
Abstract
Introduction
The aim of this sub-study was to evaluate injection success of patients with rheumatoid arthritis (RA) and their caregivers administering the adalimumab (ADL) biosimilar, PF-06410293 (ADL-PF: adalimumab-afzb; Abrilada®/Amsparity®/Xilbrilada®) by prefilled pen (PFP) during the open-label treatment period in year two (weeks 52–78) of a phase 3 multinational, double-blind, clinical study (NCT02480153) comparing ADL-PF and reference ADL (Humira®) sourced from the EU.
Methods
This sub-study included adult patients with active RA not adequately controlled by methotrexate. Patients received subcutaneous ADL-PF 40 mg by prefilled syringe (PFS) at weeks 52 and 54, then six biweekly doses (weeks 56–66) of ADL-PF 40 mg each via a single-use PFP device. Training was given on first injection at week 56; all injections were given by patients/caregivers. The primary endpoint was delivery system success rate (DSSR): the percentage of participants (i.e., actual PFP user) achieving delivery success for each of the six attempted PFP injections. Injection success was recorded by the observer (Observer Assessment Tool) and participant (Participant Assessment Tool).
Results
In total, 50 patients with no experience self-injecting with an autoinjector/injection pen were included (74.0% female; mean age at screening, 54.9 years; mean RA duration, 8.0 years). Of these, 49 (98.0%) completed the sub-study and 46 (92.0%) received all six PFP injections. Overall DSSR (n = 294 injections) across visits was 100% (95% CI 92.0–100.0%). Complete injection was confirmed following inspection of 292 used and returned PFPs. A total of 47/49 (95.9%) participants who completed the sub-study elected to continue study treatment using PFP injections, rather than switching back to the PFS.
Conclusions
All actual PFP users could safely and effectively administer ADL-PF by PFP at each visit, and nearly all participants who completed the sub-study elected to continue study treatment using PFP injections.
Trial registration
ClinicalTrials.gov identifier: NCT02480153; EudraCT number: 2014-000352-29.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? | |
Prefilled pen (PFP) devices have been developed for adalimumab (ADL) and other biologic original and biosimilar disease-modifying anti-rheumatic drugs (boDMARDs and bsDMARDs) to offer patients more convenience during dose administration. | |
In addition to studies showing that switching from a prefilled syringe to PFP device does not impact product quality, acceptability studies are conducted to support such device changes. | |
In this sub-study, we evaluated the injection success of patients with RA and their caregivers administering the ADL biosimilar PF-06410293 (ADL-PF) by PFP during the second year of a phase 3 clinical study comparing ADL-PF and reference ADL sourced from the EU. | |
What was learned from the study? | |
All PFP users were able to successfully administer ADL-PF by PFP at each visit, and PFP injections of ADL-PF by patients or their caregivers were well tolerated. | |
These findings suggest that patients with RA and their caregivers can safely and effectively administer ADL-PF by PFP. |
DIGITAL FEATURES
This article is published with digital features, including a plain language summary, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.19361315.
Introduction
Adalimumab (ADL) is a recombinant, fully human IgG1 monoclonal antibody that targets tumor necrosis factor alpha (TNF-α) and interferes with its binding to cell-surface TNF receptors [1]. The biologic original disease-modifying anti-rheumatic drug (boDMARD) ADL has been marketed as Humira® (AbbVie Inc, North Chicago, IL, USA; and AbbVie Deutschland GmbH Co. KG, Ludwigshafen, Germany) in the US and EU since 2002 and 2003, respectively [2, 3]. The boDMARD ADL reduces symptoms and radiographic progression in patients with rheumatoid arthritis (RA) [4]. boDMARD ADL is also approved for multiple indications in addition to RA [2, 3].
Biosimilars are biopharmaceuticals that are clinically equivalent to a licensed biologic (reference product), notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences in purity, potency, and safety from the reference product [5,6,7]. Access to boDMARDs, such as ADL (Humira), can be limited due to cost [8, 9]. Biosimilars may expand access potentially through lower prices [9]. The ADL biosimilar DMARD (bsDMARD) PF-06410293 (ADL-PF; adalimumab-afzb [Abrilada®/Amsparity®/Xilbrilada®]; Pfizer Inc, New York, NY, USA; Pfizer Europe MA EEIG, Brussels, Belgium; and Wyeth Indústria Farmacêutica Ltda., São Paulo, Brazil) has been approved in countries and regions such as the US, Europe, Switzerland, Canada, Argentina, and Brazil for use in all indications for which boDMARD ADL (Humira) is approved [10,11,12,13,14,15].
