
Abstract
Introduction
A fixed-dose combination (FDC) of ibuprofen and acetaminophen has been developed that provides greater analgesic efficacy than either agent alone at the same doses without increasing the risk for adverse events.
Methods
We report three clinical phase I studies designed to assess the pharmacokinetics (PK) of the FDC of ibuprofen/acetaminophen 250/500 mg (administered as two tablets of ibuprofen 125 mg/acetaminophen 250 mg) in comparison with its individual components administered alone or together, and to determine the effect of food on the PK of the FDC. Two studies in healthy adults aged 18–55 years used a crossover design in which subjects received a single dose of each treatment with a 2-day washout period between each. In the third study, the bioavailability of ibuprofen and acetaminophen from a single oral dose of the FDC was assessed in healthy adolescents aged 12–17 years, inclusive.
Results
A total of 35 and 46 subjects were enrolled in the two adult studies, respectively, and 21 were enrolled in the adolescent study. Ibuprofen and acetaminophen in the FDC were bioequivalent to the monocomponents administered alone or together. With food, the maximum concentration (Cmax) for ibuprofen and acetaminophen from the FDC was reduced by 36% and 37%, respectively, and time to Cmax (i.e. tmax) was delayed. Overall drug exposure to ibuprofen or acetaminophen in the fed versus fasted states was similar. In adolescents, overall exposure to acetaminophen and ibuprofen was comparable with that in adults, with a slightly higher overall exposure to ibuprofen. Exposure to acetaminophen and ibuprofen in adolescents aged 12–14 years was slightly higher versus those aged 15–17 years. Adverse events were similar across all treatment groups.
Conclusions
The FDC of ibuprofen/acetaminophen 250/500 mg has a PK profile similar to its monocomponent constituents when administered separately or coadministered, indicating no drug–drug interactions and no formulation effects. Similar to previous findings for the individual components, the rates of absorption of ibuprofen and acetaminophen from the FDC were slightly delayed in the presence of food. Overall, adolescents had similar exposures to acetaminophen and ibuprofen as adults, while younger adolescents had slightly greater exposure than older adolescents, probably due to their smaller body size. The FDC was generally well tolerated.
Similar content being viewed by others


The pharmacokinetic profile of a fixed-dose combination (FDC) of ibuprofen 250 mg and acetaminophen 500 mg was found to be similar to its individual components administered separately or coadministered; bioequivalence was demonstrated for both overall and maximal exposure. |
Similar to what has been previously reported for the individual ingredients, food delayed the absorption of both components but had no effect on overall exposure; exposure to both components in adolescents was similar to that in adults, with younger adolescents having slightly greater exposure. |
Since the efficacy of this FDC has been shown to be superior to the same doses of individual components and it is generally well tolerated, this new FDC may provide another analgesic treatment option to relieve pain. |
1 Introduction
Ibuprofen and acetaminophen are among the most widely used non-prescription over-the-counter (OTC) analgesic/antipyretic drugs, both in the US and globally [1, 2]. The efficacy of these agents for the treatment of mild-to-moderate acute pain and fever in the OTC setting is well established [2,3,4,5]. Ibuprofen is a nonsteroidal anti-inflammatory drug (NSAID) that inhibits the cyclooxygenase (COX)-1 and -2 isoenzymes and hence the synthesis of pro-inflammatory prostaglandins, whereas acetaminophen is believed to act through inhibition of a subclass of COX enzyme isoforms in the central nervous system [6]. Additionally, acetaminophen has been reported to have effects on descending inhibitory serotonergic pain pathways to inhibit the l-arginine nitric oxide pathway; effects on cannabinoid receptors may also be operant [7]. Both ibuprofen and acetaminophen are associated with a ceiling effect for pain relief, i.e. a point at which higher single doses of either agent provide comparable changes in pain scores versus lower doses; respective dose ceilings are 400 mg for ibuprofen [8, 9] and 1000 mg for acetaminophen [10]. Conversely, the risk of adverse events (AEs) with ibuprofen and acetaminophen, such as gastrointestinal toxicity and acute liver failure, respectively, increases with greater doses of each agent [11, 12].
Previous phase III clinical studies have demonstrated better efficacy of a fixed-dose combination (FDC) of ibuprofen and acetaminophen relative to monoactives in the same doses [13, 14]. Combining ibuprofen and acetaminophen may also allow for the effective use of lower doses of both agents, potentially reducing safety concerns associated with these drugs when administered alone at higher doses [11]. Indeed, combinations of ibuprofen and acetaminophen have been previously studied and have been shown to be effective in the management of acute pain and reduction of fever [3, 13,14,15,16]. Furthermore, these studies also demonstrated a safety profile of the ibuprofen/acetaminophen combination comparable or superior to the individual components [3, 13,14,15].
