
Abstract
Fabry disease is a progressive, X-linked inherited lysosomal storage disorder where accumulation of glycosphingolipids increases the risk for early cardiovascular complications, including heart failure, stroke, and end stage renal disease. Besides disease-specific therapy, blood pressure (BP) control is of central importance in Fabry disease to reduce disease progression and improve prognosis. Both Fabry disease and hypertension are characterized by the activation of the innate component of the immune system, with Toll-like receptor 4 (TLR4) as a common trigger to the inflammatory cascade. The renin-angiotensin system (RAS) participates in the establishment of low-grade chronic inflammation and redox unbalance that contribute to organ damage in the long term. Besides exploiting the anti-inflammatory effects of RAS blockade and enzyme replacement therapy, targeted therapies acting on the immune system represent an appealing field of research in these conditions. The aim of this narrative review is to examine the issue of hypertension in the setting of Fabry disease, focusing on the possible determinants of their reciprocal relationship, as well as on the related clinical and therapeutic implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Fabry disease is a progressive, X-linked inherited lysosomal storage disorder affecting the glycosphingolipid metabolism [1]. The underlying defect, resulting from mutations in the GLA gene, concerns the activity of the lysosomal enzyme alpha-galactosidase A, whose reduced or absent activity leads to the progressive accumulation of glycosphingolipids, mainly globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3), in the affected cells and in body fluids [2]. Cellular dysfunction and microvascular pathology that occur early in the disease history trigger a cascade of events involving impaired metabolism, immune system activation, and defective tissue perfusion that eventually induce the development of irreversible fibrosis at crucial sites (i.e. heart, kidney) [1]. Although the classical phenotype is more typical of males, it may also occur among females due to random X-inactivation [1].
Fabry disease is underdiagnosed. In the late Nineties, its estimated prevalence ranged between 1:117,000 [3] and 1:476,000 [4], but more recent evidence claims it to be reconsidered at between 1:3100 [5] and 1:8500 [6, 7], or even lower (1:1818 according to a screening campaign in South Sardinia [8]). The exclusion of positive cases related to mutations that are not disease-causing still provides an estimated prevalence of 1:5000 [7], making the chance of assisting a potential Fabry patient not so uncommon.
Patients are often diagnosed late with Fabry disease. They are typically young adults with symptomatic organ involvement, including progressive chronic kidney disease (CKD) up to the stage of renal failure, left ventricular hypertrophy (LVH) associated with myocardial fibrosis and arrhythmias, lung involvement, and neurological manifestations like sudden deafness, transient ischemic attacks (TIA), and stroke [7]. Backwards in their personal history, chronic neuropathic pain and episodic severe pain crises in childhood as well as early pathological albuminuria and glomerulosclerosis may be retrieved [7]. Diagnostic delay negatively affects prognosis: therefore, suspicion and early recognition of possible cases are crucial. On the other side, the awareness and the appropriate management of global cardiovascular risk, namely through blood pressure (BP) control, are of paramount importance against disease progression. In fact, although the prevalence of hypertension in Fabry disease was reported not being dissimilar to the general population [2, 9], with in some cases a tendency to hypotension attributable to inappropriate vascular control due to autonomic impairment [10], effective BP control is a requisite for outcome improvement in the affected individuals with hypertension.
In this narrative review, we will examine the issue of hypertension in the setting of Fabry disease, focusing on the possible determinants of their reciprocal relationship, as well as on the related clinical and therapeutic implications.
2 Search Methodology
For the purpose of this narrative review, a comprehensive search of major electronic databases (PubMed, EMBASE) was conducted for articles in the English language published in medical
journals from inception through August 2020. The search used the terms “Fabry disease”, “hypertension”, “cardiovascular system”, “inflammation”, “immunity”, “oxidative stress”, “reactive oxygen species”, and “renin-angiotensin-aldosterone system” in several combinations. In addition, review articles on the topic were searched for eligible articles. The search results were accessed, and relevant references were made for the purpose of this review.