Biosimilarity of ADL-PF and reference ADL sourced from the EU (ADL-EU; AbbVie Deutschland GmbH Co. KG, Ludwigshafen, Germany) was confirmed in a multinational, two-arm, randomized, double-blind, parallel-group phase 3 clinical trial in patients with active RA [16, 17]. The study consisted of three 26-week treatment periods (TPs) and a 16-week follow-up after the last dose of study drug [16, 17]. In TP1 (first 26 weeks of dosing), no clinically meaningful differences in efficacy, safety, immunogenicity, pharmacokinetics (PK), or pharmacodynamics (PD) were observed between ADL-PF and ADL-EU [16]. The observed week 12 ACR20 values (primary endpoint) were 68.7% (ADL-PF) versus 72.7% (ADL-EU), representing a treatment difference of – 2.98%; corresponding 95% and 90% confidence intervals (CIs) were entirely contained within the pre-specified equivalence margins [16]. Similarity between treatments was maintained up to week 52, regardless of whether patients switched from ADL-EU to ADL-PF at week 26 (start of TP2) [17]. Long-term efficacy and safety of ADL-PF following 78 weeks of treatment was demonstrated during TP3 and was unaffected by switching from ADL-EU to ADL-PF at week 26 or week 52 [18].
To provide patients with a more convenient administration, prefilled pen (PFP) devices have been developed for several boDMARDs and bsDMARDs [19,20,21,22,23,24,25,26]. In 2006, a new presentation of ADL as a single-use, disposable PFP was approved by the FDA [27]. A phase 1 study in healthy volunteers demonstrated bioequivalence and similar safety profiles of ADL administered by PFP versus prefilled syringe (PFS) [28], demonstrating that this variation does not affect the quality of the product.
Alongside confirmation that the change in device does not impact product quality, acceptability studies were undertaken for ADL to support device switching (e.g., from a PFS to PFP device) [19, 20, 22]. Acceptability studies have also been conducted for device changes for other boDMARDs and bsDMARDs [29,30,31,32,33,34]. However, these studies were based on subjective patient/healthcare provider preference surveys. To date, only a few studies have used objective measures of successful dose administration to support device changes [35,36,37,38].
An open-label, single-dose, PK study in healthy subjects demonstrated bioequivalence between ADL-PF administered by PFS versus PFP, with similar safety profiles between treatment arms [39]. The aim of the current sub-study was to assess the success of using ADL-PF PFP by patients with RA and their caregivers—using objective patient/caregiver and observer questionnaires—during the second year of the phase 3 clinical study comparing ADL-PF and ADL-EU.
Methods
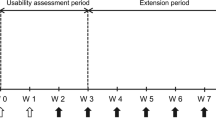
This was an optional, open-label sub-study (Fig. 1) to determine whether patients/caregivers could safely and effectively administer ADL-PF using a PFP device. The study design and inclusion criteria for the main study (NCT02480153) have been described in detail elsewhere [16,17,18]; features relevant to this sub-study of ADL-PF PFP are summarized below.

PFP device sub-study design. aPre-dose PK samples collected as specified in the main study protocol; no additional PK evaluation was performed in the sub-study. bSub-study unique visit. cOnsite and home refer to clinic-based and home-based dose administration using the PFP device. IC informed consent; N/A not applicable; OAT Observer Assessment Tool; PAT Participant Assessment Tool; PFP prefilled pen; P-PK pre-dose pharmacokinetics (main study)
Study Population
Adult (age ≥ 18 years) patients with a diagnosis of active RA ≥ 4 months, based on the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) criteria [40], and an inadequate response to methotrexate (MTX) participated in the main study [16, 17]. Patients must have been treated with MTX for ≥ 12 weeks and been on a stable dose of 10–25 mg/week for ≥ 4 weeks before the first dose of study drug. Lower doses of MTX (6 mg/week) were allowed if indicated in local guidance or standards of care.