Pharmacokinetic (PK) studies have demonstrated no alterations in plasma drug concentrations, indicating no drug–drug interactions, when ibuprofen and acetaminophen are administered concomitantly [6, 17]; therefore, no additional safety concerns are expected when ibuprofen and acetaminophen are used in combination compared with either agent alone. In this study, we report on three separate clinical PK studies of a new FDC of ibuprofen and acetaminophen containing lower doses of the two ingredients (250 mg ibuprofen/500 mg acetaminophen, administered as two tablets of 125 mg ibuprofen/250 mg acetaminophen) than the maximum recommended doses of the single-ingredient products. Analgesic studies of this FDC have demonstrated efficacy superior to the individual components and comparable to the maximal analgesic dose of ibuprofen (400 mg; Kellstein and Leyva, unpublished data). The objectives of the three studies detailed herein were (1) to determine the relative bioavailability of ibuprofen and acetaminophen from this new FDC compared with its individual monocomponents administered together or separately in adults; (2) to evaluate the effects of food on PK in adults; and (3) to determine exposure to ibuprofen and acetaminophen from the FDC in adolescents.
2 Methods
2.1 Study Design
Three clinical phase I PK studies were conducted. Study 1 (conducted from January to March 2016) and Study 2 (conducted from December 2015 to March 2016) were performed at the Pfizer Clinical Research Unit (New Haven, CT, USA) and were open-label, four-way crossover studies that enrolled healthy adults. The third study (Study 3) was carried out from August to November 2016 at three clinical research centers across the US (WCCT Global, Costa Mesa, CA; Pharmaceutical Research Associates, Salt Lake City, UT; and Altasciences/Vince and Associates Clinical Research, Overland Park, KS) and was an open-label, single-dose study in healthy adolescents. The studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonization Good Clinical Practice guidelines, as well as local regulatory requirements. All subjects (or parents or guardians in the adolescent study) provided written informed consent, and informed assent was obtained from each minor subject. The studies were managed by Pfizer Inc. and conducted by investigators employed or contracted by, and under the direction of, Pfizer Inc. Data management and analyses were conducted by Pfizer Inc., and investigators had full access and control over data interpretation for this study.
Subjects included in the two adult studies (i.e. Studies 1 and 2) were healthy male and female volunteers aged 18–55 years, inclusive. Included subjects had a body mass index (BMI) of 17.5–30.5 kg/m2, inclusive, and a total body weight of > 50 kg. For inclusion in Study 3, subjects were healthy males and females 12–17 years of age, inclusive, with at least one painful condition (e.g. headache, dysmenorrhea, or musculoskeletal pain) that required the use of an oral OTC analgesic five or more times in the previous 4 weeks. Total body weight, stature, and BMI must have been within the 10th and 90th percentile for age and sex. Female subjects of childbearing potential and males able to father children must have been willing to use a highly effective method of contraception for the duration of the study and for 28 days after the last dose of study medication.
Exclusion criteria were similar in each of the three studies. Pregnant or breastfeeding subjects were not allowed to participate in any of the studies. The presence or history of significant disease, bleeding disorder, signs of dehydration (adolescents), or any other condition in which study medication may have increased risk; any condition that could have affected drug absorption (e.g. gastrectomy); a positive urine drug screen; history of regular alcohol consumption (adults); alcohol use within 24 h (adults) or 48 h (adolescents) of dosing; or use of tobacco within 6 months of screening (adults) or within 24 h prior to dosing (adolescents) was exclusionary. Treatment with an investigational drug within 30 days, use of prescription and non-prescription drugs or dietary supplements within 7 days, or use of nutritional supplements within 14 days prior to the first dose of study drug was prohibited. Subjects with hypertension (i.e. blood pressure ≥ 140/90 mmHg in adults or ≥ 95th percentile for age and height in adolescents); QT or QRS prolongation of > 450 ms or > 120 ms, respectively; aspartate or alanine aminotransferase ≥ 3 × or total bilirubin ≥ 1.5 × the upper limit of normal; blood donation of approximately 1 pint (adults) or exceeding 130 mL or 3 mL/kg (adolescents) within 56 days; hypersensitivity to aspirin, acetaminophen, ibuprofen, or any other NSAID; consumption of grapefruit or related citrus fruits within 7 days prior to dosing; use of acetaminophen, ibuprofen, or any other NSAID within 48 h prior to the first dose of study medication; or use of caffeine or alcohol within 24 h of admission prompted study exclusion. Subjects with a positive test for hepatitis B, hepatitis C, or human immunodeficiency virus were excluded.
2.2 Determination of Sample Size
In a previous publication [6] studying the PK profile of a novel FDC tablet formulation of ibuprofen and paracetamol (ibuprofen 400 mg/acetaminophen 1000 mg) under fasted and fed conditions, it was observed that the fed versus fasted ratio of Cmax was lower than 80% for both ibuprofen and acetaminophen, suggesting that it was unlikely to achieve bioequivalence regardless of the sample size used for this parameter. Therefore, the sample size calculation in Study 1 was focused on having adequate power for declaring bioequivalence in terms of area under the concentration-time curve from time zero to infinity (AUC∞). The sample size calculation was based on the results of the log-transformed AUC∞ in acetaminophen, the analyte with the higher variability in this study.