3 Cardiovascular Changes in Fabry Disease
Cardiovascular involvement in Fabry disease spans structural and functional changes that can be only partially reverted by therapy [2]. At the cardiac level, they are responsible for systolic and diastolic dysfunction, reduced exercise tolerance, ischemic symptoms, heart rate disturbances up to malignant arrhythmias, myocardial infarction and congestive heart failure [1, 11]. The key structural changes is a typically concentric, non-obstructive LVH mimicking hypertrophic cardiomyopathy that impairs cardiac perfusion, but the involvement of the sinus node and the conduction system, the coronary tree itself, as well as a certain degree of autonomic imbalance also contribute to the observed clinical features [1, 11]. In the cerebrovascular system, a multifocal vasculopathy of the small vessels, together with a dilatative arteriopathy of the vertebrobasilar circulation can lead to early and/or recurrent TIA and strokes [1, 11]. In the kidney, early and progressive proteinuria and reduced glomerular filtration rate up to renal insufficiency are the typical manifestations of the disease [1, 11].
Histologically, lysosomal myelin-like inclusions, corresponding to glycosphingolipids deposits, occur at the affected sites, including the endothelial and smooth muscle cells of heart and arterial vessels, the glomerular mesangial and interstitial cells, the podocytes, and the epithelial cells of the renal tubule [1]. Inclusions are initially perinuclear and extend to the cytoplasm with the disease progression [12]. Myocytes hypertrophy, structural podocytes changes with reduced fenestrations, progressive segmental and global glomerulosclerosis, cellular necrosis and apoptosis, and interstitial and replacement fibrosis of various degrees are other typical features of cardiovascular involvement [1].
The major alterations observed in Fabry disease, like LVH and arterial remodeling, are not only a mechanical implication of glycosphingolipids accumulation (hampered relaxation, structural subversion, or myofilaments degradation) [12,13,14]. Indeed, one contribution is represented by plasmatic hypertrophy-promoting proliferation factors [13], later identified as the circulating form of lyso-Gb3 [15] and sphingosine-1-phosphate [16]. In fact, the exposure of smooth muscle cells to lyso-Gb3 at the plasmatic concentrations observed in Fabry disease promoted their proliferation, supporting a role for lyso-Gb3 in the pathogenesis of LVH and intima-media thickness (IMT) in these patients [15]. Circulating sphingosine-1-phosphate was significantly higher in Fabry disease patients compared with controls, it correlated with LVH and IMT, and its administration to mice induced cardiovascular remodelling similar to that observed in the human disease [16].
Noteworthy are also the metabolic, hormonal, and inflammatory events elicited by glycosphingolipids vacuoles, and their role in the disease progression. In fact, a hypothesis for the pathogenesis of Fabry vasculopathy suggests that IMT secondary to the hypertrophy of vascular smooth muscle cells reduces arterial compliance, with subsequent local upregulation of the renin angiotensin system (RAS) mediated by the action of the endothelial angiotensin-converting enzyme (ACE) that converts circulating angiotensin I into angiotensin II (Ang II) [17, 18]. In turn, by binding to the AT1 receptor, Ang II triggers a proinflammatory cascade through several mechanisms, from increasing the expression of adhesion molecules and contributing to leukocytes recruitment and cytokines/chemokines release, to inducing superoxide anion generation mediated by NADH-/NADPH-oxidases [19,20,21]. The subsequent increase in reactive oxygen species (ROS) and inflammatory mediators amplifies the proinflammatory effects of Ang II by inducing the proinflammatory transcription factor, nuclear factor-kB (NF-kB) [21]. ROS decrease nitric oxide bioavailability and ultimately cause endothelial dysfunction [11, 22]. Inflammation-induced integrins mediate qualitative and quantitative changes in the extracellular matrix towards the development of fibrosis, which is also facilitated by the RAS through the induction of the profibrotic and prothrombotic mediator, plasminogen activator inhibitor type 1 (PAI-1) [23].
Gb3 itself was found to be able to induce intracellular ROS production and to up-regulate the expression of cell adhesion molecules (intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin) in endothelial cells [24]. Interestingly, plasma from Fabry patients, but not from healthy controls, significantly increased ROS generation in the same cells [24]. It was later shown that circulating antioxidant defenses were reduced in Fabry patients, i.e. they displayed reduced glutathione and glutathione peroxidase activity as well as increased superoxide dismutase (SOD)/catalase ratios, indicating higher susceptibility to oxidative damage [25].
4 Innate Immunity in Fabry Disease and Hypertension
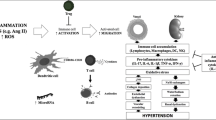
With the multitude of largely unexplored pathways that constitute its complexity, the immune system participates to a large extent to human physiopathology, and both Fabry disease and hypertension are characterized by immune system activation and low-grade chronic inflammation [26,27,28]. A role for the innate component of the immune system, in particular, has been described for both conditions (Fig. 1).