The sub-study was conducted in the second year of the main study during open-label treatment with ADL-PF (TP3). Approximately 50 patients from 19 centers in four countries (Czech Republic, Lithuania, Poland, and the USA) were planned for enrollment between October 3, 2016 and May 16, 2017. Eligible patients were active participants in year 2 of the main study who completed study week 52 and moved into the open-label period (TP3), were self-injecting their study medication or had a non-healthcare professional caregiver performing their injections, and provided written informed consent.
Study Design and Treatment
At the start (TP1) of the main study, patients were randomized 1:1 (stratified by geographic region) to ADL-EU or ADL-PF, administered by PFS, in combination with MTX, for 26 weeks [16, 17]. At the start of TP2 (week 26), patients in the ADL-EU arm were blindly re-randomized (1:1) to continue ADL-EU or switch to ADL-PF. At the start of TP3 (week 52), the remaining ADL-EU patients were switched to ADL-PF (open-label).
This optional, open-label, single arm sub-study was conducted during TP3 (weeks 52–78) of the main study (Fig. 1). After receiving two doses of subcutaneous ADL-PF 40 mg by PFS during TP3 (at weeks 52 and 54), patients switched to receive subcutaneous ADL-PF 40 mg via a single-use PFP device containing an identical PFS (Fig. S1). Patients received six additional biweekly doses (weeks 56 to 66) of ADL-PF by PFP, with doses 1, 3, and 6 administered at the study site under supervision, and doses 2, 4, and 5 administered at home.
Patients and their non-healthcare professional caregivers were trained by local site personnel to administer ADL-PF using the PFP device, with their first PFP device available to them during the training. The training adhered to the PFP instructions for use provided to the patients and their caregivers. The patient or their non-healthcare professional caregiver (i.e., actual PFP user) administered the first injection in the abdomen after all procedures and assessments were completed on sub-study day 1, in the presence of the site investigator or designated observer. The site personnel who trained the patient and/or their non-healthcare professional caregiver might have been the same as or different from the site investigator/designated observer. All injections were given by patients/caregivers. Following successful completion of the sub-study, patients had the option of continuing to use the PFP for the remainder of TP3 or returning to use the PFS.
Assessments
The primary endpoint for evaluating PFP device injection success was delivery system success rate (DSSR), defined as the percentage of participants achieving delivery success for an attempted injection at each of the six sub-study PFP injections. Success was based on participant (actual PFP user) and investigator/designated observer assessments of ADL-PF administration by PFP: ‘Yes’, for injection success reported on the Observer Assessment Tool (OAT; if performed at study site; Fig. S2), and lack of injection failure reported on the Participant Assessment Tool (PAT; Fig. S3).
ADL-PF PFP user (patient/caregiver) and investigator observations were collected over the 10-week sub-study duration (Fig. 1). At the three on-site administration visits, the investigator/designated observer witnessed the PFP injection and inspected the window on the PFP following administration to check if the blue bar on the pen had moved across the window. The observer then recorded the success of the drug administration using the PFP OAT (Fig. S2). The observer was not allowed to physically assist with the injection using the PFP (except if urgently required to prevent avoidable injury) but could provide verbal guidance in response to questions; any such guidance was recorded. The observer completed the OAT while blinded to the actual PFP user’s assessment of performance.
Following each injection using the PFP, the actual PFP user (patient or their non-healthcare professional caregiver, if applicable) inspected the window on the PFP to check if the blue bar on the pen had moved across the window and recorded the success of the drug administration using the PAT (Fig. S3). Secondary endpoints for PFP device injection success evaluations were: characterization of unsuccessful PFP injections and determination, by inspection, of the correct mechanical function of returned PFP devices.
All PFPs (both used and those whose container had been opened) were to be returned by the patient via an individual biohazard disposal container. Characterization of unsuccessful PFP injections was descriptive and correlated with the inspection of returned PFPs. A portion of the used PFPs were visually inspected centrally and underwent an industrial computed tomography scan to assess PFP function and correct operation.