In order to provide at least 85% power for declaring bioequivalence for AUC∞ in Study 1, 32 subjects were required to complete the study. This sample size estimate assumed a root mean square of error (RMSE) of 0.120 and that the true bioavailability of the test formulation was within 15% of that for the reference formulation [6]. To ensure at least 32 subjects completed all four periods of the study, approximately 36 subjects were to be enrolled.
For Study 2, acetaminophen and log-transformed Cmax, the analyte and the parameter with the highest variabilities observed in previous Pfizer Consumer Healthcare (PCH) studies, were used in the sample size calculation. Additionally, it was assumed that the true bioavailability of the test formulation (FDC ibuprofen/acetaminophen 250/500 mg) was within 5.0% of that for the reference formulation and that it was similar between the two analytes, ibuprofen and acetaminophen. This was observed in a previous publication studying the PK profiles of a novel FDC tablet of ibuprofen and acetaminophen (ibuprofen 400 mg/acetaminophen 1000 mg) [6].
Using these assumptions and an RMSE of 0.2742 (observed in a previous PCH study [#PA-96-01], which studied the PK profiles of different acetaminophen formulations of 1000 mg; unpublished data), it was estimated that a sample size of approximately 40 subjects would provide at least 85% power to establish bioequivalence in Study 2. To ensure approximately 40 subjects completed all four periods of the study, at least 44 subjects were to be enrolled.
For Study 3, review of previous PK data for ibuprofen and acetaminophen revealed that acetaminophen and the apparent oral clearance (CL), as calculated by the dose administered/AUC∞ (CL/F), were the analyte and PK parameter, respectively, with the higher variability. Taking into consideration regulatory guidance from the US FDA as well as historical PK data, and using the method proposed by Wang et al. [18], it was estimated that a sample size of nine subjects within each of the age groups (12–14 years and 15–17 years) would provide at least 90% power to target the 95% confidence interval (CI) to be within 60% and 140% of the geometric mean estimate of CL/F for FDC ibuprofen/acetaminophen in each age subgroup, assuming an approximate between-subject coefficient of variability of 30% for the untransformed CL for acetaminophen based on a previous adult PK study conducted by PCH. To allow for dropouts in this multicenter study, approximately 22 subjects were to be enrolled to ensure that a minimum of 18 subjects completed the study (at least 9 within each age subgroup).
2.3 Study Treatments
Subjects were administered a single dose of study drug at baseline as described below. In Study 1, subjects were randomized to receive a single dose of 2 × FDC ibuprofen/acetaminophen 125 mg/250 mg (i.e. FDC ibuprofen/acetaminophen 250/500 mg; ©2019 GSK group of companies or its licensor, Madison, NJ, USA) either after a 10-h fast or following a high-fat breakfast, or ibuprofen 200 mg (Advil®; ©2019 GSK group of companies or its licensor), or acetaminophen extended release (ER) 650 mg (Tylenol® 8 HR; Johnson & Johnson Consumer Inc, New Brunswick, NJ, USA), both fasted. Acetaminophen 650 mg ER and ibuprofen 200 mg were used for comparison based on regulatory requirements. In Study 2, subjects were randomized to 2 × FDC ibuprofen/acetaminophen 125/250 mg, coadministered monocomponents ibuprofen 250 mg (©2019 GSK group of companies or its licensor) and acetaminophen 500 mg (Tylenol® Extra Strength; McNeil Consumer Healthcare, Ft. Washington, PA, USA), ibuprofen 250 mg alone, or acetaminophen 500 mg alone following an overnight fast of at least 10 h. Studies 1 and 2 had a crossover design; subjects received all four treatments, with a 2-day washout between each treatment period. Subjects remained in the clinic for the duration of the study. In Study 3, all subjects received a single dose of 2 × FDC ibuprofen/acetaminophen 125/250 mg (i.e. FDC ibuprofen/acetaminophen 250/500 mg) after an overnight fast.
2.4 Phamacokinetic (PK) Sampling, Analysis, and Calculations
In each study, a predose PK sample was obtained approximately 60 min before each treatment period. PK sampling was conducted at 10, 20, 30, 40, 50, 60, 75, and 90 min and 2, 3, 4, 6, 8, 10, 12, 18, and 24 h after dosing in Study 1, and at the same time points through 12 h in Study 2. Samples were obtained at 10, 20, 30, 45, 60, 75, and 90 min and at 2, 3, 6, 9, and 12 h in Study 3.
