
Abstract
Background and Objectives
Previously published studies have suggested the lack of a pharmacokinetic interaction between ibuprofen and paracetamol when they are delivered as a fixed-dose oral combination. The aim of this study was to determine the pharmacokinetic profile and safety of a fixed-dose intravenous (IV) combination, containing 3 mg/mL ibuprofen and 10 mg/mL paracetamol, in comparison with its individual components. The study also assessed the relative bioavailability of the same doses of the active ingredients when they were administered as an oral formulation.
Methods
A single-dose, open-label, randomized, five-period cross-over sequence pharmacokinetic study was undertaken in 30 healthy volunteers. Serial plasma samples were assayed for both paracetamol and ibuprofen concentrations, using validated liquid chromatography–tandem mass spectrometry methods. Pharmacokinetic parameters were computed using standard non-compartmental analyses. Adverse events were also assessed. The ratios of the maximum measured plasma concentration (C max), the area under the plasma concentration–time curve (AUC) from time zero to the time of the last measurable plasma concentration (AUC t ) and AUC from time zero to infinity (AUC∞) were analysed for bioequivalence as determined by 90 % confidence intervals.
Results
The pharmacokinetic parameters of ibuprofen and paracetamol were very similar for the combination and monotherapy IV preparations; the ratios of the C max, AUC t and AUC∞ values fell within the 80–125 % acceptable bioequivalence range. Precise dose proportionality for both compounds was also determined for the half dose of the IV formulation in comparison with the full dose. The relative bioavailability of paracetamol (93.78 %) and ibuprofen (96.45 %) confirmed the pharmacokinetic equivalence of the oral and IV formulations of the fixed-dose combination.
Conclusion
Concomitant administration of 3 mg/mL ibuprofen and 10 mg/mL paracetamol in a fixed-dose IV combination does not alter the pharmacokinetic profiles of either drug. The IV and oral dose forms of such a combination are pharmacokinetically equivalent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Concomitant administration of 3 mg/mL ibuprofen and 10 mg/mL paracetamol in a fixed-dose intravenous combination does not alter the pharmacokinetic profile of either drug. |
The intravenous and oral dose forms of such a combination are pharmacokinetically equivalent. |
1 Introduction
Ibuprofen and paracetamol are among the most commonly used analgesics and are widely available without a prescription [1]. Ibuprofen is a non-steroidal anti-inflammatory drug (NSAID), which non-selectively inhibits cyclooxygenase isozymes 1 and 2 (COX-1 and COX-2), resulting in inhibition of prostaglandins and related compounds at peripheral sites [2]. Contrastingly, the mode of action of paracetamol is not fully understood but is thought to relate to inhibition of either prostaglandin synthesis or cannabinoid receptors [3].
A fixed-dose combination of ibuprofen 300 mg + paracetamol 1000 mg (FDC 300/1000), when formulated as oral tablets (henceforth referred to as ‘FDC-oral’), has been shown to provide superior pain relief in comparison with its individual components [4]. An intravenous (IV) formulation of FDC 300/1000 (henceforth referred to as ‘FDC-IV’) has been developed to permit its administration to patients in whom the use of oral analgesics is limited by various patient factors, such as inability to swallow, the presence of postoperative nausea and vomiting, or reduced gastric motility.
Previous studies have demonstrated that oral ibuprofen and paracetamol are rapidly absorbed after oral administration and are not subject to significant first-pass metabolism [5–9], and that concomitant administration of both compounds does not result in a pharmacokinetic interaction [10–12]. In comparison with oral dosing, the same doses of ibuprofen and paracetamol administered in an IV formulation result in twofold and 70 % increases in the maximum measured plasma concentration (C max) values, respectively [5, 13]. Furthermore, IV ibuprofen demonstrates great dose proportionality with respect to the area under the plasma concentration–time curve (AUC) and C max [6, 14].
