
Abstract
Background
Migraine is a primary headache with genetic susceptibility, but the pathophysiological mechanisms are poorly understood, and it remains an unmet medical need. Earlier we demonstrated significant differences in the transcriptome of migraineurs' PBMCs (peripheral blood mononuclear cells), suggesting the role of neuroinflammation and mitochondrial dysfunctions. Post-transcriptional gene expression is regulated by miRNA (microRNA), a group of short non-coding RNAs that are emerging biomarkers, drug targets, or drugs. MiRNAs are emerging biomarkers and therapeutics; however, little is known about the miRNA transcriptome in migraine, and a systematic comparative analysis has not been performed so far in migraine patients.
Methods
We determined miRNA expression of migraineurs’ PBMC during (ictal) and between (interictal) headaches compared to age- and sex-matched healthy volunteers. Small RNA sequencing was performed from the PBMC, and mRNA targets of miRNAs were predicted using a network theoretical approach by miRNAtarget.com™. Predicted miRNA targets were investigated by Gene Ontology enrichment analysis and validated by comparing network metrics to differentially expressed mRNA data.
Results
In the interictal PBMC samples 31 miRNAs were differentially expressed (DE) in comparison to healthy controls, including hsa-miR-5189-3p, hsa-miR-96-5p, hsa-miR-3613-5p, hsa-miR-99a-3p, hsa-miR-542-3p. During headache attacks, the top DE miRNAs as compared to the self-control samples in the interictal phase were hsa-miR-3202, hsa-miR-7855-5p, hsa-miR-6770-3p, hsa-miR-1538, and hsa-miR-409-5p. MiRNA-mRNA target prediction and pathway analysis indicated several mRNAs related to immune and inflammatory responses (toll-like receptor and cytokine receptor signalling), neuroinflammation and oxidative stress, also confirmed by mRNA transcriptomics.
Conclusions
We provide here the first evidence for disease- and headache-specific miRNA signatures in the PBMC of migraineurs, which might help to identify novel targets for both prophylaxis and attack therapy.
Similar content being viewed by others

Introduction
Migraine is a common primary headache disease with complex pathophysiological mechanisms. This neurovascular disorder involves meningeal vasodilatation, oedema formation activation and sensitisation of the trigeminal pain pathways [1]. Immune activation and the release of proinflammatory cytokines and neuropeptides, such as CGRP (calcitonin gene-related peptide) and PACAP (pituitary adenylate cyclase-activating polypeptide), seem to be critical elements [2,3,4,5,6]. Besides genetic factors, environmental influence has been described for migraine susceptibility [7]. There has not been a breakthrough in effective personalised preventive or acute treatment of migraine despite extensive efforts. It is still an unmet medical need mainly due to the heterogeneity of the disease. Therefore, it is essential to explore the complexity of the pathophysiological mechanisms by unbiased omics approaches. PBMCs, containing lymphocytes (T cells, B cells, natural killer cells) and monocytes, have become attractive blood-based marker candidates in clinical practice due to minimally invasive sampling and relatively simple isolation. Their potential value consists of reflecting pathophysiological changes in the central nervous system in various diseases. Hence neuroinflammatory processes might be studied in a specific way using PBMCs [8,9,10]. We have recently provided evidence for increased immune cell activity, oxidative stress and mitochondrial dysfunction in migraine using transcriptomics of peripheral blood mononuclear cells (PBMC) [11]. MicroRNAs (miRNAs) are short, non-coding RNAs regulating post-transcriptional gene expression, and thus controlling cell-to-cell communication and multiple cellular processes. [12,13,14]. Since miRNAs can reflect the environmental impact on gene expression modulation [15], their relevance in preclinical research and clinical applications is rapidly growing. Circulating miRNAs are stable, easily measurable and tissue-specific molecules [13, 16]; therefore, they receive increased attention as tissue-specific biomarkers for various pathological processes, drug resistance modulators, and novel drug targets. Several clinical studies suggest specific miRNAs to be indicators of disease progression, e.g. in diabetes, coronary heart disease, breast cancer, epilepsy, depressive disorder, stroke, etc. [16]. Interestingly, miRNAs can interfere with immune, vascular, and neuronal activities [17,18,19]. Thus, they might be valuable in neuro-immune-vascular interactions and alterations like neuropathic pain, Alzheimer’s and Parkinson’s disease, multiple sclerosis and migraine [20]. Their relevance in chronic pain conditions, e.g. complex regional pain syndrome, irritable bowel syndrome and fibromyalgia, have also been studied [21, 22].
Detection of miRNAs in molecular research and diagnostics is fundamental as well as challenging due to their low cell content, small size, and the similar sequences of miRNA families. Continuous development has yielded detection methods ranging from conventional techniques such as microarrays and PCR (polymerase chain reaction) to the more recent next-generation sequencing. MicroRNA microarray is a technique for miRNA profiling utilizing hybridization between target miRNAs and their corresponding complementary probes on a solid surface and eventual detection of the signal intensity of the hybridization probes. It has high throughput although low sensitivity and selectivity. PCR, on the other hand, has high sensitivity, a wider range but poor specificity for miRNAs, along with limited throughput. RNA-seq represents the last extensive approach for miRNA profiling. Its workflow involves the isolation of RNA followed by cDNA (complementary DNA) library construction and sequencing. Small RNA-Seq was chosen for our study based on its main advantages of having a non-targeted and greatly sensitive nature [23, 24].
The potential roles of miRNAs in migraine diagnosis and therapy have recently been reviewed, but the clinical relevance of the results needs to be clarified and confirmed [25, 26]. Few investigations have been performed on specific miRNAs [16,17,18,19] and microarray-based miRNA profile descriptions, with controversial results [27,28,29]. One study describes serum miRNA alterations both during attacks and pain-free periods of migraineurs, using high-content serum microRNA PCR arrays [27]. In contrast, no alterations were described in the miRNA profiles of 20 mixed gender migraine patients’ PBMC samples compared to 5 headache-free controls in a conference abstract [29]. The extension of this pilot study (MicroMIG) demonstrated a few DE miRNAs when comparing different subgroups of migraine patients to controls, but they were not specified [30]. Two-sample microarray analysis of exosomes isolated from pooled blood samples of 15 female migraineurs without aura compared to 13 healthy matched controls described 22 dysregulated circulating miRNAs, 4 of which were validated by qPCR. MiR-22 and let-7b downregulation was confirmed in PBMC samples [28]. PBMCs represent an easily accessible biological material, which can reflect pathophysiological changes in the brain and characterize neuroinflammatory processes [8, 9]. Therefore, here we determine miRNAs from PBMC samples to gather more information on disease- and headache-related mechanisms in migraineurs since their transcriptomic alterations were proven to be specific and sensitive indicators of migraine-related processes [11]. To our knowledge, this is the first study to perform small RNA sequencing on PBMCs isolated from interictal and ictal samples of migraine patients compared with healthy controls. Combining network theoretical miRNA-target prediction and an already analyzed mRNA data set provides a unique and valuable platform for discovering potential novel biomarkers and/or drug targets in migraine.
Methods
Study design
The study was approved by the National Public Health Center, Ministry of Human Capacities (28324–5/2019/EÜIG). Informed written consent was obtained from each participant following the Declaration of Helsinki.
Sixteen episodic migraine patients with (n = 1) or without aura (n = 15) and 12 healthy controls, aged between 23 and 59 years, were enrolled in the present study. Out of the 16 migraineurs, 8 patients applied for blood sampling during headache attacks (one patient applied twice). Therefore, 9 ictal (self-controlled paired) samples were further investigated. In total, 37 samples from 28 participants were selected from the cohort collected between September 2018 and December 2019. The inclusion criteria were female gender, aged 20–60 years, diagnosed with migraine with or without aura by the third edition of the International Classification of Headache Disorders [31]. Exclusion criteria were chronic inflammatory diseases and depression. A detailed questionnaire was used to assess the characteristics of the headache concerning the following: prophylactic or attack medication before sampling, the number of attacks in the previous month, the time of the last episode, the beginning of the current attack, other known diseases, applied drugs and contraceptives, the relation of migraine attacks to the menstrual cycle, the presence of allodynia, attack frequency, duration of migraine, severity of pain during attacks as measured on a visual analogue scale, co-morbidities with other chronic diseases, the familial manifestation of migraine, regular sport activity and the time of the last meal were recorded, as described earlier [11]. Besides, patients were examined by Hamilton’s depression scale to exclude participants with an assessment of depression. There were no restrictions regarding food and drink intake. Blood samples were collected from migraine sufferers in an attack-free period and during an attack. Healthy controls were screened for non-reported/non-treated headaches and matched all demographic characteristics.