Safety evaluations were conducted in accordance with the main study protocol, as reported in detail elsewhere [16, 17]; no additional safety assessments were conducted for the PFP sub-study. Safety endpoints included the type, incidence, severity, timing, seriousness, and relatedness of treatment-emergent adverse events (TEAEs), abnormalities, and laboratory parameters. Injection site reactions (ISRs) were among pre-specified events of special interest in the main study [16, 17] and continued to be captured as in the main study.
Safety was assessed in all patients who received at least a portion of a dose of ADL-PF using the PFP device. Any medical device complaints, including failure of the PFP to perform, regardless of whether the complaint was associated with an adverse event (AE), were recorded. PFP device injection success was evaluated in sub-study patients who attempted ≥ 1 injection using the PFP device (intent-to-treat population). If a participant engaged in multiple PFP injection attempts at a given visit, only the first attempt was to be included in the DSSR calculation.
Statistical Analysis
The DSSR at each visit was accompanied by an exact, two-sided 95% CI using the Clopper–Pearson method [41]. No formal statistical hypothesis was tested for the sub-study data; all data were summarized using descriptive statistics.
The sample size of 50 sub-study patients would have allowed for description of the success of a projected ≤ 300 PFP injections. The patient dropout rate for the sub-study was anticipated to be minimal as patients had already been maintained in the study for > 12 months before enrollment.
Compliance with Ethics Guidelines
This sub-study was conducted in compliance with the ethical principles originating in, or derived from, the Declaration of Helsinki and in compliance with all International Conference on Harmonisation Good Clinical Practice Guidelines. The sub-study protocol and informed consent documentation were reviewed and approved by the Institutional Review Boards and/or Independent Ethics Committees at each of the investigational centers participating in the sub-study. In addition, all local regulatory requirements were followed; in particular, those affording greater protection of the safety of trial participants.
All patients provided informed consent before undergoing any screening procedures. The study was sponsored by Pfizer and registered on ClinicalTrials. Gov identifier: NCT02480153 and EudraCT number: 2014–000352-29.
Results
Patient Disposition and Demographics
The PFP device sub-study included 50 patients with previously highly active RA. Demographic characteristics are summarized in Table 1. The mean age of patients (n = 37 [74.0%] female; n = 13 [26.0%] male) at screening was 54.9 years. Most patients (46/50 [92.0%]) were right-handed. No patients experienced use of an autoinjector or injection pen before participation in the sub-study. Mean RA duration was 8.0 years among sub-study patients. The mean Disease Activity Score 28-joint count using C-reactive protein (DAS28-CRP) was 6.11 at baseline; this decreased to 2.87 at week 52 (i.e., sub-study screening).
Of the 50 patients treated in the sub-study, 49 (98.0%) completed the sub-study. In total, 46/50 (92.0%) patients received all six PFP injections (i.e., full compliance); of the remaining four patients: one discontinued from both the main and PFP sub-study after three PFP injections (withdrew consent); two missed the week 58 PFP injection, one due to an AE of upper respiratory tract inflammation and one due to hospitalization from pelvic fracture and lack of access to study drug; and one missed the week 64 PFP injection due to a grade 3 serious AE (SAE) of RA flare. No patients needed to make a second attempt at injection using a second PFP during the sub-study.
PFP Device Injection Success
All actual PFP users successfully administered ADL-PF by PFP at each visit. The DSSR at each visit was 100.0%, with the lower bound of the two-sided 95% CI exceeding 92% and the upper bound being 100.0%. All 294 attempted injections were successfully administered, resulting in an overall DSSR (95% CI) across visits of 100% (92.0%, 100.0%) (Table 2). OAT responses were recorded at the three onsite clinic visits at weeks 56, 60, and 66. Observers recorded all injections as successful. Five patients could not attend the onsite visit at week 60 and instead performed the injection with OAT assessment at week 62. These five OAT assessments were successful and included in the OAT contribution to overall success, but were not included in the OAT summary by scheduled visit. Three (6.0%) patients requested verbal instructions at the week 56 visit (first PFP injection) and one (2.3%) at the week 60 visit.
