resource87. The threshold for linear B-cell epitopes was more than or equal to the average predicted residue score of the protein. The HLA-class II-binding peptides were predicted by using the IEDB recommended 2.22 algorithm with a default 12–18 residues option88. The criterion for being HLA-class II-binding peptides included the percentile rank ≤ 10 and the IC50 threshold for MHC binding affinity ≤ 1000 nM43. The common HLA class II haplotypes among Thai population were based on allele frequency ≥ 0.1 according to the previous report44.
Ethical approval
The study protocol was approved by the Institutional Review Board on Human Research of Faculty of Medicine, Chulalongkorn University (IRB No. 384/60 and COA No. 805/2018). Written informed consent was obtained from participants or from parents or guardians prior to blood sample collections. All procedures were performed in accordance to the relevant guidelines and regulations.
Data availability
Thirty-five complete coding sequences of PmMSP1 have been deposited in NCBI GenBank under accession numbers OM525734–OM525768. The datasets generated during and/or analyses during the current study are available from the corresponding author upon request.
References
Betson, M., Clifford, S., Stanton, M., Kabatereine, N. B. & Stothard, J. R. Emergence of nonfalciparum Plasmodium infection despite regular artemisinin combination therapy in an 18-month longitudinal study of Ugandan children and their mothers. J. Infect. Dis. 217, 1099–1109 (2018).
Groger, M. et al. Prospective clinical and molecular evaluation of potential Plasmodium ovale curtisi and wallikeri relapses in a high-transmission setting. Clin. Infect. Dis. 69, 2119–2126 (2019).
Yman, V. et al. Persistent transmission of Plasmodium malariae and Plasmodium ovale species in an area of declining Plasmodium falciparum transmission in eastern Tanzania. PLoS Negl. Trop. Dis. 13, e0007414 (2019).
Hawadak, J., Dongang Nana, R. R. & Singh, V. Global trend of Plasmodium malariae and Plasmodium ovale spp. malaria infections in the last two decades (2000–2020): A systematic review and meta-analysis. Parasit. Vectors 14, 297 (2021).
Gilles, H. M. & Hendrickse, R. G. Nephrosis in Nigerian children. Role of Plasmodium malariae, and effect of antimalarial treatment. Br. Med. J. 2, 27–31 (1963).
Ward, P. A. & Kibukamusoke, J. W. Evidence for soluble immune complexes in the pathogenesis of the glomerulonephritis of quartan malaria. Lancet 1, 283–285 (1969).
Hendrickse, R. G. & Adeniyi, A. Quartan malarial nephrotic syndrome in children. Kidney Int. 16, 64–74 (1979).
Silva, G. B. D. J., Pinto, J. R., Barros, E. J. G., Farias, G. M. N. & Daher, E. F. Kidney involvement in malaria: An update. Rev. Inst. Med. Trop. Sao Paulo 59, e53 (2017).
Maguire, J. D. et al. Chloroquine-resistant Plasmodium malariae in south Sumatra, Indonesia. Lancet 360, 58–60 (2002).
Collins, W. E. & Jeffery, G. M. Extended clearance time after treatment of infections with Plasmodium malariae may not be indicative of resistance to chloroquine. Am. J. Trop. Med. Hyg. 67, 406–410 (2002).
Collins, W. E. & Jeffery, G. M. Plasmodium malariae: Parasite and disease. Clin. Microbiol. Rev. 20, 579–592 (2007).
Verra, F. et al. A systematic review of transfusion-transmitted malaria in non-endemic areas. Malar. J. 17, 36 (2018).
Aschar, M. et al. The hidden Plasmodium malariae in blood donors: A risk coming from areas of low transmission of malaria. Rev. Inst. Med. Trop. Sao Paulo 62, e100 (2020).
Putaporntip, C., Buppan, P. & Jongwutiwes, S. Improved performance with saliva and urine as alternative DNA sources for malaria diagnosis by mitochondrial DNA-based PCR assays. Clin. Microbiol. Infect. 17, 1484–1491 (2011).
Putaporntip, C. et al. Cryptic Plasmodium inui and P. fieldi infections among symptomatic malaria patients in Thailand. Clin. Infect. Dis. (In press) (2022).
Cunha, M. G. et al. Mixed Plasmodium malariae infections were underdetected in a malaria endemic area in the Amazon Region, Brazil. Am. J. Trop. Med. Hyg. 105, 1184–1186 (2021).