Sample collection
Human blood samples were collected from participants, after which processing started within 1 h. Tubes containing anticoagulant EDTA (ethylenediaminetetraacetic acid) were used for sample collection. PBMCs were isolated using Ficoll-Paque PREMIUM (GE Healthcare, Budapest, Hungary) according to the manufacturer's instructions, as previously described [11]. After removing the liquid phase, cells were resuspended with 1 ml of TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) and stored at -80 °C until further investigations.
RNA extraction and quality control
Total RNA was extracted by applying the phenol–chloroform-based TRI Reagent procedure (Molecular Research Center, Cincinnati, OH, USA), followed by extraction and purification using the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA, USA). The manufacturer’s protocol was followed, including the optional on-column DNase digestion, as described earlier [11, 32]. RNA concentrations were measured with Qubit 3.0 (Invitrogen, Carlsbad, CA, USA), and quality was checked on TapeStation 4200 using RNA ScreenTape (Agilent Technologies, Santa Clara, CA, USA). High-quality (RIN > 8) RNA samples were further used for library preparation.
Small RNA library preparation and sequencing
Small RNA Libraries were generated using the SmallRNA-Seq Library Prep Kit (Lexogen, Vienna, Austria). Briefly, 200 ng of total RNA was used as input, and 3’ and 5’ adapters were ligated, followed by column purifications. Thereafter, the ligation products were reverse transcribed, and the input RNA, flanked by 5’ and 3’ adapters, were converted into cDNA. Finally, the libraries were amplified and barcoded using 16 cycles of PCR. All libraries were assessed on the TapeStation 4200 (Agilent Technologies, Santa Clara, CA, USA) to examine if adapter dimers formed during PCR. The libraries were sequenced on an Illumina NextSeq550 platform generating 75 bp single-end reads. All RNA-Seq datasets generated as part of this study will be publicly available at the European Nucleotide Archive (https://www.ebi.ac.uk/ena), under accession number PRJEB46142. A total of 37 blood samples were used for PBMC small RNA sequencing.
Bioinformatics – miRNA-Seq, miRNA target prediction, and miRNA-mRNA target network analysis
The sequencing reads were aligned against the Homo sapiens reference genome (GRCh37 Ensembl release) with STAR v2.5.3a [33]. After alignment, the reads were associated with known protein-coding genes, and the number of reads aligned within each gene was counted using Rsubread package v2.0.0 [34]. Gene count data were normalized using the TMM (trimmed mean of M values) normalization method of the edgeR R/Bioconductor package (v3.28, R v3.6.0, Bioconductor v3.9) [35]. The data were further log transformed using the voom approach [36] in the limma package [37] for statistical testing. Normalized counts were represented as TPM (transcripts per million) values. FC (fold change) values between the compared groups resulting from the linear modelling process and modified t-test p-values were produced by the limma package. The Benjamini–Hochberg method was used to control the FDR (False Discovery Rate), and adjusted p-values were calculated by limma. In the case of paired ictal and interictal samples, the correlation between samples originating from the same patient was taken into account using the duplicateCorrelation function of limma.
After statistical filtering for DE (differentially expressed) miRNAs in each comparison (|FC|> 1.2 and p < 0.05), the DIANA-miRPath v3.0 web tool [38] was used to perform GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis (p < 0.01) using the microTCDS predicted targets of the DE miRNA list for each comparison with default settings, accessed on November 12th, 2021. BP (biological process) ontology GO results were further filtered for redundant terms using Revigo [39], with default settings accessed on November 14th, 2021. To highlight specific terms, GO BP terms with frequency values less than 0.15 were taken into account, where the frequency is defined as the proportion of gene products annotated by the selected GO term in the European Bioinformatics Institute Gene Ontology Annotation database for human. Higher frequency value implies more general terms, and lower implies more specific ones.
The network theoretical miRNAtarget.com™ software (mirnatarget.com; Pharmahungary, Szeged, Hungary) was used to predict target genes and their expected expression changes based on the lists of DE miRNAs. For this analysis, similarly to previous works [18, 19, 40,41,42,43], data from the manually curated, experimentally validated miRTarBase [44] (v7.0), the predicted miRDB [45] (v5.0 with score > 80.0) and microRNA.org [46] (release of August 2010 with mirSVR score < -1.2), miRNA-target interaction databases were integrated by miRNAtarget. Positive and negative edge weights (1, -1) in the network, referring to predicted interaction with up- and downregulated miRNAs, respectively, were summed to calculate node strength values, which estimate the extent of the predicted expression change of the targets. To achieve adequate visual representation of the information content of our networks, we used the EntOptLayout plugin [47] (version 2.1) for the Cytoscape [48] (version 3.7.2) software and further improved the visualisation by graphically highlighting node strength values. To elucidate the role of predicted target genes in biological processes, GO enrichment analysis [49, 50] was performed for each comparison, separately for up-and downregulated targets. The online PANTHER Overrepresentation Test ([51], version released on July 28th, 2020) was used with default settings (Fisher’s Exact test with FDR calculation). From the GO Ontology Database ([52], version released on October 9th, 2020) Homo sapiens “GO biological process complete” annotation dataset was used for the analysis.
Validation of predictions at mRNA level based on RNA sequencing data
To validate the small RNA sequencing-based miRNA-target predictions at the mRNA level, we used mRNA-Seq data from our previous study [11]. We included samples from the same cohort mentioned above in the present investigation with minimal changes to ensure a more homogenous patient group. We validated the in silico predicted miRNA-target interactions at the mRNA level by matching the predicted mRNAs from the present study to the differentially expressed mRNAs from our previous study cited above. We compared the predicted node strength values to the mRNA binary logarithm of the FC values with respect to the direction of changes. We considered genes to be validated that showed expression change consistent with the predictions.
Statistical analysis
The demographic and clinical characteristics of the studied population are presented in Table 1. Numerical data were summarized with their arithmetic means and 95% confidence intervals. Continuous parameters were tested for normality with Shapiro–Wilk’s tests and visual inspections. Homogeneity of variance across groups was studied using Levene's tests. If the resulting p-values from these two tests were less than the significance level of 0.05, non-parametric tests were used for response variables. The differences between the mean values of each parameter and its distribution within the study population (migraineurs and healthy) were tested using the non-parametric Kruskal–Wallis rank-sum test or ANOVA (analysis of variance), if applicable followed by Benjamini–Hochberg multiple-testing correction. To compare the parameters between the migraineurs group and its two phases: interictal and ictal, paired-samples Wilcoxon test (or paired t-test, if applicable) was used. The statistical significance level was set at 0.05 for all two-sided tests. The data were analysed using R version 4.0.2.
Results
Clinical characteristics of the patient population
The patients’ demographic and main clinical features for the total number of 28 participants are provided in Table 1. The interictal group consists of 16 migraine patients, with blood sampling in a headache-free period. Ictal samples were collected from 8 of them for self-control comparison. The 12 control samples consisted of healthy volunteers. Demographic characteristics were similar across the subgroups. No significant alterations were found when comparing different subgroups' demographic and clinical characteristics, e.g. age, attack frequency, pain severity, etc. The only significant difference between the interictal and ictal groups was the number of attacks in the previous month and the date of the last attack before sampling (Table 1).
microRNA signatures of PBMC samples
When interictal PBMC samples were compared to healthy ones, 31 miRNAs were DE, when significance was tested against a fold change threshold of 1.2 and a p-value threshold of 0.05. 14 miRNAs were upregulated, and 17 were downregulated. Based on the average of fold change and p-value ranks (average rank), hsa-miR-5189-3p, hsa-miR-96-5p, hsa-miR-3613-5p, hsa-miR-99a-3p, and hsa-miR-542-3p were found at the top of the DE miRNA list (Table 2; for further details please see Table S1).