Possible mechanisms for the activation of innate immunity in Fabry disease and hypertension. Exogenous (e.g. LPS) and endogenous (e.g., glycosphingolipids) ligands elicit innate immunity response by binding to TLR4. In turn, TLR4 stimulates NADPH oxidase and the transcription of pro-inflammatory cytokines. The resulting pro-oxidant, pro-inflammatory environment leads to anatomical changes (e.g., smooth muscle cells hypertrophy; fibrosis) and functional impairment (e.g., impaired vasoreactivity; glomerular dysfunction). See text for further details. PAMPs pathogen-associated molecular patterns, DAMPs damage-associated molecular patterns, LPS lipopolysaccharide, LDL low-density lipoproteins, Ang II angiotensin II, HSP heat shock proteins, ECM extracellular matrix, Gb3 globotriaosylceramide, lyso-Gb3 globotriaosylsphingosine, TLR4 toll-like receptor 4, NADPH nicotinamide adenine dinucleotide phosphate, MyD88 myeloid differentiation primary response 88, MAPK mitogen-activated protein kinase, NF-kB nuclear factor-kB, ROS reactive oxygen species
Since lysosomes play a central role in several immune processes, including phagocytosis, antigens processing and presentation, perforins secretion, and release of proinflammatory mediators, the diseases that affect their function can have consequences on the immune system [26]. Also, lysosomal deposition of unmetabolized glycosphingolipids stimulates the continuous activation of the inflammatory response [26]. Storage diseases have been classified as a subgroup of autoinflammatory disorders [29], which are primarily diseases of the innate immunity with predominance of neutrophil and monocyte/macrophage-driven inflammation and inappropriate cytokine-mediated pathology [29, 30]. It was hypothesized that Fabry disease also belongs to the category of autoinflammatory disorders [26, 31]. In fact, increased constitutive expression and secretion of tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β and IL-6 were observed in peripheral blood mononuclear cells (PBMC) from Fabry patients, in keeping with the finding of increased TNF-α and IL-6 plasma levels in Fabry disease [25, 31]. The basal proinflammatory cytokine production profile was particularly evident for dendritic cells and monocytes, and it was further exacerbated following an inflammatory stimulus (lipopolysaccharide, LPS) [31]. Interestingly, Gb3 was shown to induce the same cytokine profile on normal monocyte-derived macrophages and dendritic cells, indicating that it could be responsible for triggering the proinflammatory events observed in Fabry disease, while the blockade of Toll-like receptor 4 (TLR4) prevented the Gb3-mediated production of TNF-α and IL-1β [31]. TLR4 is a member of the TLR family that responds to factors derived from pathogens (pathogen-associated molecular patterns, PAMPs) or cellular damage (damage-associated molecular patterns, DAMPs) with innate immunity activation [32]. Altogether, the mentioned findings demonstrate the involvement of innate immunity in Fabry disease pathology, with evidence of Gb3 having a direct role in triggering a proinflammatory state. In this setting, the chronic release of chemokines, adhesion molecules, and other proinflammatory mediators that occurs over time leads to organ damage (Fig. 1) [26].