Thimasarn, K., Jatapadma, S., Vijaykadga, S., Sirichaisinthop, J. & Wongsrichanalai, C. Epidemiology of malaria in Thailand. J. Travel Med. 2, 59–65 (1995).
Putaporntip, C. et al. Differential prevalence of Plasmodium infections and cryptic Plasmodium knowlesi malaria in humans in Thailand. J. Infect. Dis. 199, 1143–1150 (2009).
Jongwutiwes, S. et al. Plasmodium knowlesi malaria in humans and macaques, Thailand. Emerg. Infect. Dis. 17, 1799–1806 (2011).
Putaporntip, C. et al. Plasmodium cynomolgi co-infections among symptomatic malaria patients, Thailand. Emerg. Infect. Dis. 27, 590–593 (2021).
Blackman, M. J. & Carruthers, V. B. Recent insights into apicomplexan parasite egress provide new views to a kill. Curr. Opin. Microbiol. 16, 459–464 (2013).
Das, S. et al. Processing of Plasmodium falciparum merozoite surface protein msp1 activates a spectrin-binding function enabling parasite egress from RBCs. Cell Host Microbe. 18, 433–444 (2015).
Holder, A. A. The precursor to major merozoite surface antigens: Structure and role in immunity. Prog. Allergy 41, 72–97 (1988).
Tanabe, K., Mackay, M., Goman, M. & Scaife, J. G. Allelic dimorphism in a surface antigen gene of the malaria parasite Plasmodium falciparum. J. Mol. Biol. 195, 273–287 (1987).
Blackman, M. J., Heidrich, H. G., Donachie, S., McBride, J. S. & Holder, A. A. A single fragment of a malaria merozoite surface protein remains on the parasite during red cell invasion and is the target of invasion-inhibiting antibodies. J. Exp. Med. 172, 379–382 (1990).
Egan, A. F. et al. Clinical immunity to Plasmodium falciparum malaria is associated with serum antibodies to the 19-kDa C-terminal fragment of the merozoite surface antigen, PfMSP-1. J. Infect. Dis. 173, 765–769 (1996).
Conway, D. J. et al. A principal target of human immunity to malaria identified by molecular population genetic and immunological analyses. Nat. Med. 6, 689–692 (2000).
Goel, V. K. et al. Band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc. Natl. Acad. Sci. USA 100, 5164–5169 (2003).
Boyle, M. J., Richards, J. S., Gilson, P. R., Chai, W. & Beeson, J. G. Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites. Blood 115, 4559–4568 (2010).
Baldwin, M. R., Li, X., Hanada, T., Liu, S. C. & Chishti, A. H. Merozoite surface protein 1 recognition of host glycophorin A mediates malaria parasite invasion of red blood cells. Blood 125, 2704–2711 (2015).
Dijkman, P. M. et al. Structure of the merozoite surface protein 1 from Plasmodium falciparum. Sci. Adv. 7, eabg0465 (2021).
Putaporntip, C. et al. Mosaic organization and heterogeneity in frequency of allelic recombination of the Plasmodium vivax merozoite surface protein-1 locus. Proc. Natl. Acad. Sci. USA 99, 16348–16353 (2002).
Putaporntip, C., Thongaree, S. & Jongwutiwes, S. Differential sequence diversity at merozoite surface protein-1 locus of Plasmodium knowlesi from humans and macaques in Thailand. Infect. Genet. Evol. 18, 213–219 (2013).
Putaporntip, C., Hughes, A. L. & Jongwutiwes, S. Low level of sequence diversity at merozoite surface protein-1 locus of Plasmodium ovale curtisi and P. ovale wallikeri from Thai isolates. PLoS One 8, e58962 (2013).
Fandeur, T., Volney, B., Peneau, C. & de Thoisy, B. Monkeys of the rainforest in French Guiana are natural reservoirs for P. brasilianum/P. malariae malaria. Parasitology 120(I), 11–21 (2000).
Birkenmeyer, L., Muerhoff, A. S., Dawson, G. J. & Desai, S. M. Isolation and characterization of the MSP1 genes from Plasmodium malariae and Plasmodium ovale. Am. J. Trop. Med. Hyg. 82, 996–1003 (2010).
Araújo, M. S. et al. Natural Plasmodium infection in monkeys in the state of Rondônia (Brazilian Western Amazon). Malar. J. 12, 180 (2013).