In ictal vs interictal comparison, 25 miRNAs were DE (significance was tested against fold change 1.2 and p-value: 0.05), 15 were upregulated, and 10 were downregulated. Top 5 miRNAs were as follows: hsa-miR-3202, hsa-miR-7855-5p, hsa-miR-6770-3p, hsa-miR-1538, hsa-miR-409-5p (Table 3; for further details please see Table S2).
In ictal samples compared to the healthy group, 31 miRNAs were DE (fold change: 1.2, p-value: 0.05), 22 were upregulated, 9 were downregulated, hsa-miR-1277-5p being at the top of the list (Table S3).
DE miRNAs were visualised on heat maps (Figs. 1 and 2), and samples were clustered based on correlation. They were partitioned into major groups, largely overlapping with the original patient groups. DE miRNAs for ictal vs. healthy interaction were represented on a heat map in Figure S1.

Heat map representation of differentially expressed genes in the interictal vs healthy PBMC comparison. Columns represent samples, and rows represent genes. Pearson correlation was respectively calculated between samples and genes, visualised by dendrograms

Heat map representation of differentially expressed genes in the ictal vs interictal comparison. Columns represent samples, and rows represent genes. Pearson correlation was respectively calculated between samples and genes, visualised by dendrograms. Samples from patient “A” in different ictal and interictal periods are marked with respective colors
Functional enrichment analysis of DE miRNAs (DE list enrichment) was carried out. GO, and KEGG analysis was performed to link information with functions and utilities of the biological system (Tables 4 and 5). Interictal samples compared to healthy ones show a relation to several GO categories (pathways and processes), including several toll-like receptor signalling pathways, platelet degranulation, cellular response to glucagon stimulus, and alpha-linolenic acid metabolic process.
In the ictal PBMC samples compared to the interictal group, several toll-like receptor signalling pathways and positive regulation of protein insertion into the mitochondrial membrane involved in the apoptotic signalling pathway were altered. Interestingly, regulation of rhodopsin mediated signalling pathway and phototransduction were significantly affected.
KEGG pathway mapping revealed ECM (extracellular matrix)—receptor interaction and GABAergic synapse involvement, characterising interictal samples compared to control samples. TGF-beta) Transforming growth factor β and ErbB (epidermal growth factor receptor) signalling pathways appeared in interictal vs healthy and ictal vs interictal comparisons.
Predicted targets with the highest absolute node strength values in interictal vs healthy comparison
7012 mRNA targets of the 31 DE miRNAs were predicted when comparing interictal with healthy control samples. Predicted targets with the highest absolute node strength values are enlisted in Table 6. and the intersect of predicted targets and DE mRNAs in S4. As compared to the control group, targets like CADM2 (cell adhesion molecule 2), PLEKHM3 (pleckstrin homology domain-containing M3), MEF2C (myocyte enhancer factor 2C), BBX (BBX high mobility group box domain containing), ribosomal modification protein RIMKLB (rimK like family member B) and HACE1 (HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1) were significantly downregulated. In contrast, CCN T2 (cyclin T2) and KLHL15 (kelch like family member 15) showed significant upregulation in the interictal group (Table 5). DE miRNAs with their predicted interactions are shown in Fig. 3.

Visualisation of miRNA-mRNA interaction network (EntOptLayout) and target prediction analysis by miRNAtarget.com™ (interictal vs healthy). Rectangle and oval-shaped nodes represent miRNAs and mRNA targets of miRNAs, respectively. The node size and colour intensity of mRNA targets change according to node strength values. Down (blue)- and upregulated (red) interacting miRNAs suggest a central role of upregulated (orange) cyclin T2 (CCNT2) and kelch like family member 15 (KLHL15) and downregulated (light-blue) cell adhesion molecule 2 (CADM2) mRNAs. Whole predicted miRNA-target interaction network is shown on panel A. To highlight important mRNA targets, targets with an absolute node strength value less than or equal to 1 (i.e. -1, 0 or 1) presented uniformly smaller and fainter. On panel B, a subnetwork of the whole predicted miRNA-target interaction network is shown, containing only those target mRNAs (marked with a red oval on panel A) and their interacting miRNAs that were validated by RNA sequencing. The same arrangement as in the whole network with a proportional magnification of the target mRNAs was applied
Predicted targets with the highest absolute node strength values in ictal vs interictal comparison
In ictal PBMC samples compared to the interictal subgroup, 5665 mRNAs were predicted to be regulated by the 25 significantly DE miRNAs. Among the targets with the highest absolute node strength values, NR3C1 (nuclear receptor subfamily 3 group C member 1) appears with a strength value of 7, GRIA2 (glutamate ionotropic receptor AMPA type subunit 2), and MLLT3 (MLLT3 super elongation complex subunit) with a strength value of 6. 32 upregulated targets with a node strength of -2, like FN1 (Fibronectin 1) and CBX5 (Chromobox 5) were detected in ictal-interictal comparison (Table 6; the intersect of predicted targets and DE mRNAs in S5). DE miRNAs with their predicted interactions are shown in Fig. 4.

Visualisation of miRNA-mRNA interaction network (EntOptLayout) and target prediction analysis (ictal vs interictal) by miRNAtarget.com™. Rectangle and oval-shaped nodes represent miRNAs and mRNA targets of miRNAs, respectively. The node size and colour intensity of mRNA targets change according to node strength values. Down (blue)- and upregulated (red) miRNAs, suggesting a central role of downregulated (light blue) nuclear receptor subfamily 3 group C member 1 (NR3C1) and multiple upregulated (orange) mRNAs. Whole predicted miRNA-target interaction network is shown on panel A. To highlight important mRNA targets, targets with an absolute node strength value less than or equal to 1 (i.e. -1, 0 or 1) presented uniformly smaller and fainter. On panel B, a subnetwork of the whole predicted miRNA-target interaction network is shown, containing only those target mRNAs (marked with a red oval on panel A) and their interacting miRNAs that were validated by RNA sequencing. The same arrangement as in the whole network with a proportional magnification of the target mRNAs was applied
In addition, the intersect of 6781 predicted targets 31 DE miRNAs data in ictal vs healthy comparison and DE mRNAs are enlisted in Table S6. The visualisation of DE miRNAs with their predicted interactions in ictal vs. healthy interaction can be found in Figure S2.
Gene Ontology enrichment analysis of predicted mRNAs
To explore biological processes modified by migraine in headache-free (interictal) period and during headache (ictal), GO enrichment analyses of all predicted mRNAs were performed. The result of the GO enrichment analysis showed that DE miRNA-targeted mRNAs were significantly associated with several metabolic regulation processes and negative regulation of platelet activation when the interictal group was compared to healthy samples. Besides, regulation of nuclear-transcribed mRNA catabolic process, neuron development, and neuron differentiation seem to be affected by migraine (Table 7).
Validation of predicted mRNA targets miRNAs based on mRNA sequencing data
In interictal PBMC samples compared to healthy samples, out of 7012 predicted targets, 54 can also be found in DE mRNA data. In this comparison, 31 targets showed expression changes in the predicted direction, like TNF (tumor necrosis factor), SOCS3 (suppressor of cytokine signalling 3), numerous chemokines such as IL6 (interleukin-6), and SOD2 (superoxide dismutase 2), with a role in oxidative phosphorylation (Table 8). Of 5665 predicted targets, 26 can also be found in DE mRNA data in the ictal-interictal analysis. In this settlement, 12 targets showed expression changes in the predicted direction, e.g., RAB3B. (Ras-related protein Rab-3B), LRRTM2 (Leucine-Rich Repeat Transmembrane Neuronal 2) (Table 9).
Discussion
The present results obtained from a self-controlled study design provide the first evidence for disease and headache-specific miRNA patterns in PBMC of migraineurs. Our findings can help to identify key elements of the pathophysiological pathways, determine potential novel diagnostic and prognostic biomarkers and drug targets. The discovery of the small non-coding RNAs [25], together with the use of PBMCs, has offered new opportunities in this area [10].