For all three studies, plasma samples were analyzed for total ibuprofen and acetaminophen using a validated analytical assay employing a high-performance liquid chromatography tandem mass spectrometric method by PPD (Middleton, WI, USA). The lower limit of quantitation (LLOQ) of the assay for ibuprofen and acetaminophen was 0.2 and 0.1 μg/mL, respectively. Clinical specimens with plasma ibuprofen or acetaminophen concentrations below the LLOQ were reported as below the LLOQ. In Studies 1 and 2, PK metrics were calculated for each subject using non-compartmental analysis of plasma concentration-time data using the internally validated software system electronic non-compartmental analysis (eNCA) version 2.2.4. In Study 3, PK metrics were calculated using Phoenix WinNonlin version 6.4 (Pharsight Corporation, Mountain View, CA, USA).
PK metrics calculated included Cmax, time to maximum concentration (tmax), area under the concentration-time curve from time zero to the last quantifiable concentration (AUClast), AUC∞, and terminal half-life (t½). The comparability of PK metrics in Studies 1 and 2 was determined by constructing 90% CIs around the estimated difference between test and reference treatments using a mixed-effects model based on natural log-transformed data. The mixed-effects model was implemented using SAS PROC MIXED (SAS Institute, Inc., Cary, NC, USA) with the restricted maximum likelihood estimation method and the Kenward–Roger degrees of freedom algorithm. Because the monocomponent doses in Study 1 were different from those of the FDC, PK metrics were dose normalized to ibuprofen 250 mg and acetaminophen 500 mg for the purposes of comparison.
Safety, including AEs, was monitored throughout the in-patient portion of the studies and during a follow-up phone call 14 days after the last investigational drug dose in each study.
3 Results
3.1 Baseline Characteristics
Baseline characteristics for the three studies are shown in Table 1. A total of 35 subjects were randomized in Study 1 and 46 in Study 2. In Study 3, 21 subjects were assigned to treatment. In all three studies, the proportion of males and females was approximately 50%. In Study 3, the majority (62%) of subjects were White, whereas in Studies 1 and 2, the largest proportion of subjects were Black (54% and 41%, respectively). One subject in Study 1 discontinued study drug during the first treatment period due to an inability to swallow study medication, and two subjects in Study 2 discontinued study drug during the first treatment period due to difficulties in collecting PK samples; no PK profiling was possible for these two subjects. All 21 subjects in Study 3 completed treatment and were analyzed for PK metrics and safety.
3.2 PK Profiles
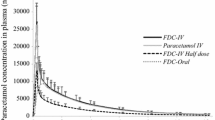
PK concentration-time profiles for ibuprofen and acetaminophen in the three studies are shown in Figs. 1 and 2, respectively.

Median plasma ibuprofen concentration over time following a single oral dose: a Study 1; b Study 2; c Study 3. Note that plasma concentrations for IBU 200 mg in Study 1 are dose-normalized. APAP acetaminophen, FDC fixed-dose combination, IBU ibuprofen

Median plasma acetaminophen concentration over time following a single oral dose: a Study 1; b Study 2; c Study 3. Note that plasma concentrations for APAP ER 650 mg in Study 1 are dose-normalized. APAP acetaminophen, ER extended-release, FDC fixed-dose combination, IBU ibuprofen
3.2.1 Study 1: Comparison of Fixed-Dose Combination (FDC) to Single-Ingredient Products and Food Effects
PK metrics for ibuprofen and acetaminophen in Study 1 are summarized in Table 2. Under fasted conditions, the median plasma ibuprofen dose-normalized concentration-time profiles were similar for FDC ibuprofen/acetaminophen 250/500 mg and ibuprofen 200 mg alone (Fig. 1a) except for a shorter ibuprofen tmax for the FDC (1.38 h vs. 2.00 h, respectively) (Table 2). As seen in Table 3, the ratios of AUC∞ (99.93%), AUClast (100.63%), and Cmax (102.44%) of FDC ibuprofen/acetaminophen 250/500 mg versus ibuprofen 200 mg under fasted conditions, and their 90% CIs, were completely contained within the limits of 80–125%, indicating bioequivalence under fasted conditions.
There was no meaningful food effect on the overall extent of absorption of ibuprofen from the FDC. The ratios for FDC fed/FDC fasted of the adjusted dose-normalized geometric means for AUC∞ and AUClast were 86.61% and 85.96%, respectively. The 90% CIs for these AUC values (Table 3) were within the acceptance range for bioequivalence of 80–125%. However, the rate of absorption was delayed with food compared with fasting; the ratio for dose-normalized Cmax for ibuprofen was 63.86% (Table 3), indicative of an approximately 36% lower peak concentration of ibuprofen when the FDC was administered in the fed state. Similarly, the tmax was delayed for ibuprofen when the FDC was administered with food: 3.00 h with food and 1.38 h fasted. The t½ was similar with or without food (2.4 h vs. 2.2 h, respectively) (Table 2).