The concept that also hypertension has an immunological basis dates back to the 1960s [27, 33]. Since then, in the wake of pioneering studies defining the role of immune cells in atherosclerosis, several studies advanced our understanding of the role of immunity and inflammation in hypertension [27]. Besides the prominent role of adaptive immunity, dysfunctional innate immune stimulation, primarily by means of TLR4, contributes to the pathogenesis of high BP (Fig. 1) [27, 28]. Findings from mouse models of hypertension consistently indicate that pharmacological blockade or genetic deletion of TLR4 exert benefit on BP, with evidence of reduced oxidative stress and inflammation in the cardiovascular and central nervous systems [34]. For instance, administration of anti-TLR4 antibodies reduced BP and serum levels of IL-6 in spontaneously hypertensive rats (SHR) [35], while treatment with a TLR4 antagonist inhibited inflammatory cytokines and decreased BP in the aldosterone and salt administration (Aldo-salt) hypertensive model [36]. Also, in a model of Ang II-induced hypertension, TLR4 deficiency was associated with reduced oxidative stress (ROS, NADPH oxidase), reduced inflammation (TNF-α, IL-6), increased antioxidant capacity (SOD, catalase activity), and with decreased transforming growth factor (TGF)-β-induced extracellular matrix protein deposition in the kidney [37]. In parallel, in the G-nitro-L-arginine methyl ester (L-NAME) model of hypertension, TLR4 knockout mice were protected against BP increases [38]. Mechanistically, it was described that TLR4 activation in RAS-dependent hypertension models might be triggered by Ang II [20], while it depends on DAMP release in a RAS-independent model of hypertension like that induced by L-NAME [38]. Following Ang II binding to AT1 in vascular smooth muscle cells, TLR4 activation induces inflammation through NF-kB and, in parallel, promotes ROS formation by inhibiting the expression and activity of the antioxidant enzyme SOD, as well as by activating NADPH oxidase [39, 40]. Pharmacological or genetic blockade of TLR4 in SHR and Ang II-infused mice decreased BP, reduced vascular inflammation and oxidative stress, normalized the increased contractile response of resistance arteries and prevented their remodeling due to collagen deposition and altered elastin structure [35, 41, 42]. In SHR, treatment with losartan reverted the increase in TLR4 mRNA levels in aortic smooth muscle cells [42]. In L-NAME-induced hypertension, L-NAME leads to cellular damage and DAMP release, with consequent TLR4-dependent vascular ROS production and impaired vasodilation that can be prevented by TLR4 silencing [38].
Based on the reported evidence, the innate immunity is involved in both Fabry disease and hypertension, thus broadening our understanding of the mechanistic events behind their occurrence.
5 Cardiovascular Risk Management in Fabry Disease: Focus on Hypertension
Hypertension contributes to the disease burden in Fabry disease. Despite susceptibility to hypertension among Fabry disease patients may not exceed that of the general population [2], similar to what is described in other clinical settings [43], its presence was shown to negatively impact the disease progression and prognosis [44]. In fact, it aggravates the disease burden by worsening the cardiac involvement and by increasing the risk of end stage renal disease (ESRD) and stroke [2]. Further, since many mechanisms can intervene in the determination of BP levels [45], hypertension might be difficult to control in Fabry disease due to the underlying proinflammatory environment, similar to what is widely observed in the presence of low-grade systemic inflammation of other sources [46,47,48,49,50]. In agreement with this, a 2006 report from the Fabry Outcome Survey (FOS) indicated at around 50% (men: 54%; women: 47%) the prevalence of office BP values higher than 130/80 mmHg [51].
It is also possible that the renal impairment that occurs early in Fabry disease promotes the development of hypertension and worsens BP control in the long term. The mechanisms that mediate this relationship include increased RAS activity, salt and water retention, overactivity of the sympathetic nervous system, endothelial dysfunction, oxidative stress, and increased intracellular calcium levels following secondary hyperparathyroidism [52]. Altogether, these events lead to increased vasoconstriction, impaired vasodilation, volume expansion, and arterial stiffness, with consequent increase in systemic BP [52].
In order to reduce cardiovascular morbidity and mortality in Fabry disease, an expert consensus statement on therapeutic goals in Fabry disease was recently published [2]. The document refers to the existing guidelines in cardiology and hypertension, given the lack of specific recommendations for the management of cardiac manifestations in Fabry disease [2]. Hypertension and LVH were identified as the risk factors with the strongest association with the occurrence of cardiovascular events in Fabry disease, with an odds ratio (OR) for hypertension of 7.8 in men (95% confidence interval [CI] 2.1–28.6, p = 0.0019) and of 4.5 in women (95% CI 1.6–12.3, p = 0.0037) [53].
Targeted therapies hold promise for genetic-related diseases [54]. A recent systematic literature review examining the effect of enzyme replacement therapy (ERT) on clinical outcomes in male patients with Fabry disease concluded that strong and substantiated evidence support the dose-dependent benefit of ERT on renal and cardiac outcomes, with consistent preservation in eGFR and reduction/stabilization of left ventricular mass and cardiac wall thickness, especially when treatment was initiated early [55]. 2018 Canadian Fabry Disease Initiative treatment guidelines emphasized the importance of an aggressive control of hypertension in patients with nephropathy in addition to ERT [56]). Hypertension of at least one year duration was listed as a minor criteria for renal indications to disease-specific therapy, and adequate BP control for at least 1 year was encouraged in patients with cardiac manifestations [56]. ACE inhibitors and angiotensin-receptor blockers are considered the preferred treatment for high BP in Fabry disease. Further, given that ERT does not reduce proteinuria to a significant extent, blockade of the RAS is also paramount for reducing urine protein excretion and the consequent risk of CKD progression [2]. In addition, the effects of the RAS components on the immune system, the redox balance, and fibrosis, which can be counteracted by RAS blockers, are of particular importance in the setting of Fabry disease [18, 57,58,59,60]. Thus, RAS blockers represent a pivotal therapy in these patients [2].