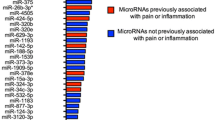
In the interictal PBMC samples, 31 miRNAs were DE compared to healthy controls, including hsa-miR-5189-3p, hsa-miR-96-5p, hsa-miR-3613-5p, hsa-miR-99a-3p, and hsa-miR-542-3p. hsa-miR-5189-3p closely correlates with spinal cord injury and is part of a set of transcripts proposed to have a role in classifying the disease [85]. The miR-96 and its miR-183 family have been observed to be regulated in diverse regions of the somatosensory nociceptive pathway in different chronic pain conditions in rodents [86]. The miR-183 cluster is distinctly enriched in sensory organs. It may influence the neuronal changes associated with the development and maintenance of chronic pain conditions by coordinately regulating multiple diverse nociceptive genes [87]. Circulating hsa-miR-3613-5p increased in severe axial pain after accident [88] and decreased in endometriosis [89, 90]. Serum hsa-miR-542-3p was found to be a part of miRNA signatures for endometriosis diagnosis [91]. The hsa-miR-99a-3p was downregulated in salivary exosomal miRNAs of burning mouth syndrome patients [92].
Our results revealed GABAergic synaptic changes in both interictal and ictal samples of migraineurs compared to healthy controls. The abnormalities of GABAergic signalling in migraine pathophysiology have been widely investigated in the literature [93]. Serum miRNA results also have suggested that targeting the GABA system might have therapeutical relevance [27]. Interestingly, but not surprisingly, the rhodopsin-mediated signalling pathway and phototransduction were involved as attack specific alterations (ictal-interictal comparison). Photophobia is a light-induced phenomenon linked with migraine and intensifying headache pain [94]. Although migraine research primarily focuses on the affected melanopsin-related pathways [95], it is clear that abnormal light processing is present in these patients. However, the reflection of these alterations in peripheral blood samples could be an exciting question in further research.
Similar to our PBMC results, differentially expressed miR-155 and let-7 g levels related to endothelial functions were described in migraineurs’ serum samples during ictal and interictal periods [96]. Another member of the let-7 family, let-7b, was found to be downregulated in migraine patients’ PBMC samples [28].
The top DE miRNAs during headache attacks compared to the self-control interictal samples were the following: hsa-miR-3202, hsa-miR-7855-5p, hsa-miR-6770-3p, hsa-miR-1538, hsa-miR-409-5p. Among exosomal miRNAs, hsa-miR-3202 was dysregulated in mild traumatic brain disorders [97], and hsa-miR-409-5p was DE in complex regional pain syndrome [98]. The latest was downregulated after contusion spinal cord injury, while its overexpression could promote recovery [91]. hsa-miR-7855-5p is known to be hypoxia-responsive [99], suggesting a relation to oxidative stress, and hsa-miR-1538 modifies cell response to oxidative stress [100]. hsa-miR-6770-3p decreased in chronic periodontitis as a potential biomarker [92]
In both comparisons, the list of top predicted targets with the highest absolute node strength value consists of a few mRNAs linked to the pathophysiological mechanisms of migraine. GRIA2 and NR3C1, involved in glutamate and glucocorticoid signalling, respectively, appear to be specific to the headache phase in our data set. Our results are supported by the demonstration of glutamatergic neurotransmission changes in trigeminovascular activation and central sensitisation [101, 102]. However, a case–control study did not find an association between migraine and GRIA2 polymorphisms [103]. DNA methylation of the NR3C1 gene is the focus of epigenetic literature [104, 105]. GWAS studies link the transcription factor MEF2, which was downregulated in our study, to neuroinflammatory processes [106], epilepsy [107], psychological and metabolic features [108, 109].
Pathway analysis pointed out TLR (toll-like receptor) signalling pathways involved in both the headache-free state and headache. TLRs are transmembrane receptors playing a crucial role in innate immune response and inflammation, which activate the NF-κB and interferon regulatory factors resulting in proinflammatory cytokine production like TNFα, IL1, IL6, and IL12. Altered interleukin and TNFα levels in migraine have been demonstrated in the literature [110,111,112]. Our miRNA target prediction results are in line with these data. IL-6 and TNFα were validated by comparing to mRNA levels demonstrated in our previous paper [11]. Although the involvement of TLR4 signalling in initiating and maintaining migraine-like behaviour in mice and inducing hyperalgesia in rats has been proposed [113,114,115,116,117], this is the first study that links the TLR pathway specifically to migraineurs. In addition, there is evidence suggesting a strong TLR-4/miRNA interplay, which might be a possible target for modern immunotherapy [118].
Among validated targets describing the migraine disease in the attack-free period, EGR1 (early growth response gene 1) was enlisted, which can regulate multiple aspects of synaptic plasticity [119] [120]. Several validated targets line up as regulators and participants of the immune system and inflammatory pathways, like NFKBIA (NFKB inhibitor alpha) and IER3 (early response 3) [121, 122]. TNFAIP6 (TNF Alpha Induced Protein 6) might be a potential miRNA target with anti-inflammatory properties [59]. NR4A2 (Nuclear Receptor Subfamily 4 Group A Member 2), through the regulation of various signals, inhibits the expression of proinflammatory mediators and plays a neuroprotective role [63]. EGR3 (Early Growth Response 3) is essential for controlling inflammation and antigen-induced immune cell proliferation [123]; it suppresses SOCS1, SOCS3 (cytokine signalling-1 and 3), regulating cytokine or hormone signalling. The enzyme encoded by the SAT1 (Spermidine/Spermine N1-Acetyltransferase 1) gene catalyses the acetylation of spermidine and spermine, thus mediating polyamine metabolism. This data underlines our previous findings [11], where we described alterations in spermin and spermidine metabolites in migraineurs plasma samples. The role of oxidative stress and mitochondrial dysfunction in migraine susceptibility and headache generation has also been demonstrated in our recent work [11]. Sgk (serum- and glucocorticoid-inducible protein kinase) [124] and SOD (superoxide dismutase) [125] increased in the interictal samples when compared to controls, underlining the importance of these pathways. In addition, the ictal vs interictal comparison enlisted downregulation of the transmembrane signalling enzyme NOX5 (NADPH oxidase 5) [119].
Conclusions
We provide here the first miRNA profile of migraineurs in headache-free periods and during attacks compared to healthy controls suggesting potential disease- and pain-specific pathophysiological mechanisms. Predicted mRNA targets of differentially expressed miRNAs confirmed and validated by the transcriptomics analysis reveal the involvement of inflammatory and immune mechanisms, cytokine and chemokine signalling, and oxidative stress.
Availability of data and materials
All RNA-Seq data sets generated as part of this study will be publicly available at the European Nucleotide Archive (https://www.ebi.ac.uk/ena), under accession number PRJEB46142.