PAT responses were recorded for all six sub-study injections by the actual PFP users. Actual PFP users for the first injection at week 56 consisted of 48 (96.0%) sub-study patients themselves and two (4.0%) non-healthcare professional caregivers (a relative or friend). No actual PFP user switched from patient to non-healthcare professional caregiver, or vice versa, for the subsequent sub-study visits. Of the two patients who received sub-study injections from a non-healthcare professional caregiver, one received all injections from the non-healthcare professional caregiver and one received the first five injections from a non-healthcare professional caregiver and the sixth injection from a healthcare professional caregiver (presumably due to absence of the non-healthcare professional caregiver at the clinic visit).
There were no medical device complaints. Inspection of the 292 used and returned PFPs demonstrated complete injection of the full volume for all PFPs. Upon completion of the sub-study, 47/49 (95.9%) of the sub-study completers elected to continue study treatment using PFP injections and two (4.1%) elected to continue treatment using PFS injections.
Safety
There were no deaths, ISRs, or permanent discontinuations from treatment or study due to TEAEs reported during the sub-study (Table 3). A total of 20 all-causality TEAEs were reported by 17 (34.0%) patients during the sub-study. The most frequently reported TEAE was viral upper respiratory tract infection (five [10.0%] patients). All other TEAEs were reported by ≤ 2 patients. There were three (6.0%) SAEs (tonsillitis, RA flare, and pelvic fracture, n = 1 each) reported during the sub-study, all of which were also grade 3.
Discussion
Studies of ADL (Humira) autoinjector pens in patients with RA and other chronic autoimmune disorders (e.g., Crohn’s disease) have demonstrated that they are suitable for biologic therapies and are preferred by many patients [19, 20, 22,23,24]. Autoinjection/PFP devices for delivery of ADL (Humira) are well tolerated in patients with RA, with no new safety signals compared with PFS delivery systems containing the same active drug [22, 23]. PFP devices are generally preferred by patients due to their ease of use and convenience [19, 20, 22, 23], and achieve serum drug levels similar to that of PFS [24].
Autoinjector devices have also been developed for other boDMARDs, including etanercept and sarilumab [21, 25, 26]. The use of these pen devices compares favorably with delivery by PFS (based on patient surveys) [21, 25, 26]. The experiences and preferences (PFS or PFP device) of patients with RA and other chronic autoimmune conditions is largely based on subjective surveys, and device changes for boDMARDs and bsDMARDs are often based on patient/healthcare professional preference studies [19, 20, 22, 29,30,31,32,33,34]. Few studies have used objective assessments to evaluate injection success [35,36,37,38]. Use of objective patient/site personnel questionnaires in a study to evaluate the success of self-injection with an ADL biosimilar (BI695501) demonstrated the vast majority (99%) of autoinjections were successful [37]. As studies using an objective assessment when changing device are lacking, the PAT and OAT were developed to perform a more objective assessment in evaluating usefulness of the PFP device to administer ADL-PF.
All actual PFP users successfully administered ADL-PF by PFP at each visit, as determined using the OAT and PAT. Injections of ADL-PF by the patient or their non-healthcare professional caregiver using the PFP device were well tolerated. The AEs (including SAEs) reported for the sub-study population were consistent with those reported for the first year of the overall study population [17], and there were no new safety findings to alter the benefit–risk profile of ADL-PF. Furthermore, a high proportion of patients who completed the sub-study (95.9%) selected to continue PFP injections for the remainder of the study treatment.
There are several limitations identified in this sub-study. One limitation was that it was conducted during the second year of treatment in the main study and not in treatment-naïve patients. Although participants were naïve to autoinjectors or PFPs, they had 13 months of experience with ADL-PF administration via a PFS during the first year of the study and may not be representative of a treatment-naïve RA population or an RA population with less than 13 months of treatment with ADL. In addition, in the current sub-study, patients and their caregivers were trained by healthcare professionals to administer ADL-PF using the PFP device before their first injection. It is possible that patients/caregivers in a real-world setting may not receive training or the same extent of training as participants in this trial, which could limit generalizability of the current findings.