The results of this study, which was conducted with aims to (1) describe the pharmacokinetic profile of the IV formulation of FDC-IV; (2) confirm the lack of a pharmacokinetic interaction between ibuprofen and paracetamol following IV administration; (3) determine whether dose proportionality is apparent from comparison of the full-dose combination of FDC-IV and the half-dose combination; and (4) establish the relative bioavailability of FDC-oral versus that of FDC-IV, are reported here.
2 Methods
2.1 Trial Design
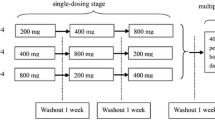
This study was a phase I, single-centre, single-dose, open-label, randomized, five-way cross-over trial in 30 healthy adult participants. After an initial screening period of up to 28 days, there were five study periods, separated by washout periods of at least 48 h, and a subsequent final follow-up period of up to 7 days. The research was conducted in accordance with Good Clinical Practice (GCP), including the Declaration of Helsinki and all applicable regulatory requirements. The study protocol received approval by the Health and Disability Ethics Committee, Ministry of Health, New Zealand, and the trial was registered with the Australian New Zealand Clinical Trial Registry (trial ID: ACTRN12614000809639).
2.2 Study Population
Healthy volunteers of both genders, 18–50 years of age with a body mass index (BMI) of 18.0–32.0 kg/m2, were recruited from the Christchurch Clinical Studies Trust databases. Informed consent was obtained by the principal investigator from all individual participants included in the study prior to the screening visit. Screening involved a physical examination and recording of demographic data, vital signs, the medical history and concomitant medications. A blood sample was taken for haematology, biochemistry and serology screening, and a urine sample was collected for urinalysis, and illicit-drug and alcohol breath screening tests were performed. Suitable participants had to comply with all study inclusion and exclusion criteria (presented in Table 1).
2.3 Treatment and Study Procedures
Participants were randomized in a cross-over fashion to a study sequence of four IV doses and one oral dose, using a computer-generated list prior to commencement. For all doses, participants were confined to the study centre from the previous evening to approximately 12 h after study drug administration. The study drug was administered in the morning, following a 10-h fast, and a standard lunch and snacks were provided at 4 and 8 h after study drug administration, respectively.
Patients received each of the following five treatments in a randomized order:
-
Treatment A (FDC-IV: 3 mg/mL ibuprofen + 10 mg/mL paracetamol, 100 mL IV)
-
Treatment B (10 mg/mL paracetamol, 100 mL IV)
-
Treatment C (3 mg/mL ibuprofen, 100 mL IV)
-
Treatment D (FDC-IV half dose: 1.5 mg/mL ibuprofen + 5 mg/mL paracetamol, 100 mL IV)
-
Treatment E (FDC-oral: ibuprofen 150 mg + paracetamol 500 mg per tablet, 2 tablets)
Treatments A, B, C and D were manufactured by SM Farmaceutici SRL, Italy, and administered as a slow IV infusion over 15 min into an indwelling cannula. Treatment E was manufactured by Sigma Laboratories, India, and administered orally with 240 mL of water.
2.4 Sampling Schedule
Blood samples (approximately 5 mL) for pharmacokinetic analysis were collected into lithium–heparin tubes. The blood sampling schedules were different for the IV and tablet formulations, as outlined below:
-
Sampling time points for the intravenous formulations Blood samples were drawn pre-dose, on completion of the 15-min IV infusion, at 5, 10, 15, 20, 30 and 45 min, and at 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10 and 12 h post-completion of the infusion.
-
Sampling time points for the tablet formulation Blood samples were drawn pre-dose, at 5, 10, 20, 30 and 45 min, and at 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10 and 12 h after study drug administration.
2.5 Bioanalytical Methods
All haematology, biochemistry and urinary analyses were conducted using standard methodologies within a single laboratory. Plasma concentrations of paracetamol and ibuprofen in human plasma (lithium–heparin) were determined using validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) procedures.