Abbreviations
- ANOVA:
-
Analysis of variance
- BMI body:
-
Mass index
- CGRP:
-
Calcitonin gene-related peptide
- DE:
-
Differentially expressed
- EDTA:
-
Ethylenediaminetetraacetic acid
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GO:
-
Gene Ontology
- GWAS:
-
Genome-wide association studies
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PACAP:
-
Pituitary adenylate cyclase-activating polypeptide
- PBMC:
-
Peripheral blood mononuclear cells
- TNF:
-
Tumor necrosis factor
- VAS:
-
Visual analogue scale
References
Bernstein C, Burstein R (2012) Sensitization of the Trigeminovascular Pathway: Perspective and Implications to Migraine Pathophysiology. J Clin Neurol 8:89. https://doi.org/10.3988/jcn.2012.8.2.89
Bruno PP, Carpino F, Carpino G, Zicari A (2007) An overview on immune system and migraine. Eur Rev Med Pharmacol Sci 11:245–248
Conti P, D’Ovidio C, Conti C, Gallenga CE, Lauritano D, Caraffa A et al (2019) Progression in migraine: Role of mast cells and pro-inflammatory and anti-inflammatory cytokines. Eur J Pharmacol 844:87–94. https://doi.org/10.1016/j.ejphar.2018.12.004
Tajti J, Tuka B, Botz B, Helyes Z, Vecsei L (2015) Role of pituitary adenylate cyclase-activating polypeptide in nociception and migraine. CNS Neurol Disord Drug Targets 14:540–553. https://doi.org/10.2174/1871527314666150429114234
Tuka B, Szabó N, Tóth E, Kincses ZT, Párdutz Á, Szok D et al (2016) Release of PACAP-38 in episodic cluster headache patients - an exploratory study. J Headache Pain 17:69. https://doi.org/10.1186/s10194-016-0660-7
Edvinsson L, Haanes KA, Warfvinge K (2019) Does inflammation have a role in migraine? Nat Rev Neurol 15:483–490. https://doi.org/10.1038/s41582-019-0216-y
Lisicki M, D’Ostilio K, Erpicum M, Schoenen J, Magis D (2018) Sunlight irradiance and habituation of visual evoked potentials in migraine: The environment makes its mark. Cephalalgia 38:1351–1360. https://doi.org/10.1177/0333102417730128
Rollins B, Martin MV, Morgan L, Vawter MP (2010) Analysis of whole genome biomarker expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet 153B:919–936. https://doi.org/10.1002/ajmg.b.31062
Sullivan PF, Fan C, Perou CM (2006) Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet 141B:261–268. https://doi.org/10.1002/ajmg.b.30272
Mosallaei M, Ehtesham N, Rahimirad S, Saghi M, Vatandoost N, Khosravi S (2020) PBMCs: a new source of diagnostic and prognostic biomarkers. Arch Physiol Biochem 0:1–7. https://doi.org/10.1080/13813455.2020.1752257
Aczél T, Körtési T, Kun J, Urbán P, Bauer W, Herczeg R et al (2021) Identification of disease- and headache-specific mediators and pathways in migraine using blood transcriptomic and metabolomic analysis. J Headache Pain 22:117. https://doi.org/10.1186/s10194-021-01285-9
Bär C, Thum T, de Gonzalo-Calvo D (2019) Circulating miRNAs as mediators in cell-to-cell communication. Epigenomics 11:111–113. https://doi.org/10.2217/epi-2018-0183
Velu VK, Ramesh R, Srinivasan AR (2012) Circulating MicroRNAs as Biomarkers in Health and Disease. J Clin Diagn Res 6:1791–1795. https://doi.org/10.7860/JCDR/2012/4901.2653
Witkos TM, Koscianska E, Krzyzosiak WJ (2011) Practical Aspects of microRNA Target Prediction. Curr Mol Med 11:93–109. https://doi.org/10.2174/156652411794859250
Vrijens K, Bollati V, Nawrot TS (2015) MicroRNAs as Potential Signatures of Environmental Exposure or Effect: A Systematic Review. Environ Health Perspect 123:399–411. https://doi.org/10.1289/ehp.1408459
Hanna J, Hossain GS, Kocerha J (2019) The Potential for microRNA Therapeutics and Clinical Research. Front Genet 10:478. https://doi.org/10.3389/fgene.2019.00478
Perrino C, Barabási A-L, Condorelli G, Davidson SM, De Windt L, Dimmeler S et al (2017) Epigenomic and transcriptomic approaches in the post-genomic era: path to novel targets for diagnosis and therapy of the ischaemic heart? Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc Res 113:725–736. https://doi.org/10.1093/cvr/cvx070
Makkos A, Ágg B, Petrovich B, Varga ZV, Görbe A, Ferdinandy P (2021) Systematic review and network analysis of microRNAs involved in cardioprotection against myocardial ischemia/reperfusion injury and infarction: Involvement of redox signalling. Free Radic Biol Med 172:237–251. https://doi.org/10.1016/j.freeradbiomed.2021.04.034
Bencsik P, Kiss K, Ágg B, Baán JA, Ágoston G, Varga A et al (2019) Sensory Neuropathy Affects Cardiac miRNA Expression Network Targeting IGF-1, SLC2a-12, EIF-4e, and ULK-2 mRNAs. Int J Mol Sci 20:E991. https://doi.org/10.3390/ijms20040991
Kress M, Hüttenhofer A, Landry M, Kuner R, Favereaux A, Greenberg DS et al (2013) microRNAs in nociceptive circuits as predictors of future clinical applications. Front Mol Neurosci 6:33. https://doi.org/10.3389/fnmol.2013.00033
Andersen HH, Duroux M, Gazerani P (2014) MicroRNAs as modulators and biomarkers of inflammatory and neuropathic pain conditions. Neurobiol Dis 71:159–168. https://doi.org/10.1016/j.nbd.2014.08.003
López-González MJ, Landry M, Favereaux A (2017) MicroRNA and chronic pain: From mechanisms to therapeutic potential. Pharmacol Ther 180:1–15. https://doi.org/10.1016/j.pharmthera.2017.06.001
Cheng Y, Dong L, Zhang J, Zhao Y, Li Z (2018) Recent advances in microRNA detection. Analyst 143:1758–1774. https://doi.org/10.1039/C7AN02001E
Benesova S, Kubista M, Valihrach L (2021) Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 11:964. https://doi.org/10.3390/diagnostics11060964
Gazerani P (2019) Current Evidence on Potential Uses of MicroRNA Biomarkers for Migraine: From Diagnosis to Treatment. Mol Diagn Ther 23:681–694. https://doi.org/10.1007/s40291-019-00428-8
Ahmad L, Demartini C, Corrado M, Vaghi G, Piella EM, Allena M et al (2021) Expression of Selected microRNAs in Migraine: A New Class of Possible Biomarkers of Disease? Processes 9:2199. https://doi.org/10.3390/pr9122199
Andersen HH, Duroux M, Gazerani P (2016) Serum MicroRNA Signatures in Migraineurs During Attacks and in Pain-Free Periods. Mol Neurobiol 53:1494–1500. https://doi.org/10.1007/s12035-015-9106-5
Tafuri E, Santovito D, de Nardis V, Marcantonio P, Paganelli C, Affaitati G et al (2015) MicroRNA profiling in migraine without aura: Pilot study. Ann Med 47:468–473. https://doi.org/10.3109/07853890.2015.1071871
Vila-Pueyo M, Fernández-Morales J, Torres-Ferrus M, Álvarez-Sabin J, Pozo-Rosich P (2014) EHMTI-0361. Lack of differences in microrna expression profiles of blood cells in migraine. J Headache Pain 15:H3. https://doi.org/10.1186/1129-2377-15-S1-H3
Pozo-Rosich P (2022) microRNA expression profile in migraine: the microMIG study. Available: https://www.migrainedisorders.org/wp-content/uploads/2019/09/microMIG-MRF-report-2019.pdf [cited 28 Mar 2022].
Headache Classification Committee of the International HeadacheSociety (IHS) (2018) The international classification of headache disorders, 3rd edition. Cephalalgia 38:1–211. https://doi.org/10.1177/0333102417738202
Aczél T, Kecskés A, Kun J, Szenthe K, Bánáti F, Szathmary S et al (2020) Hemokinin-1 Gene Expression Is Upregulated in Trigeminal Ganglia in an Inflammatory Orofacial Pain Model: Potential Role in Peripheral Sensitization. Int J Mol Sci 21(8):2938. https://doi.org/10.3390/ijms21082938
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Liao Y, Smyth GK, Shi W (2019) The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47:e47. https://doi.org/10.1093/nar/gkz114
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15:R29. https://doi.org/10.1186/gb-2014-15-2-r29
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. https://doi.org/10.1093/nar/gkv007
Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T et al (2015) DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res 43:W460–W466. https://doi.org/10.1093/nar/gkv403
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6:e21800. https://doi.org/10.1371/journal.pone.0021800
Ágg B, Baranyai T, Makkos A, Vető B, Faragó N, Zvara Á et al (2018) MicroRNA interactome analysis predicts post-transcriptional regulation of ADRB2 and PPP3R1 in the hypercholesterolemic myocardium. Sci Rep 8:10134. https://doi.org/10.1038/s41598-018-27740-3
Schreckenberg R, Klein J, Kutsche HS, Schulz R, Gömöri K, Bencsik P et al (2020) Ischaemic post-conditioning in rats: responder and non-responder differ in transcriptome of mitochondrial proteins. J Cell Mol Med 24:5528–5541. https://doi.org/10.1111/jcmm.15209
Huang C, Deng H, Zhao W, Xian L (2021) Knockdown of miR-384-3p protects against myocardial ischemia-reperfusion injury in rats through targeting HSP70. Heart Surg Forum 24:E143–E150. https://doi.org/10.1532/hsf.3449
Makkos A, Ágg B, Varga ZV, Giricz Z, Gyöngyösi M, Lukovic D et al (2021) Molecular network approach reveals rictor as a central target of cardiac protectomiRs. Int J Mol Sci 22:9539. https://doi.org/10.3390/ijms22179539
Huang H-Y, Lin Y-C-D, Li J, Huang K-Y, Shrestha S, Hong H-C et al (2020) miRTarBase 2020 updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res 48:D148–D154. https://doi.org/10.1093/nar/gkz896
Chen Y, Wang X (2020) miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 48:D127–D131. https://doi.org/10.1093/nar/gkz757
Betel D, Wilson M, Gabow A, Marks DS, Sander C (2008) The microRNA.org resource: targets and expression. Nucleic Acids Res 36:D149-153. https://doi.org/10.1093/nar/gkm995
Ágg B, Császár A, Szalay-Bekő M, Veres DV, Mizsei R, Ferdinandy P et al (2019) The EntOptLayout Cytoscape plug-in for the efficient visualization of major protein complexes in protein-protein interaction and signalling networks. Bioinformatics 35:4490–4492. https://doi.org/10.1093/bioinformatics/btz257
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al (2000) Gene ontology: tool for the unification of biology. Gene Ontol Consortium Nat Genet 25:25–29. https://doi.org/10.1038/75556
Gene Ontology Consortium (2021) The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49:D325–D334. https://doi.org/10.1093/nar/gkaa1113
Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47:D419–D426. https://doi.org/10.1093/nar/gky1038
Carbon S, Mungall C. Gene Ontology Data Archive. Zenodo. 2018. https://doi.org/10.5281/zenodo.4081749
Human Pain Genetics Database (HPGDB). Available: https://humanpaingenetics.org/hpgdb/ [cited 15 Jul 2021].