Under fasted conditions, the median plasma acetaminophen dose-normalized concentration-time profiles were similar for FDC ibuprofen/acetaminophen 250/500 mg and acetaminophen ER 650 mg alone (Fig. 2a), with the exception of a shorter tmax for the FDC (0.58 h vs. 1.50 h, respectively) (Table 2). The overall relative bioavailability of the two treatments was equivalent based on dose-normalized test/reference ratios for AUC∞ (108.60) and AUClast (108.27) and their associated 90% CIs (Table 4). The ratio for dose-normalized Cmax was 177.48%, indicating an approximately 77% higher peak concentration of acetaminophen derived from the FDC ibuprofen/acetaminophen 250/500 mg fasted treatment compared with that of acetaminophen ER 650 mg fasted treatment (Table 4), a finding that was not unexpected given use of the ER acetaminophen formulation in this comparison.
There was no effect of food on the extent of absorption of the acetaminophen from the FDC. The ratios of acetaminophen fed/acetaminophen fasted (Table 4) of dose-normalized geometric means for AUC∞ and AUClast were 95.01% and 94.29%, respectively; the 90% CIs for each were within the acceptance range for bioequivalence of 80–125%, indicating no relevant effect of food on these metrics. As with ibuprofen, the absorption rate of acetaminophen was delayed when the FDC was taken with food compared with fasting. The ratio for dose-normalized Cmax was 63.22%, indicating an approximately 37% lower peak concentration for acetaminophen after administration of the FDC in the fed state compared with the fasted state (Table 4). Similarly, tmax was 2.49 h with food and 0.58 h when fasted. The t½ for acetaminophen was similar with and without food (4.7 h vs. 4.6 h, respectively) (Table 2).
3.2.2 Study 2: Comparison of FDC with Individual Components and Formulation Effects
PK metrics for ibuprofen and acetaminophen in Study 2 are summarized in Table 5. As shown in Fig. 1b, the median plasma ibuprofen concentration-time profiles were similar for FDC ibuprofen/acetaminophen 250/500 mg, monocomponent ibuprofen 250 mg + acetaminophen 500 mg administered together, and ibuprofen 250 mg alone. The Cmax was 12% higher following the FDC ibuprofen/acetaminophen 250/500 mg treatment than following treatment with either the coadministered monocomponents or ibuprofen 250 mg (Table 5). The relative bioavailability of treatments was similar based on AUC∞, AUClast, and Cmax (Table 6), where the respective ratios of adjusted geometric means of ibuprofen were 102.53%, 103.00%, and 112.42% after administration of the FDC relative to the coadministered monocomponents. Similar results were obtained for the FDC relative to ibuprofen 250 mg alone. The ratios of adjusted geometric means of AUC∞, AUClast, and Cmax were 102.60%, 103.08%, and 112.02%, respectively, following administration of FDC relative to ibuprofen 250 mg alone. The 90% CIs for each of these test/reference ratios reported were contained within the bioequivalence acceptance range of 80–125%, indicating bioequivalence.
As shown in Fig. 2b, the median plasma acetaminophen concentration-time curves for FDC ibuprofen/acetaminophen 250/500 mg, monocomponent ibuprofen 250 mg + acetaminophen 500 mg, and acetaminophen 500 mg alone were similar for all treatments, and the bioavailability of treatments was also similar based on AUC∞, AUClast, and Cmax and their associated 90% CIs (Table 6). The ratios of adjusted geometric means of acetaminophen AUC∞, AUClast, and Cmax were 99.79%, 100.00%, and 94.32 after administration of FDC relative to the coadministered monocomponents; each respective 90% CI fell within the limits indicative of bioequivalence. For the comparison of FDC with acetaminophen 500 mg alone, the ratios of adjusted geometric means of acetaminophen AUC∞, AUClast, and Cmax were 104.00%, 104.12%, and 101.64%, respectively; all CIs were contained within the bioequivalence acceptance range of 80–125% (Table 6), also indicating bioequivalence.
3.2.3 Study 3: Evaluation of PK in Adolescents
PK metrics for ibuprofen and acetaminophen following administration of the FDC to adolescents in Study 3 are summarized in Table 7. Results are presented for all subjects and for the age groups 12–14 years and 15–17 years separately. The overall ibuprofen exposure (AUC values) following administration of the FDC was similar for both the younger and older age groups. However, the younger group had a Cmax that was approximately 23% higher and occurred 1 h earlier relative to the older subjects (tmax: 1 vs. 2 h, respectively). Overall acetaminophen exposure was approximately 30% higher in the younger age group compared with the older age group. As with ibuprofen, Cmax for acetaminophen was higher in the younger group (approximately 42%) and tmax was 0.5 h faster relative to the older group (0.5 vs. 1 h, respectively). The mean t½ values were similar across all age groups.
Although not designed for a direct comparison, a numerical comparison of the results for the FDC in the fasted state in adolescents (Table 7) with those in adults (Table 2) indicated that the overall (AUC) and maximal (Cmax) exposures to ibuprofen and acetaminophen in the adolescent group were similar to those in adults; overall exposure (AUC) to ibuprofen was slightly higher (95 µg∙h/mL vs. 77–78 µg∙h/mL) in adolescents.