Guimarães, L. O. et al. Merozoite surface protein-1 genetic diversity in Plasmodium malariae and Plasmodium brasilianum from Brazil. BMC Infect. Dis. 15, 529 (2015).
Li, P. et al. Plasmodium malariae and Plasmodium ovale infections in the China-Myanmar border area. Malar. J. 15, 557 (2016).
Guimarães, L. O. et al. The genetic diversity of Plasmodium malariae and Plasmodium brasilianum from human, simian and mosquito hosts in Brazil. Acta Trop. 124, 27–32 (2012).
Lalremruata, A. et al. Natural infection of Plasmodium brasilianum in humans: Man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine 2, 1186–1192 (2015).
Andreatta, M. et al. An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics 34, 1522–1528 (2018).
Paul, S., Grifoni, A., Peters, B. & Sette, A. Major histocompatibility complex binding, eluted ligands, and immunogenicity: Benchmark testing and predictions. Front. Immunol. 10, 3151 (2020).
Satapornpong, P. et al. Genetic diversity of HLA class I and class II alleles in Thai populations: Contribution to genotype-guided therapeutics. Front. Pharmacol. 11, 78 (2020).
Jongwutiwes, S., Tanabe, K. & Kanbara, H. Sequence conservation in the C-terminal part of the precursor to the major merozoite surface proteins (MSP1) of Plasmodium falciparum from field isolates. Mol. Biochem. Parasitol. 59, 95–100 (1993).
Jongwutiwes, S., Putaporntip, C. & Hughes, A. L. Bottleneck effects on vaccine-candidate antigen diversity of malaria parasites in Thailand. Vaccine 28, 3112–3117 (2010).
Sawai, H. et al. Lineage-specific positive selection at the merozoite surface protein 1 (msp1) locus of Plasmodium vivax and related simian malaria parasites. BMC Evol. Biol. 10, 52 (2010).
Tanabe, K. et al. Allelic dimorphism-associated restriction of recombination in Plasmodium falciparum msp1. Gene 397, 153–160 (2007).
Tanabe, K. et al. Within-population genetic diversity of Plasmodium falciparum vaccine candidate antigens reveals geographic distance from a Central sub-Saharan African origin. Vaccine 31, 1334–1339 (2013).
Tanabe, K. et al. Plasmodium falciparum: Genetic diversity and complexity of infections in a isolated village in Western Thailand. Parasitol. Int. 64, 260–266 (2015).
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge University Press, 1983).
Hughes, A. L. Positive selection and interallelic recombination at the merozoite surface antigen-1 (MSA-1) locus of Plasmodium falciparum. Mol. Biol. Evol. 9, 381–393 (1992).
Hughes, A. L. & Verra, F. Extensive polymorphism and ancient origin of Plasmodium falciparum. Trends Parasitol. 18, 348–351 (2002).
Putaporntip, C., Jongwutiwes, S., Iwasaki, T., Kanbara, H. & Hughes, A. L. Ancient common ancestry of the merozoite surface protein 1 of Plasmodium vivax as inferred from its homologue in Plasmodium knowlesi. Mol. Biochem. Parasitol. 146, 105–108 (2006).
Putaporntip, C. et al. Ecology of malaria parasites infecting Southeast Asian macaques: Evidence from cytochrome b sequences. Mol. Ecol. 19, 3466–3476 (2010).
Withers-Martinez, C. et al. Plasmodium subtilisin-like protease 1 (SUB1): Insights into the active-site structure, specificity and function of a pan-malaria drug target. Int. J. Parasitol. 42, 597–612 (2012).
Child, M. A., Epp, C., Bujard, H. & Blackman, M. J. Regulated maturation of malaria merozoite surface protein-1 is essential for parasite growth. Mol. Microbiol. 78, 187–202 (2010).
Das, S. et al. Processing of Plasmodium falciparum merozoite surface protein msp1 activates a spectrin-binding function enabling parasite egress from RBCs. Cell Host Microbe 18, 433–444 (2015).
Sanders, P. R. et al. Identification of protein complexes in detergent-resistant membranes of Plasmodium falciparum schizonts. Mol. Biochem. Parasitol. 154, 148–157 (2007).
Lin, C. S. et al. The merozoite surface protein 1 complex is a platform for binding to human erythrocytes by Plasmodium falciparum. J. Biol. Chem. 289, 25655–25669 (2014).