Pain Gene Resource | Pain Research Forum. Available: https://www.painresearchforum.org/resources/pain-gene-resource [cited 15 Jul 2021].
Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, Ronzano F, Centeno E, Sanz F et al (2020) The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res 48:D845–D855. https://doi.org/10.1093/nar/gkz1021
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Current Protocols Bioinformatics. 2016;54:1.30.1–1.30.33. doi:https://doi.org/10.1002/cpbi.5
Pazmandi K, Agod Z, Kumar BV, Szabo A, Fekete T, Sogor V et al (2014) Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radical Biol Med 77:281–290. https://doi.org/10.1016/j.freeradbiomed.2014.09.028
McGarry T, Biniecka M, Veale DJ, Fearon U (2018) Hypoxia, oxidative stress and inflammation. Free Radical Biol Med 125:15–24. https://doi.org/10.1016/j.freeradbiomed.2018.03.042
Day AJ, Milner CM (2019) TSG-6: A multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol 78–79:60–83. https://doi.org/10.1016/j.matbio.2018.01.011
Bensellam M, Montgomery MK, Luzuriaga J, Chan JY, Laybutt DR (2015) Inhibitor of differentiation proteins protect against oxidative stress by regulating the antioxidant–mitochondrial response in mouse beta cells. Diabetologia 58:758–770. https://doi.org/10.1007/s00125-015-3503-1
Nie H, Liu B, Yin C, Chen R, Wang J, Zeng D et al (2021) Gene Expression Profiling of Contralateral Dorsal Root Ganglia Associated with Mirror-Image Pain in a Rat Model of Complex Regional Pain Syndrome Type-I. J Pain Res 14:2739–2756. https://doi.org/10.2147/JPR.S322372
Guo Y-P, Zhi Y-R, Liu T-T, Wang Y, Zhang Y (2019) Global Gene Knockout of Kcnip3 Enhances Pain Sensitivity and Exacerbates Negative Emotions in Rats. Front Mol Neurosci 12:5. https://doi.org/10.3389/fnmol.2019.00005
Jakaria M, Haque ME, Cho D-Y, Azam S, Kim I-S, Choi D-K (2019) Molecular Insights into NR4A2(Nurr1): an Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death. Mol Neurobiol 56:5799–5814. https://doi.org/10.1007/s12035-019-1487-4
Doi Y, Oki S, Ozawa T, Hohjoh H, Miyake S, Yamamura T (2008) Orphan nuclear receptor NR4A2 expressed in T cells from multiple sclerosis mediates production of inflammatory cytokines. Proc Natl Acad Sci U S A 105:8381–8386. https://doi.org/10.1073/pnas.0803454105
Qiao YQ, Huang ML, Zheng Q, Wang TR, Xu AT, Cao Y et al (2015) CNTNAP3 Associated ATG16L1 Expression and Crohn’s Disease. Mediators Inflamm 2015:e404185. https://doi.org/10.1155/2015/404185
Rappaport N, Fishilevich S, Nudel R, Twik M, Belinky F, Plaschkes I et al (2017) Rational confederation of genes and diseases: NGS interpretation via GeneCards. MalaCards VarElect BioMed Eng OnLine 16:72. https://doi.org/10.1186/s12938-017-0359-2
Luis-Delgado OE, Barrot M, Rodeau J-L, Ulery PG, Freund-Mercier M-J, Lasbennes F (2006) The transcription factor ΔFosB is recruited by inflammatory pain. J Neurochem 98:1423–1431. https://doi.org/10.1111/j.1471-4159.2006.03970.x
Wang X, Jiang Y, Li J, Wang Y, Tian Y, Guo Q et al (2021) DUSP1 Promotes Microglial Polarization toward M2 Phenotype in the Medial Prefrontal Cortex of Neuropathic Pain Rats via Inhibition of MAPK Pathway. ACS Chem Neurosci 12:966–978. https://doi.org/10.1021/acschemneuro.0c00567
Liu L-P, Zhang J, Pu B, Li W-Q, Wang Y-S (2020) Upregulation of JHDM1D-AS1 alleviates neuroinflammation and neuronal injury via targeting miR-101-3p-DUSP1 in spinal cord after brachial plexus injury. Int Immunopharmacol 89:106962. https://doi.org/10.1016/j.intimp.2020.106962
Zahedi K, Brooks M, Barone S, Rahmati N, Murray Stewart T, Dunworth M et al (2020) Ablation of polyamine catabolic enzymes provokes Purkinje cell damage, neuroinflammation, and severe ataxia. J Neuroinflammation 17:301. https://doi.org/10.1186/s12974-020-01955-6
Gu J, Wei Y-L, Wei JCC, Huang F, Jan M-S, Centola M et al (2009) Identification of RGS1 as a candidate biomarker for undifferentiated spondylarthritis by genome-wide expression profiling and real-time polymerase chain reaction. Arthritis Rheum 60:3269–3279. https://doi.org/10.1002/art.24968
Leong MML, Cheung AKL, Kwok TCT, Lung ML (2020) Functional characterization of a candidate tumor suppressor gene, Mirror Image Polydactyly 1, in nasopharyngeal carcinoma. Int J Cancer 146:2891–2900. https://doi.org/10.1002/ijc.32732
Uluçkan Ö, Wagner EF (2016) Role of IL-17A signalling in psoriasis and associated bone loss. Clin Exp Rheumatol 34:17–20
Liu B, Li N, He Z, Zhang X, Duan G (2021) Emerging Role of Serum Glucocorticoid-Regulated Kinase 1 in Pathological Pain. Front Mol Neurosci 14:683527. https://doi.org/10.3389/fnmol.2021.683527
Niu L, Guo W, Song X, Song X, Xie L (2021) Tumor-educated leukocytes mRNA as a diagnostic biomarker for non-small cell lung cancer. Thorac Cancer 12:737–745. https://doi.org/10.1111/1759-7714.13833
Knyazev E, Maltseva D, Raygorodskaya M, Shkurnikov M (2021) HIF-Dependent NFATC1 Activation Upregulates ITGA5 and PLAUR in Intestinal Epithelium in Inflammatory Bowel Disease. Front Genet 12:791640. https://doi.org/10.3389/fgene.2021.791640
Wei W, Jiao Y, Postlethwaite A, Stuart JM, Wang Y, Sun D et al (2013) Dual-specificity phosphatases 2: surprising positive effect at the molecular level and a potential biomarker of diseases. Genes Immun 14:1–6. https://doi.org/10.1038/gene.2012.54
Barnicle A, Seoighe C, Greally JM, Golden A, Egan LJ (2017) Inflammation-associated DNA methylation patterns in epithelium of ulcerative colitis. Epigenetics 12:591–606. https://doi.org/10.1080/15592294.2017.1334023
Lee S, Xu L, Shin Y, Gardner L, Hartzes A, Dohan FC et al (2011) A potential link between autoimmunity and neurodegeneration in immune-mediated neurological disease. J Neuroimmunol 235:56–69. https://doi.org/10.1016/j.jneuroim.2011.02.007
Ko JS, Pramanik G, Um JW, Shim JS, Lee D, Kim KH et al (2015) PTPσ functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. PNAS 112:1874–1879. https://doi.org/10.1073/pnas.1410138112
Bhouri M, Morishita W, Temkin P, Goswami D, Kawabe H, Brose N et al (2018) Deletion of LRRTM1 and LRRTM2 in adult mice impairs basal AMPA receptor transmission and LTP in hippocampal CA1 pyramidal neurons. PNAS 115:E5382–E5389. https://doi.org/10.1073/pnas.1803280115
Touyz RM, Anagnostopoulou A, Rios F, Montezano AC, Camargo LL (2019) NOX5: Molecular biology and pathophysiology. Exp Physiol 104:605–616. https://doi.org/10.1113/EP086204