3.3 Safety
AEs across the three trials were all mild or moderate in intensity; treatment-emergent AEs are summarized in Table 8. In Study 1, seven subjects (20%) reported 10 AEs; three AEs in two subjects were determined to be treatment-related: one subject experienced nausea (after ibuprofen 200 mg) and somnolence (after FDC fasted), and another reported headache (after FDC fed). All of these treatment-related AEs were mild and resolved. In Study 2, 10 subjects (21.7%) experienced 15 AEs. Six of these AEs in three subjects were determined to be treatment-related: two subjects experienced treatment-related constipation, and a third experienced abdominal distention, upper abdominal pain, dyspepsia, and nausea. Two of these AEs occurred during treatment with the monocomponents administered together, one during treatment with ibuprofen 250 mg, and three during treatment with acetaminophen 500 mg. All of these treatment-related AEs were mild and resolved. In Study 3, two subjects (9.5%) experienced five mild treatment-emergent AEs. One AE of dizziness was considered to be related to treatment with the FDC. No dose reductions or discontinuations due to AEs, no serious AEs, and no deaths occurred in any of the three studies. Likewise, no clinically significant changes in vital signs were apparent in any study. The safety profile of the FDC observed during each study was consistent with the known safety profile of the individual components.
4 Discussion
This series of PK studies demonstrates that the FDC ibuprofen/acetaminophen 250/500 mg (administered as 2 × ibuprofen/acetaminophen 125/250 mg) is bioequivalent to its individual monocomponents when administered separately or together. These data substantiate the lack of drug–drug PK interactions or formulation effects, respectively, with the combination. Acetaminophen in the FDC is bioequivalent to dose-normalized acetaminophen ER 650 mg for AUC. However, an increase in acetaminophen Cmax was seen with the FDC compared with the acetaminophen ER 650 mg comparator. This result was expected as the FDC is an immediate-release formulation, whereas the acetaminophen 650 mg used was an ER formulation with delayed absorption. Ibuprofen in the FDC was also bioequivalent to dose-normalized ibuprofen 200 mg for AUC and Cmax. These results therefore demonstrate that the exposure to ibuprofen and acetaminophen in the FDC is similar to those of commercially available formulations of the individual components, indicating there should be no increased safety concerns.
A food effect on the rate of absorption was observed with the FDC, with Cmax for ibuprofen and acetaminophen decreased 36% and 37%, respectively, in the presence of food. Tmax was also delayed by approximately 1.6 h for ibuprofen and 1.9 h for acetaminophen in fed, compared with fasting, conditions. This is consistent with a previous study in which an FDC of ibuprofen/acetaminophen at a total dose of 400/1000 mg, respectively, exhibited decreased Cmax values in the fed versus fasted state (ratios of 76% and 61% for ibuprofen and acetaminophen, respectively) and delayed median tmax by 0.75 h and 1 h, respectively [6]. Studies of ibuprofen alone and acetaminophen alone have also demonstrated decreased Cmax and delayed tmax when either is administered with food [19, 20]. Although the overall extent of absorption (AUC) of ibuprofen from the FDC was slightly reduced (14%) in the presence of food compared with ibuprofen alone, both AUC∞ and AUClast met the bioequivalence standard of 80–125%. The overall exposure to acetaminophen was not affected by food, therefore it is unlikely that the slightly reduced AUC of ibuprofen observed in the fed state would be clinically meaningful in terms of analgesic efficacy. However, the effect of food on efficacy has not been evaluated as the dental pain studies of the FDC were conducted with food restrictions, as is the standard methodology.
The majority of PK studies of ibuprofen and acetaminophen have focused on adults and younger subjects, with limited data available in adolescents aged 12–17 years [6, 21,22,23,24,25,26,27,28]. As a result, population PK estimates and allometric adjustments or scaling have typically provided the rationale for dosing in this younger age group [27, 28]. The present study of FDC ibuprofen/acetaminophen 250/500 mg therefore adds new and relevant information by reporting ibuprofen and acetaminophen PK in this age group. Importantly, no clinically important differences in PK exposure were observed for adolescents in comparison with adult subjects 18 years of age and older. Indeed, while these studies were not designed to be compared, the overall plasma concentration-time curves for both components of the FDC (in the fasted state) were similar across all three studies, and comparison of AUC and Cmax values between the adolescent and adult studies also indicates similar exposures to both components, with a slightly higher overall exposure to ibuprofen in adolescents. This provides support for similar dosing recommendations for adolescents as in adults.
In the younger age subgroup of subjects aged 12–14 years, ibuprofen and acetaminophen exposures were slightly increased following a single dose of the FDC compared with the older age group aged 15–17 years. This difference was as expected given the smaller body surface area and lower body weight of the younger subjects, and the differences were not clinically meaningful.