On the other side, careful consideration should be made for avoiding hypotension when using antiproteinuric drugs for Fabry nephropathy, or concomitant agents with hypotensive effects [2, 61, 62]. It has been reported, however, that the benefits of reducing proteinuria outweigh the risks of reducing BP in patients receiving ERT, with evidence of sustained improvement in renal outcomes, no serious complications from hypotension, and restoration of baseline BP levels in patients treated with RAS blockers and agalsidase beta [63]. In addition, ERT was shown to improve baroreflex function, with consequent improvement in cardiovascular responses to orthostatic challenge in Fabry patients [64].
ERT was also investigated for its effects on the immune system activation in Fabry disease, with some encouraging, though inconclusive, evidence that needs to be further investigated in rigorous trials [26]. In parallel, whether anti-inflammatory agents targeting specific components of the immune system might impact on disease prognosis is a possibility that deserves consideration, particularly in the light of the cardiovascular implications of controlling inflammation [65].
6 Conclusions
Low-grade chronic inflammation occurs in both Fabry disease and hypertension, and common pathways mediated by innate immunity contribute to explain their pathogenesis. RAS blockers are of particular benefit in the setting of Fabry disease not only as antihypertensive agents for cardiovascular risk management through BP control, but also for their nephroprotective, antiproteinuric effects; in parallel, additional benefits might derive from their anti-inflammatory, antifibrotic, and antioxidant properties. Targeted therapies acting on the immune system represent an appealing perspective for the management of chronic conditions sharing common pathogenetic events centered on inflammation, like Fabry disease and hypertension.
References
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
Wanner C, Arad M, Baron R, Burlina A, Elliott PM, Feldt-Rasmussen U, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124:189–203.
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–54.
Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–6.
Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40.
Burton BK, Charrow J, Hoganson GE, Waggoner D, Tinkle B, Braddock SR, et al. Newborn screening for lysosomal storage disorders in Illinois: the initial 15-month experience. J Pediatr. 2017;190:130–5.
Germain DP, Fouilhoux A, Decramer S, Tardieu M, Pillet P, Fila M, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019;96:107–17.
Bolasco P, Sitzia I, Murtas S. Fabry disease in Southern Sardinia: epidemiological results from screening in an extensive area. G Ital Nefrol. 2017;34:83–102.
Del Pinto R, Pagliacci S, De Feo M, Grassi D, Ferri C. Italian Society of hypertension and federfarma. Prevalence of hypertension and associated cardiovascular risk factors among pharmacies customers: an Italian nationwide epidemiological survey. Eur J Prev Cardiol. 2020;27:1228–30.
Jain G, Warnock DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract. 2011;118:c43–8.
Tuttolomondo A, Pecoraro R, Simonetta I, Miceli S, Pinto A, Licata G. Anderson-Fabry disease: a multiorgan disease. Curr Pharm Des. 2013;19:5974–96.
Pieroni M, Chimenti C, Ricci R, Sale P, Russo MA, Frustaci A. Early detection of Fabry cardiomyopathy by tissue Doppler imaging. Circulation. 2003;107:1978–84.
Barbey F, Brakch N, Linhart A, Rosenblatt-Velin N, Jeanrenaud X, Qanadli S, et al. Cardiac and vascular hypertrophy in Fabry disease: evidence for a new mechanism independent of blood pressure and glycosphingolipid deposition. Arterioscler Thromb Vasc Biol. 2006;26:839–44.
Desnick RJ, Blieden LC, Sharp HL, Hofschire PJ, Moller JH. Cardiac valvular anomalies in Fabry disease. Clinical, morphologic, and biochemical studies. Circulation. 1976;54:818–25.
Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci USA. 2008;105:2812–7.
Brakch N, Dormond O, Bekri S, Golshayan D, Correvon M, Mazzolai L, et al. Evidence for a role of sphingosine-1 phosphate in cardiovascular remodelling in Fabry disease. Eur Heart J. 2010;31:67–76.
Rombach SM, Twickler TB, Aerts JMFG, Linthorst GE, Wijburg FA, Hollak CEM. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010;99:99–108.