Tolle, R. et al. A prospective study of the association between the human humoral immune response to Plasmodium falciparum blood stage antigen gp190 and control of malarial infections. Infect. Immun. 61, 40–47 (1993).
Elizardez, Y. B. et al. Recombinant proteins of Plasmodium malariae merozoite surface protein 1 (PmMSP1): Testing immunogenicity in the BALB/c model and potential use as diagnostic tool. PLoS One 14, e0219629 (2019).
Monteiro, E. F. et al. Naturally acquired humoral immunity against malaria parasites in non-human primates from the Brazilian Amazon, Cerrado and Atlantic Forest. Pathogens 9, 525 (2020).
Monteiro, E. F. et al. Antibody profile comparison against MSP1 antigens of multiple Plasmodium species in human serum samples from two different Brazilian populations using a multiplex serological assay. Pathogens 10, 1138 (2021).
Locher, C. P., Tam, L. Q., Chang, S. P., McBride, J. S. & Siddiqui, W. A. Plasmodium falciparum: gp195 tripeptide repeat-specific monoclonal antibody inhibits parasite growth in vitro. Exp. Parasitol. 84, 74–83 (1996).
McBride, J. S., Walliker, D. & Morgan, G. Antigenic diversity in the human malaria parasite Plasmodium falciparum. Science 217, 254–257 (1982).
Pascarella, S. & Argos, P. Analysis of insertions/deletions in protein structures. J. Mol. Biol. 224, 461–471 (1992).
Montgomery, S. B. et al. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 23, 749–761 (2013).
Streisinger, G. et al. Frameshift mutations and the genetic code. Cold Spring Harb. Symp. Quant. Biol. 31, 77–84 (1966).
Levinson, G. & Gutman, G. A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 (1987).
Chu, G. Double strand break repair. J. Biol. Chem. 272, 24097–24100 (1997).
McVey, M., Larocque, J. R., Adams, M. D. & Sekelsky, J. J. Formation of deletions during double-strand break repair in Drosophila DmBlm mutants occurs after strand invasion. Proc. Natl. Acad. Sci. USA 101, 15694–15699 (2004).
Lee, J. A., Carvalho, C. M. & Lupski, J. R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247 (2007).
Hastings, P. J., Ira, G. & Lupski, J. R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5, e1000327 (2009).
Jongwutiwes, S., Tanabe, K., Hughes, M. K., Kanbara, H. & Hughes, A. L. Allelic variation in the circumsporozoite protein of Plasmodium falciparum from Thai field isolates. Am. J. Trop. Med. Hyg. 51, 659–668 (1994).
Seethamchai, S. et al. Variation in intronic microsatellites and exon 2 of the Plasmodium falciparum chloroquine resistance transporter gene during modification of artemisinin combination therapy in Thailand. Infect. Genet. Evol. 65, 35–42 (2018).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Nei, M. Molecular Evolutionary Genetics (Columbia University Press, 1987).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Jukes, T. H. & Cantor, C. R. Evolution of protein molecules. In Mammalian Protein Metabolism (ed. Munro, H. N.) 21–132 (Academic Press, 1969).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Murrell, B. et al. FUBAR: A fast, unconstrained Bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205 (2013).
Weaver, S. et al. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 35, 773–777 (2018).
Kosakovsky Pond, S. L. & Frost, S. D. W. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533 (2005).
Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003 (2015).
Jespersen, M. C., Peters, B., Nielsen, M. & Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 45, W24–W29 (2017).
Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Acknowledgements
We are grateful to all patients who donated their blood samples for this study. This study received financial support from Ratchadapiseksompotch Fund, Faculty of Medicine, Chulalongkorn University (Grant no. RA60/126) to S.J. and C.P.
Author information
Authors and Affiliations
Contributions
S.J. and C.P. designed the study and funding acquisition. C.P., S.S., N.K. and S.J. contributed to sample collection. C.P., N.K. and S.S. performed the experiments. C.P. retrieved GenBank sequences. R.R. prepared Fig. 1 and Supplemental Fig. S3. C.P. and S.J. performed data analysis. C.P. drafted the manuscript. S.J. reviewed and finalized the manuscript. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Putaporntip, C., Kuamsab, N., Rojrung, R. et al. Structural organization and sequence diversity of the complete nucleotide sequence encoding the Plasmodium malariae merozoite surface protein-1. Sci Rep 12, 15591 (2022). https://doi.org/10.1038/s41598-022-19049-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19049-z
- Springer Nature Limited