Conclusions
Using objective patient/caregiver and observer questionnaires (PAT and OAT), our findings demonstrate that all actual PFP users could safely and effectively administer ADL-PF by PFP at each visit. In addition, nearly all participants who completed the sub-study elected to continue study treatment using PFP injections. Finally, PFP injections of ADL-PF by patients or their caregivers were well tolerated.
References
Lapadula G, Marchesoni A, Armuzzi A, et al. Adalimumab in the treatment of immune-mediated diseases. Int J Immunopathol Pharmacol. 2014;27:33–48.
AbbVie. HUMIRA (adalimumab): prescribing information. 2021. http://www.rxabbvie.com/pdf/humira.pdf. Accessed 17 May 2021.
European Medicines Agency. HUMIRA (adalimumab): summary of product characteristics. 2021. https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf. Accessed 17 May 2021.
Armuzzi A, Lionetti P, Blandizzi C, et al. Anti-TNF agents as therapeutic choice in immune-mediated inflammatory diseases: focus on adalimumab. Int J Immunopathol Pharmacol. 2014;27:11–32.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. https://www.fda.gov/media/82647/download. Accessed 17 May 2021.
European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies– non-clinical and clinical issues. 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf. Accessed 17 May 2021.
World Health Organization. Application to add anti-TNFs to the World Health Organization’s Essential Medicines List. https://www.who.int/selection_medicines/committees/expert/22/applications/s8.1_TNF-alfa-inhibitors.pdf. Accessed 19 October 2021.
Putrik P, Ramiro S, Kvien TK, et al. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis. 2014;73:198–206.
Smolen JS, Goncalves J, Quinn M, Benedetti F, Lee JY. Era of biosimilars in rheumatology: reshaping the healthcare environment. RMD Open. 2019;5:e000900.
European Medicines Agency. Amsparity (adalimumab): summary of production characteristics. 2020. https://www.ema.europa.eu/en/documents/product-information/amsparity-epar-product-information_en.pdf. Accessed 26 February 2020.
Pfizer Inc. Abrilada™ (adalimumab-afzb): prescribing information. 2019. http://labeling.pfizer.com/ShowLabeling.aspx?id=12780. Accessed 26 February 2020.
Pfizer Canada ULC. Abrilada (adalimumab injection): product monograph including patient medication information. 2021. https://www.pfizer.ca/sites/default/files/202107/Abrilada_PM_EN_252317_29-Jun-2021.pdf. Accessed 13 August 2021.
Wyeth. Xilbrilada® (adalimumabe): prescribing information. 2021. https://www.pfizer.com.br/sites/default/files/inline-files/Xilbrilada_Profissional_de_Saude_08.pdf. Accessed 15 October 2021.
SwissMedic. Abrilada®, solution for injection in pre-filled pen. https://www.swissmedicinfo.ch/ShowText.aspx?textType=PI&lang=DE&authNr=67831. Accessed 25 October 2021.
The National Administration of Drugs FaMDAA. Referencia: EX-2019–51618513-APN-DGA#ANMAT. 2020. http://www.anmat.gov.ar/boletin_anmat/Febrero_2020/Dispo_0755-20.pdf. Accessed 26 October 2021.
Fleischmann RM, Alten R, Pileckyte M, et al. A comparative clinical study of PF-06410293, a candidate adalimumab biosimilar, and adalimumab reference product (Humira(R)) in the treatment of active rheumatoid arthritis. Arthritis Res Ther. 2018;20:178.
Fleischmann RM, Alvarez DF, Bock AE, et al. Randomised study of PF-06410293, an adalimumab (ADL) biosimilar, compared with reference ADL for the treatment of active rheumatoid arthritis: results from weeks 26–52, including a treatment switch from reference ADL to PF-06410293. RMD Open. 2021;7:e001578.
Fleischmann RM, Alvarez DF, Bock AE, et al. Long-term efficacy, safety, and immunogenicity of the adalimumab biosimilar, PF-06410293, in patients with rheumatoid arthritis after switching from reference adalimumab (Humira(R)) or continuing biosimilar therapy: week 52–92 data from a randomized, double-blind, phase 3 trial. Arthritis Res Ther. 2021;23:248.