Khayer N, Mirzaie M, Marashi S-A, Jalessi M (2020) Rps27a might act as a controller of microglia activation in triggering neurodegenerative diseases. PLoS ONE 15:e0239219. https://doi.org/10.1371/journal.pone.0239219
Minster RL, Sanders JL, Singh J, Kammerer CM, Barmada MM, Matteini AM et al (2015) Genome-Wide Association Study and Linkage Analysis of the Healthy Aging Index. The Journals of Gerontology: Series A 70:1003–1008. https://doi.org/10.1093/gerona/glv006
Chen Q, Zhao Z, Yin G, Yang C, Wang D, Feng Z et al (2021) Identification and analysis of spinal cord injury subtypes using weighted gene co-expression network analysis. Ann Transl Med 9:466. https://doi.org/10.21037/atm-21-340
Bali KK, Kuner R (2014) Noncoding RNAs: key molecules in understanding and treating pain. Trends Mol Med 20:437–448. https://doi.org/10.1016/j.molmed.2014.05.006
Aldrich BT, Frakes EP, Kasuya J, Hammond DL, Kitamoto T (2009) Changes in expression of sensory organ-specific microRNAs in rat dorsal root ganglia in association with mechanical hypersensitivity induced by spinal nerve ligation. Neuroscience 164:711–723. https://doi.org/10.1016/j.neuroscience.2009.08.033
Linnstaedt SD, Walker MG, Parker JS, Yeh E, Sons RL, Zimny E, et al. MicroRNA Circulating in the Early Aftermath of Motor Vehicle Collision Predict Persistent Pain Development and Suggest a Role for microRNA in Sex-Specific Pain Differences. Mol Pain. 2015;11: s12990–015–0069–3. doi:https://doi.org/10.1186/s12990-015-0069-3
Borghese B, Zondervan KT, Abrao MS, Chapron C, Vaiman D (2017) Recent insights on the genetics and epigenetics of endometriosis. Clinical Genet 91:254–264. https://doi.org/10.1111/cge.12897
Adam Worku D. The Role of Biomarkers in the Early Diagnosis of Endometriosis. IGRWH. 2017;1. https://doi.org/10.31031/IGRWH.2017.01.000512
Wang W-T, Zhao Y-N, Han B-W, Hong S-J, Chen Y-Q (2013) Circulating MicroRNAs Identified in a Genome-Wide Serum MicroRNA Expression Analysis as Noninvasive Biomarkers for Endometriosis. J Clin Endocrinol Metab 98:281–289. https://doi.org/10.1210/jc.2012-2415
Kim K-Y, Byun J-S, Jung J-K, Choi J-K (2019) Profiling of Salivary Exosomal Micro RNAs in Burning Mouth Syndrome Patients. J Oral Med Pain 44:25–30. https://doi.org/10.14476/jomp.2019.44.1.25
D’Andrea G, Granella F, Cataldini M, Verdelli F, Balbi T (2001) GABA and glutamate in migraine. J Headache Pain 2:s57–s60. https://doi.org/10.1007/s101940170011
Noseda R, Copenhagen D, Burstein R (2019) Current understanding of photophobia, visual networks and headaches. Cephalalgia 39:1623–1634. https://doi.org/10.1177/0333102418784750
Zele AJ, Dey A, Adhikari P, Feigl B (2021) Melanopsin hypersensitivity dominates interictal photophobia in migraine. Cephalalgia 41:217–226. https://doi.org/10.1177/0333102420963850
Cheng C-Y, Chen S-P, Liao Y-C, Fuh J-L, Wang Y-F, Wang S-J (2018) Elevated circulating endothelial-specific microRNAs in migraine patients: a pilot study. Cephalalgia 38:1585–1591. https://doi.org/10.1177/0333102417742375
Devoto C, Lai C, Qu B-X, Guedes VA, Leete J, Wilde E et al (2020) Exosomal MicroRNAs in military personnel with mild traumatic brain injury: preliminary results from the chronic effects of neurotrauma consortium biomarker discovery project. J Neurotrauma 37:2482–2492. https://doi.org/10.1089/neu.2019.6933
McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R et al (2014) Functional significance of macrophage-derived exosomes in inflammation and pain. Pain 155:1527–1539. https://doi.org/10.1016/j.pain.2014.04.029
Shih C-H, Chuang L-L, Tsai M-H, Chen L-H, Chuang EY, Lu T-P et al (2021) Hypoxia-Induced MALAT1 Promotes the Proliferation and Migration of Breast Cancer Cells by Sponging MiR-3064–5p. Front Oncol 11:658151. https://doi.org/10.3389/fonc.2021.658151
Pallarès-Albanell J, Zomeño-Abellán MT, Escaramís G, Pantano L, Soriano A, Segura MF et al (2019) A High-Throughput Screening Identifies MicroRNA Inhibitors That Influence Neuronal Maintenance and/or Response to Oxidative Stress. Mol Ther Nucleic Acids 17:374–387. https://doi.org/10.1016/j.omtn.2019.06.007
Hoffmann J, Charles A (2018) Glutamate and Its Receptors as Therapeutic Targets for Migraine. Neurotherapeutics 15:361–370. https://doi.org/10.1007/s13311-018-0616-5
Ramadan NM (2003) The link between glutamate and migraine. CNS Spectr 8:446–449. https://doi.org/10.1017/s1092852900018757
Gasparini CF, Sutherland HG, Haupt LM, Griffiths LR (2014) Genetic Analysis of GRIA2 and GRIA4 Genes in Migraine. Headache J Head Face Pain 54:303–312. https://doi.org/10.1111/head.12234
Aroke EN, Joseph PV, Roy A, Overstreet DS, Tollefsbol TO, Vance DE et al (2019) Could epigenetics help explain racial disparities in chronic pain? J Pain Res 12:701–710. https://doi.org/10.2147/JPR.S191848
Berretta E, Guida E, Forni D, Provenzi L (2021) Glucocorticoid receptor gene (NR3C1) methylation during the first thousand days: Environmental exposures and developmental outcomes. Neurosci Biobehav Rev 125:493–502. https://doi.org/10.1016/j.neubiorev.2021.03.003
Zhang Z-G, Li Y, Ng CT, Song Y-Q (2015) Inflammation in Alzheimer’s Disease and Molecular Genetics: Recent Update. Arch Immunol Ther Exp (Warsz) 63:333–344. https://doi.org/10.1007/s00005-015-0351-0
Borlot F, Whitney R, Cohn RD, Weiss SK (2019) MEF2C-related epilepsy: Delineating the phenotypic spectrum from a novel mutation and literature review. Seizure 67:86–90. https://doi.org/10.1016/j.seizure.2019.03.015
Morris J, Bailey MES, Baldassarre D, Cullen B, de Faire U, Ferguson A et al (2019) Genetic variation in CADM2 as a link between psychological traits and obesity. Sci Rep 9:7339. https://doi.org/10.1038/s41598-019-43861-9
Yan X, Wang Z, Schmidt V, Gauert A, Willnow TE, Heinig M et al (2018) Cadm2 regulates body weight and energy homeostasis in mice. Molecular Metabolism 8:180–188. https://doi.org/10.1016/j.molmet.2017.11.010
Covelli V, Massari F, Fallacara C, Munno I, Pellegrino NM, Jirillo E et al (1991) Increased spontaneous release of tumor necrosis factor-α/cachectin in headache patients. A possible correlation with plasma endotoxin and hypothalamic-pituitary-adrenal axis. Int J Neurosci 61:53–60. https://doi.org/10.3109/00207459108986270
Perini F, D’andrea G, Galloni E, Pignatelli F, Billo G, Alba S et al (2005) Plasma cytokine levels in migraineurs and controls. Headache J Head Face Pain 45:926–931
Uzar E, Evliyaoglu O, Yucel Y, Cevik MU, Acar A, Guzel I, et al. (2011) Serum cytokine and pro-brain natriuretic peptide (BNP) levels in patients with migraine. Eur Rev Med Pharmacol Sci 15:1111-1116.