Dandona P, Dhindsa S, Ghanim H, Chaudhuri A. Angiotensin II and inflammation: the effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J Hum Hypertens. 2007;21:20–7.
Zhang H. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc Res. 1999;44:215–22.
Biancardi VC, Bomfim GF, Reis WL, Al-Gassimi S, Nunes KP. The interplay between Angiotensin II, TLR4 and hypertension. Pharmacol Res. 2017;120:88–96.
Ruiz-Ortega M, Lorenzo O, Rupérez M, Esteban V, Suzuki Y, Mezzano S, et al. Role of the renin-angiotensin system in vascular diseases: expanding the field. Hypertension. 2001;38:1382–7.
Del Pinto R, Ferri C. Inflammation-accelerated senescence and the cardiovascular system: mechanisms and perspectives. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19123701.
Nakamura S, Nakamura I, Ma L, Vaughan DE, Fogo AB. Plasminogen activator inhibitor-1 expression is regulated by the angiotensin type 1 receptor in vivo1. Kidney Int. 2000. https://doi.org/10.1046/j.1523-1755.2000.00160.x.
Shen J-S, Meng X-L, Moore DF, Quirk JM, Shayman JA, Schiffmann R, et al. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab. 2008;95:163–8.
Biancini GB, Vanzin CS, Rodrigues DB, Deon M, Ribas GS, Barschak AG, et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim Biophys Acta. 2012;1822:226–32.
Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122:19–27.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–40.
Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H. Immune mechanisms in arterial hypertension. J Am Soc Nephrol. 2016;27:677–86.
Galon J, Aksentijevich I, McDermott MF, O’Shea JJ, Kastner DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol. 2000;12:479–86.
Hoffman HM, Broderick L. The role of the inflammasome in patients with autoinflammatory diseases. J Allergy Clin Immunol. 2016;138:3–14.
De Francesco PN, Mucci JM, Ceci R, Fossati CA, Rozenfeld PA. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab. 2013;109:93–9.
McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H184–96.
White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron. 1964;1:93–102.
Nunes KP, de Oliveira AA, Lima VV, Webb RC. Toll-like receptor 4 and blood pressure: lessons from animal studies. Front Physiol. 2019;10:655.
Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, et al. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci. 2012;122:535–43.
Zhang Y, Peng W, Ao X, Dai H, Yuan L, Huang X, et al. TAK-242, a toll-like receptor 4 antagonist, protects against aldosterone-induced cardiac and renal injury. PLoS One. 2015;10:e0142456.
Pushpakumar S, Ren L, Kundu S, Gamon A, Tyagi SC, Sen U. Toll-like receptor 4 deficiency reduces oxidative stress and macrophage mediated inflammation in hypertensive kidney. Sci Rep. 2017;7:6349.
Sollinger D, Eißler R, Lorenz S, Strand S, Chmielewski S, Aoqui C, et al. Damage-associated molecular pattern activated toll-like receptor 4 signalling modulates blood pressure in L-NAME-induced hypertension. Cardiovasc Res. 2014;101:464–72.
Ji Y, Liu J, Wang Z, Liu N. Angiotensin II induces inflammatory response partly via toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem. 2009;23:265–76.
Nakashima T, Umemoto S, Yoshimura K, Matsuda S, Itoh S, Murata T, et al. TLR4 is a critical regulator of angiotensin II-induced vascular remodeling: the roles of extracellular SOD and NADPH oxidase. Hypertens Res. 2015;38:649–55.
Hernanz R, Martínez-Revelles S, Palacios R, Martín A, Cachofeiro V, Aguado A, et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol. 2015;172:3159–76.
De Batista PR, Palacios R, Martín A, Hernanz R, Médici CT, Silva MASC, et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS One. 2014;9:e104020.
Del Pinto R, Pietropaoli D, Russomando U, Evangelista P, Ferri C. Health status of Afro-Asian refugees in an Italian urban area: a cross-sectional monocentric study. Public Health. 2018;158:176–82.
Krämer J, Bijnens B, Störk S, Ritter CO, Liu D, Ertl G, et al. Left ventricular geometry and blood pressure as predictors of adverse progression of Fabry cardiomyopathy. PLoS One. 2015;10:e0140627.
Del Pinto R, Wright JT, Monaco A, Pietropaoli D, Ferri C. Vitamin D and blood pressure control among hypertensive adults: results from NHANES 2001–2014. J Hypertens. 2020;38:150–8.