These studies confirm the results of previous studies indicating no drug–drug PK interaction between ibuprofen and acetaminophen [6, 17]; therefore, no additional safety concerns are expected compared with the administration of either agent alone. The data also indicate no formulation effect when combining the two ingredients into one tablet. Consistent with these findings, the FDC ibuprofen/acetaminophen 250/500 mg was safe and generally well tolerated in these studies. All AEs were mild or moderate in severity, with no discontinuations due to AEs. AEs were equally distributed between treatment arms, with no new safety concerns compared with the individual components. Importantly, exposure in adolescents was similar to that in adults, and the FDC was generally well tolerated, indicating that this formulation can be safely used in this population. However, it should be noted that these were small, short-duration studies, and rare and serious AEs such as gastrointestinal bleeding and liver failure would not be expected. However, a large, longer-duration study (13 weeks) evaluating a similar FDC of ibuprofen/acetaminophen (200/500 mg) found a safety profile that was at least as favorable as maximum single OTC doses of the individual components (ibuprofen 400 mg, acetaminophen 1000 mg) [15].
5 Conclusions
The FDC ibuprofen/acetaminophen 250/500 mg has a PK profile similar to its monocomponent constituents when administered alone or coadministered. Overall exposure to ibuprofen and acetaminophen was bioequivalent under fed versus fasted conditions, although, as expected, food delayed absorption, similar to what has previously been observed for each individual monocomponent. In adolescents, overall exposure to ibuprofen from the FDC, as measured by AUC, was similar in individuals 12–14 years of age and 15–17 years of age, but Cmax was 23% higher and tmax was achieved earlier in the younger age group of patients. In contrast, acetaminophen exposure (i.e. AUC) was 30% higher in the younger group after administration of the FDC; Cmax was 42% higher and tmax occurred earlier in the younger age group of patients than in older adolescents. Exposure to ibuprofen and acetaminophen in the overall group of adolescents was similar to that in adults, supporting the same dosing in that population.
Data Sharing Statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU, or (2) in programs that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer. On 1 August 2019, PCH became part of GSK Consumer Healthcare.
References
Dickman A. Choosing over-the-counter analgesics. Pharm J. 2008;281:631.
Perrott DA, Piira T, Goodenough B, Champion GD. Efficacy and safety of acetaminophen vs ibuprofen for treating children’s pain or fever: a meta-analysis. Arch Pediatr Adolesc Med. 2004;158:521–6.
Moore RA, Wiffen PJ, Derry S, Maguire T, Roy YM, Tyrrell L. Non-prescription (OTC) oral analgesics for acute pain—an overview of Cochrane reviews. Cochrane Database Syst Rev. 2015;(11):CD010794.
Goldman RD, Ko K, Linett LJ, Scolnik D. Antipyretic efficacy and safety of ibuprofen and acetaminophen in children. Ann Pharmacother. 2004;38:146–50.
Pierce CA, Voss B. Efficacy and safety of ibuprofen and acetaminophen in children and adults: a meta-analysis and qualitative review. Ann Pharmacother. 2010;44:489–506.
Tanner T, Aspley S, Munn A, Thomas T. The pharmacokinetic profile of a novel fixed-dose combination tablet of ibuprofen and paracetamol. BMC Clin Pharmacol. 2010;10:10.
Anderson BJ. Paracetamol (acetaminophen): mechanisms of action. Paediatr Anaesth. 2008;18:915–21.
Laska EM, Sunshine A, Marrero I, Olson N, Siegel C, McCormick N. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin Pharmacol Ther. 1986;40:1–7.
Seymour RA, Ward-Booth P, Kelly PJ. Evaluation of different doses of soluble ibuprofen and ibuprofen tablets in postoperative dental pain. Br J Oral Maxillofac Surg. 1996;34:110–4.
Skoglund LA, Skjelbred P, Fyllingen G. Analgesic efficacy of acetaminophen 1000 mg, acetaminophen 2000 mg, and the combination of acetaminophen 1000 mg and codeine phosphate 60 mg versus placebo in acute postoperative pain. Pharmacotherapy. 1991;11:364–9.
Moore N, Scheiman JM. Gastrointestinal safety and tolerability of oral non-aspirin over-the-counter analgesics. Postgrad Med. 2018;130:188–99.
Becker DE. Pain management: Part 1: managing acute and postoperative dental pain. Anesth Prog. 2010;57:67–78 (quiz 9–80).
Mehlisch DR, Aspley S, Daniels SE, Bandy DP. Comparison of the analgesic efficacy of concurrent ibuprofen and paracetamol with ibuprofen or paracetamol alone in the management of moderate to severe acute postoperative dental pain in adolescents and adults: a randomized, double-blind, placebo-controlled, parallel-group, single-dose, two-center, modified factorial study. Clin Ther. 2010;32:882–95.
Mehlisch DR, Aspley S, Daniels SE, Southerden KA, Christensen KS. A single-tablet fixed-dose combination of racemic ibuprofen/paracetamol in the management of moderate to severe postoperative dental pain in adult and adolescent patients: a multicenter, two-stage, randomized, double-blind, parallel-group, placebo-controlled, factorial study. Clin Ther. 2010;32:1033–49.