Borras-Blasco J, Gracia-Perez A, Rosique-Robles JD, Castera MD, Abad FJ. Acceptability of switching adalimumab from a prefilled syringe to an autoinjection pen. Expert Opin Biol Ther. 2010;10:301–7.
Corominas H, Garcia-Diaz S, Sanchez-Eslava L, Figuls R. Switching adalimumab from syringe to pen. Expected outcomes. Expert Opin Biol Ther. 2012;12:805–6.
Kivitz A, Baret-Cormel L, van Hoogstraten H, et al. Usability and patient preference phase 3 study of the sarilumab pen in patients with active moderate-to-severe rheumatoid arthritis. Rheumatol Ther. 2018;5:231–42.
Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28:1619–29.
Kivitz A, Segurado OG. HUMIRA pen: a novel autoinjection device for subcutaneous injection of the fully human monoclonal antibody adalimumab. Expert Rev Med Devices. 2007;4:109–16.
Little RD, Chu IE, van der Zanden EP, et al. Comparison of adalimumab serum drug levels when delivered by pen versus syringe in patients with inflammatory bowel disease. An international, multicentre cohort analysis. J Crohns Colitis. 2019;13:1527–36.
Muller-Ladner U, Flipo RM, Vincendon P, Brault Y, Kielar D. Comparison of patient satisfaction with two different etanercept delivery systems. A randomised controlled study in patients with rheumatoid arthritis. Z Rheumatol. 2012;71:890–9.
Rekaya N, Vicik SM, Hulesch BT, McDonald LL. Enhancement of an auto-injector device for self-administration of etanercept in patients with rheumatoid arthritis confers emotional and functional benefits. Rheumatol Ther. 2020;7:537–52.
Fierce Biotech. Press release: HUMIRA receives FDA approval for treatment of Crohn's disease. 2007. https://www.fiercebiotech.com/biotech/press-release-humira-receives-fda-approval-for-treatment-of-crohn-s-disease. Accessed 25 October 2021.
Paulson S. Assessment of relative bioavailability, safety, and tolerability of single doses of adalimumab administered via an autoinjector pen and a prefilled syringe. J Am Acad Derm. 2007;56:AB9.
Barbosa CM, Rodriguez de Castro B, Labeaga Beramendi Y, Terroba Alonso P, Barbazan Vazquez J. Patient satisfaction survey: substitution of reference etanercept with a biosimilar product. Eur J Hosp Pharm. 2021;28:109–11.
Blauvelt A, Gordon KB, Lee P, et al. Efficacy, safety, usability, and acceptability of risankizumab 150 mg formulation administered by prefilled syringe or by an autoinjector for moderate to severe plaque psoriasis. J Dermatolog Treat. 2021. https://www.tandfonline.com/doi/full/10.1080/09546634.2021.1914812.
Fettner S, Mela C, Wildenhahn F, et al. Evidence of bioequivalence and positive patient user handling of a tocilizumab autoinjector. Expert Opin Drug Deliv. 2019;16:551–61.
Rho YH, Rychlewska-Hanczewska A, Sliwowska B, Kim TH. Usability of prefilled syringe and autoinjector for SB4 (an etanercept biosimilar) in patients with rheumatoid arthritis. Adv Ther. 2019;36:2287–95.
Fenwick S, Thakur K, Munro D. Nurse and patient perceptions and preferences for subcutaneous autoinjectors for inflammatory joint or bowel disease: findings from a European survey. Rheumatol Ther. 2019;6:195–206.
Ghil J, Zielinska A, Lee Y. Usability and safety of SB5 (an adalimumab biosimilar) prefilled syringe and autoinjector in patients with rheumatoid arthritis. Curr Med Res Opin. 2019;35:497–502.
Bernstein D, Pavord ID, Chapman KR, et al. Usability of mepolizumab single-use prefilled autoinjector for patient self-administration. J Asthma. 2020;57:987–98.
Callis Duffin K, Bukhalo M, Bobonich MA, et al. Usability of a novel disposable autoinjector device for ixekizumab: results from a qualitative study and an open-label clinical trial, including patient-reported experience. Med Devices (Auckl). 2016;9:361–9.