Ashayeri Ahmadabad R, Khaleghi Ghadiri M, Gorji A (2020) The role of Toll-like receptor signaling pathways in cerebrovascular disorders: the impact of spreading depolarization. J Neuroinflammation 17:108. https://doi.org/10.1186/s12974-020-01785-6
Bruno K, Woller SA, Miller YI, Yaksh TL, Wallace M, Beaton G et al (2018) Targeting Toll-like receptor-4 (TLR4) – emerging therapeutic target for persistent pain states. Pain 159:1908–1915. https://doi.org/10.1097/j.pain.0000000000001306
Lacagnina MJ, Watkins LR, Grace PM (2018) Toll-like receptors and their role in persistent pain. Pharmacol Ther 184:145–158. https://doi.org/10.1016/j.pharmthera.2017.10.006
Ramachandran R, Wang Z, Saavedra C, DiNardo A, Corr M, Powell SB et al (2019) Role of Toll-like receptor 4 signaling in mast cell-mediated migraine pain pathway. Mol Pain 15:1744806919867842. https://doi.org/10.1177/1744806919867842
Su M, Ran Y, He Z, Zhang M, Hu G, Tang W et al (2018) Inhibition of toll-like receptor 4 alleviates hyperalgesia induced by acute dural inflammation in experimental migraine. Mol Pain 14:1744806918754612. https://doi.org/10.1177/1744806918754612
Litak J, Grochowski C, Litak J, Osuchowska I, Gosik K, Radzikowska E et al (2020) TLR-4 Signaling vs. immune checkpoints, mirnas molecules, cancer stem cells, and wingless-signaling interplay in glioblastoma multiforme-future perspectives. Int J Mol Sci 21(9):3114. https://doi.org/10.3390/ijms21093114
Duclot F, Kabbaj M (2017) The Role of Early Growth Response 1 (EGR1) in brain plasticity and neuropsychiatric disorders. Front Behav Neurosci 11:35. https://doi.org/10.3389/fnbeh.2017.00035
Sosthenes MCK, Diniz DG, Roodselaar J, Abadie-Guedes R, de Siqueira Mendes FdCC, Fernandes TN et al (2019) Stereological Analysis of Early Gene Expression Using Egr-1 Immunolabeling After Spreading Depression in the Rat Somatosensory Cortex. Front Neurosci 13:1020. https://doi.org/10.3389/fnins.2019.01020
Arlt A, Schäfer H (2011) Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur J Cell Biol 90:545–552. https://doi.org/10.1016/j.ejcb.2010.10.002
Mitchell S, Vargas J, Hoffmann A (2016) Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med 8:227–241. https://doi.org/10.1002/wsbm.1331
Taefehshokr S, Key Y, Khakpour M, Dadebighlu P, Oveisi A (2017) Early growth response 2 and Egr3 are unique regulators in immune system. Central Eur J Immunol 42:205–209. https://doi.org/10.5114/ceji.2017.69363
Leong MLL, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL (2003) Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem 278:5871–5882. https://doi.org/10.1074/jbc.M211649200
Gross EC, Lisicki M, Fischer D, Sándor PS, Schoenen J (2019) The metabolic face of migraine - from pathophysiology to treatment. Nat Rev Neurol 15:627–643. https://doi.org/10.1038/s41582-019-0255-4
Acknowledgements
Not applicable
Funding
Open access funding provided by University of Pécs. This research was supported by National Brain Research Program 2017–1.2.1-NKP-2017–00002 (NAP-2; Chronic Pain Research Group), Gazdaságfejlesztési és Innovációs Operatív Program (Economy Development and Innovation Operative Programme) (GINOP)-2.3.2–15-2016–00050 (Peptidergic Signaling in Health and Disease; PEPSYS), GINOP 2.3.2–15-2016–0034, Emberi Erőforrás Operatív Program (Human Resource Operative Programme) (EFOP) 3.6.2–16-2017–00008 (2017–2019), EFOP-3.6.1–16-2016–00004, TUDFO/47138–1/2019-ITM and National Research Development and Innovation Office of Hungary (NKFIA) grant OTKA FK132587, OTKA-138125-K and NVKP-16–1-2016–0017 National Heart Program. This study was also financed by the Thematic Excellence Programme (2020–4.1.1.-TKP2020) of the Ministry for Innovation and Technology in Hungary, within the framework of the Therapeutic Development and Bioimaging thematic programmes of the Semmelweis University. A.G. and J.K. were supported by the grants GINOP-2.3.4–15-2020–00010, GINOP-2.3.1–20-2020–00001 and Educating Experts of the Future: Developing Bioinformatics and Biostatistics competencies of European Biomedical Students (BECOMING, 2019–1-HU01-KA203-061251). B.Á. was supported by the New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund (ÚNKP-20–4-I-SE-7, ÚNKP-21–4-II-SE-18). Bioinformatics infrastructure was supported by ELIXIR Hungary (http://elixir-hungary.org/).
Author information
Authors and Affiliations
Contributions
Conceptualisation: Z.H., K.B., J.K., A.G, L.V., J.T., P.F. Formal Analysis: J.K., B.Á., B.B., W.B., A.G. Investigation: A.T., B.B., Á.B., T.K., J.K., B.T., U.P. Resources: L.V, J.T., A.G., K.B., Z.H. Writing – Original Draft Preparation: A.T., B.B., Á.B., J.K., U.P., W.B., K.B., Z.H. Writing – Review and Editing: A.T., T.K., J.K., P.U., W.B., A.G., B.T., J.T., P.F., L.V., P.F., K.B., Z.H. Visualization: A.T., B.B., Á.B., J.K. Supervision: Z.H., K.B., A.G, P.F., L.V., J.T. Funding Acquisition: Z.H., K.B., A.G., P.F., L.V., J.T. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the National Public.
Health Center, Ministry of Human Capacities of Hungary (28324–5/2019/EÜIG). All study.
participants gave their written informed consent in accordance with the Declaration of Helsinki.
Consent for publication
not applicable.
Competing interests
The authors disclose no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additionalfile 1: Supplementary Table 1.
Differentially expressed (DE)miRNA list of PBMC small RNA-seq data of the interictal vs healthy comparison. Supplementary Table 2. Differentially expressed (DE) miRNA list of PBMC small RNA-seq data of the ictal vs interictal comparison. Supplementary Table 3. Differentially expressed (DE) miRNA list ofPBMC small RNA-seq data of the ictal vs healthy comparison. Supplementary Table 4. List of intersectof predicted targets up- or downregulated by DE miRNAs and DE mRNAs ininterictal vs healthy comparison. Supplementary Table 5. List of intersect of predicted targets up- ordownregulated by DE miRNAs and DE mRNAs in ictal vs interictal comparison. Supplementary Table 6. List of intersect of predicted targets up- ordownregulated by DE miRNAs and DE mRNAs in ictal vs healthy comparison. Supplementary Figure 1. Heat map visualization of DE miRNAs in ictal vs. healthy comparison. Supplementary Figure 2. Visualization of DE miRNAs with predicted interactions in ictal vs. healthy interaction.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Aczél, T., Benczik, B., Ágg, B. et al. Disease- and headache-specific microRNA signatures and their predicted mRNA targets in peripheral blood mononuclear cells in migraineurs: role of inflammatory signalling and oxidative stress. J Headache Pain 23, 113 (2022). https://doi.org/10.1186/s10194-022-01478-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-022-01478-w