Doherty M, Hawkey C, Goulder M, et al. A randomised controlled trial of ibuprofen, paracetamol or a combination tablet of ibuprofen/paracetamol in community-derived people with knee pain. Ann Rheum Dis. 2011;70:1534–41.
Malya RR. Does combination treatment with ibuprofen and acetaminophen improve fever control? Ann Emerg Med. 2013;61:569–70.
Wright CE 3rd, Antal EJ, Gillespie WR, Albert KS. Ibuprofen and acetaminophen kinetics when taken concurrently. Clin Pharmacol Ther. 1983;34:707–10.
Wang Y, Jadhav PR, Lala M, Gobburu JV. Clarification on precision criteria to derive sample size when designing pediatric pharmacokinetic studies. J Clin Pharmacol. 2012;52:1601–6.
Koenigsknecht MJ, Baker JR, Wen B, et al. In vivo dissolution and systemic absorption of immediate release ibuprofen in human gastrointestinal tract under fed and fasted conditions. Mol Pharm. 2017;14:4295–304.
Divoll M, Greenblatt DJ, Ameer B, Abernethy DR. Effect of food on acetaminophen absorption in young and elderly subjects. J Clin Pharmacol. 1982;22:571–6.
Davies NM. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clin Pharmacokinet. 1998;34:101–54.
Walson PD, Mortensen ME. Pharmacokinetics of common analgesics, anti-inflammatories and antipyretics in children. Clin Pharmacokinet. 1989;17(Suppl 1):116–37.
Wang C, Allegaert K, Tibboel D, et al. Population pharmacokinetics of paracetamol across the human age-range from (pre)term neonates, infants, children to adults. J Clin Pharmacol. 2014;54:619–29.
Zuppa AF, Hammer GB, Barrett JS, et al. Safety and population pharmacokinetic analysis of intravenous acetaminophen in neonates, infants, children, and adolescents with pain or fever. J Pediatr Pharmacol Ther. 2011;16:246–61.
Nahata MC, Durrell DE, Powell DA, Gupta N. Pharmacokinetics of ibuprofen in febrile children. Eur J Clin Pharmacol. 1991;40:427–8.
Peterson RG, Rumack BH. Pharmacokinetics of acetaminophen in children. Pediatrics. 1978;62:877–9.
Troconiz IF, Armenteros S, Planelles MV, Benitez J, Calvo R, Dominguez R. Pharmacokinetic–pharmacodynamic modelling of the antipyretic effect of two oral formulations of ibuprofen. Clin Pharmacokinet. 2000;38:505–18.
Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45:1013–34.
Acknowledgements
Medical writing support was provided by John H. Simmons, MD, of Peloton Advantage, LLC, an OPEN Health company, and was funded by Pfizer. On 1 August 2019, PCH became part of GSK Consumer Healthcare. The authors would like to thank Zhongwei Zhou, lead programmer, and all the programmers who supported these studies, as well as the study participants.
Author information
Authors and Affiliations
Contributions
Study design: All authors; Study investigator: Sanela Tarabar, Debra Kelsh; Enrolled subjects: Sanela Tarabar, Debra Kelsh; Collection and assembly of data: All authors; Data analysis: Rina Leyva (Lead Statistician); Data interpretation: All authors; Manuscript preparation: All authors; Manuscript review and revisions: All authors; Final approval of manuscript: All authors.
Corresponding author
Ethics declarations
Funding
These studies were funded by PCH. On 1 August 2019, PCH became part of GSK Consumer Healthcare.
Conflict of interest
Sanela Tarabar, Rina Leyva, Dongweon Song, and Kyle Matschke are employees of and may own stock/stock options in Pfizer Inc. David E. Kellstein and Mario Cruz-Rivera were employees of Pfizer Inc. at the time this research was conducted and may own stock/stock options in Pfizer Inc. Suzanne Meeves was an employee of Pfizer Inc. at the time this research was conducted. Debra Kelsh and Bradley Vince declare they have no conflicts of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Pfizer Clinical Research Unit, New Haven, CT, USA (Studies 1 and 2); WCCT Global, Costa Mesa, CA, USA, Pharmaceutical Research Associates, Salt Lake City, UT, USA, and Altasciences/Vince and Associates Clinical Research, Overland Park, KS, USA (Study 3) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants (or parents or guardians in the adolescent study) included in the studies; informed assent was obtained from each minor subject.
Additional information
The authors David E. Kellstein, Suzanne Meeves and Mario Cruz-Rivera were Employees of Pfizer Consumer Healthcare, Madison, NJ, USA at the time this research was conducted.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tarabar, S., Kelsh, D., Vince, B. et al. Phase I Pharmacokinetic Study of Fixed-Dose Combinations of Ibuprofen and Acetaminophen in Healthy Adult and Adolescent Populations. Drugs R D 20, 23–37 (2020). https://doi.org/10.1007/s40268-020-00293-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00293-5