Cohen S, Klimiuk PA, Krahnke T, Assudani D. Successful administration of BI 695501, an adalimumab biosimilar, using an autoinjector (AI): results from a phase II open-label clinical study (VOLTAIRE((R))-RL). Expert Opin Drug Deliv. 2018;15:545–8.
Collier DH, Bitman B, Coles A, Liu L, Kumar S, Judd C. A novel electromechanical autoinjector, AutoTouch, for self-injection of etanercept: real-world use and benefits. Postgrad Med. 2017;129:118–25.
Cox DS, Alvarez DF, Bock AE, Cronenberger CL. Randomized, open-label, single-dose, parallel-group pharmacokinetic study of PF-06410293 (adalimumab-afzb), an adalimumab biosimilar, by subcutaneous dosing using a prefilled syringe or a prefilled pen in healthy subjects. Clin Pharmacol Drug Dev. 2021;10:1166–73.
Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81.
Clopper CJP, Pearson ES. The use of confidence intervals or fiducial limits illustrated in the case of binomial. Biometrika. 1934;26:404–13.
Acknowledgements
We thank the participants (patients and their caregivers) of the sub-study.
Funding
This study, medical writing support, and the Rapid Service Fee were sponsored by Pfizer (New York, NY, USA).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
AEB, CC, CMG, and IV contributed to conception or design of the study; RMF and ED contributed to the acquisition of data; and WZ contributed to data analysis. All authors participated in the interpretation of the data, contributed to the drafting or revision of the manuscript, read and gave final approval of the submitted manuscript, were involved in the decision to submit the manuscript for publication, and accept accountability for all aspects of the work.
Medical Writing, Editorial, and Other Assistance
The authors thank K. Lea Sewell, formerly of Pfizer, for valuable contributions to the PF-06410293 clinical development program. Medical writing support was provided by Elyse Smith, PhD, of Engage Scientific Solutions and was funded by Pfizer (New York, NY, USA).
Disclosures
Roy M Fleischmann reports personal fees from Pfizer. Eva Dokoupilová reports research grants from AbbVie, Affibody AB, Eli Lilly, Galapagos, Gilead, GSK, Hexal AG, MSD, Novartis, Pfizer, R-Pharm, Sanofi-Aventis, and UCB. Charles M Godfrey is a full-time employee of Pfizer. Wuyan Zhang is a full-time employee of Pfizer and declares shareholdings, stock holdings, and/or stock options from Abbott, AbbVie, and Pfizer. Amy E Bock, Carol Cronenberger, and Ivana Vranic are full-time employees of, and declare shareholdings, stock holdings, and/or stock options from Pfizer.
List of Investigators
Eva Dokoupilová (Czech Republic); Roma Milasiene and Margarita Pileckyte (Lithuania); Barbara Grabowicz-Wasko, Artur Racewicz, Sabina Hajduk-Kubacka, and Piotr Klimiuk (Poland); Jacob Aelion, William Edwards, Roy M Fleischmann, Michael Grisanti, Wassim Saikali, Paul DeMarco, Asad Fraser, Alastair Kennedy, and Suman Thakker (United States).
Compliance with Ethics Guidelines
This sub-study was conducted in compliance with the ethical principles originating in, or derived from, the Declaration of Helsinki and in compliance with all International Conference on Harmonisation Good Clinical Practice Guidelines. The sub-study protocol and informed consent documentation were reviewed and approved by the Institutional Review Boards and/or Independent Ethics Committees at each of the investigational centers participating in the sub-study. In addition, all local regulatory requirements were followed; in particular, those affording greater protection of the safety of trial participants.
All patients provided informed consent before undergoing any screening procedures. The study was sponsored by Pfizer and registered on ClinicalTrials. Gov identifier: NCT02480153 and EudraCT number: 2014–000,352-29.
Data Availability
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fleischmann, R.M., Bock, A.E., Zhang, W. et al. Usability Study of PF-06410293, an Adalimumab Biosimilar, by Prefilled Pen: Open-Label, Single-Arm, Sub-Study of a Phase 3 Trial in Patients with Rheumatoid Arthritis. Rheumatol Ther 9, 839–850 (2022). https://doi.org/10.1007/s40744-022-00439-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-022-00439-8