Pietropaoli D, Del Pinto R, Ferri C, Ortu E, Monaco A. Definition of hypertension-associated oral pathogens in NHANES. J Periodontol. 2019;90:866–76.
Del Pinto R, Pietropaoli D, Munoz-Aguilera E, D’Aiuto F, Czesnikiewicz-Guzik M, Monaco A, et al. Periodontitis and hypertension: is the association causal? High Blood Press Cardiovasc Prev. 2020;27:281–9.
Mai F, Del Pinto R, Ferri C. COVID-19 and cardiovascular diseases. J Cardiol. 2020. https://doi.org/10.1016/j.jjcc.2020.07.013.
Pietropaoli D, Del Pinto R, Ferri C, Wright JT Jr, Giannoni M, Ortu E, et al. Poor oral health and blood pressure control among US hypertensive adults. Hypertension. 2018;72:1365–73.
Pietropaoli D, Monaco A, D’Aiuto F, Muñoz Aguilera E, Ortu E, Giannoni M, et al. Active gingival inflammation is linked to hypertension. J Hypertens. 2020. doi:https://doi.org/10.1097/HJH.0000000000002514.
Kleinert J, Dehout F, Schwarting A, de Lorenzo AG, Ricci R, Kampmann C, et al. Prevalence of uncontrolled hypertension in patients with Fabry disease. Am J Hypertens. 2006;19:782–7.
Ku E, Lee BJ, Wei J, Weir MR. Hypertension in CKD: core curriculum 2019. Am J Kidney Dis. 2019;74:120–31.
Patel MR, Cecchi F, Cizmarik M, Kantola I, Linhart A, Nicholls K, et al. Cardiovascular events in patients with fabry disease natural history data from the fabry registry. J Am Coll Cardiol. 2011;57:1093–9.
Del Pinto R, Grassi D, Properzi G, Desideri G, Ferri C. Low density lipoprotein (LDL) cholesterol as a causal role for atherosclerotic disease: potential role of PCSK9 inhibitors. High Blood Press Cardiovasc Prev. 2019;26:199–207.
Germain DP, Elliott PM, Falissard B, Fomin VV, Hilz MJ, Jovanovic A, et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol Genet Metab Rep. 2019;19:100454.
(2020). http://garrod.ca/wp-content/uploads/2019/04/Canadian-Fabry-Treatment-Guidelines-2018-final.pdf. Accessed 13 Aug 2020
Romero CA, Orias M, Weir MR. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol. 2015;11:242–52.
Desideri G, Grassi D, Croce G, Bocale R, Tiberti S, Evangelista S, et al. Different effects of angiotensin converting enzyme inhibitors on endothelin-1 and nitric oxide balance in human vascular endothelial cells: evidence of an oxidant-sensitive pathway. Mediators Inflamm. 2008;2008:305087.
Brasier AR, Recinos A 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–66.
Weidemann F, Sanchez-Niño MD, Politei J, Oliveira J-P, Wanner C, Warnock DG, et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013;8:116.
Del Pinto R, Ferri C. Hypertension management at older age: an update. High Blood Press Cardiovasc Prev. 2019;26:27–36.
Townsend RR, Chang TI, Cohen DL, Cushman WC, Evans GW, Glasser SP, et al. Orthostatic changes in systolic blood pressure among SPRINT participants at baseline. J Am Soc Hypertens. 2016;10:847–56.
Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol. 2007;18:2609–17.
Hilz MJ, Marthol H, Schwab S, Kolodny EH, Brys M, Stemper B. Enzyme replacement therapy improves cardiovascular responses to orthostatic challenge in Fabry patients. J Hypertens. 2010;28:1438–48.
Zanoli L, Briet M, Empana JP, Cunha PG, Mäki-Petäjä KM, Protogerou AD, et al. Vascular consequences of inflammation: a position statement from the ESH working group on vascular structure and function and the ARTERY society. J Hypertens. 2020;38:1682–98.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by Università degli Studi dell’Aquila within the CRUI-CARE Agreement.
Conflict of interest
The authors declare that they have no conflict of interest statement.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Del Pinto, R., Ferri, C. The role of Immunity in Fabry Disease and Hypertension: A Review of a Novel Common Pathway. High Blood Press Cardiovasc Prev 27, 539–546 (2020). https://doi.org/10.1007/s40292-020-00414-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40292-020-00414-w