Paracetamol plasma concentrations were analysed using API 3000 and Quattro premier mass spectrometry in multiple reaction monitoring (MRM) mode, using a turbo ion spray with positive ionization. The chromatographic separation of paracetamol employed a C18 column, using a mobile phase consisting of de-ionized water, formic acid and acetonitrile. The calibration curves were linear over the working range of 50–20,000 ng/mL, with a regression coefficient (R 2) of ≥0.99937. The lower limit of quantification (LLOQ) was 50 ng/mL (precision 3.44 %, accuracy 93.69 %).
Ibuprofen plasma concentrations were analysed using a Sciex API 3000 and API 4000 triple quadrupole mass spectrometer in MRM mode, using a turbo ion spray with negative ionization. Selective analysis of ibuprofen was achieved on a Symmetry C18 column by using a mobile phase consisting of ammonium formate, methanol and acetonitrile. The calibration curves were linear over the concentration range of 50–35,000 ng/mL, with an R 2 of ≥0.9975. The LLOQ was 50 ng/mL (precision 8.85 %, accuracy 104.25 %). These methods have also been described elsewhere [10].
2.6 Pharmacokinetic Analysis
The pharmacokinetic parameters of paracetamol and ibuprofen were calculated using Excel 2013. C max and the time to reach C max (T max) were determined directly from the raw data. The elimination rate constant (k e) was calculated from the slope of the linear relationship between the loge concentration and the time during the terminal elimination phase. AUC from time zero to the time of the last measurable plasma concentration (AUC t ) was calculated by the linear trapezoidal rule, and AUC from time zero to infinity (AUC∞) was calculated after extrapolation from time t to infinity as the ratio of the last measurable plasma concentration (C last) to k e. The elimination half-life (t ½) was estimated from the elimination rate constant as ln(2)/k e.
2.7 Statistical Methods
Statistical analysis was performed using the validated program SPSS v22.0. The ratios used to test bioequivalence were calculated from loge-transformed data for C max, AUC t and AUC∞. The differences between the loge means and the 90 % confidence intervals (CIs) of the differences, derived from the residual variance from the analysis of variance (ANOVA) model, were back-transformed to estimate the ratios of the two formulations and the 90 % CIs of these ratios.
2.8 Safety
Safety was assessed in terms of the overall proportion of subjects with adverse events (AEs) and by haematological and biochemical assessment of blood samples. AEs were evaluated for their severity (mild, moderate or severe) according to the subjective impact they had on the performance of daily activities and their likely relationship to the medication (not related, unlikely, possibly, probably or definitely related) according to the likelihood of a temporal association between the onset of the event and the administration of the medicinal product.
3 Results
3.1 Participants
Thirty subjects were enrolled in this study; 23 (77 %) were male, and all were Caucasian. The mean (± standard deviation) age and BMI were 29.9 (±19.4) years and 24.5 (±2.9) kg/m2, respectively. Twenty-nine participants completed the study, each of whom received all five treatments and was included in the pharmacokinetic analysis.
3.2 Pharmacokinetic Results
The mean paracetamol plasma concentration–time curves for ibuprofen and paracetamol are presented in Figs. 1 and 2, respectively. A tabulated summary of the pharmacokinetic data for both compounds is also presented in Table 2.

Mean (standard error of the mean) paracetamol plasma concentrations after single administration of FDC-IV (treatment A: 3 mg/mL ibuprofen + 10 mg/mL paracetamol, 100 mL IV), paracetamol IV (treatment B: 10 mg/mL paracetamol, 100 mL IV), FDC-IV half dose (treatment D: 1.5 mg/mL ibuprofen + 5 mg/mL paracetamol, 100 mL IV) and FDC-oral (treatment E: ibuprofen 300 mg + paracetamol 1000 mg, 2 tablets). FDC fixed-dose combination, IV intravenous

Mean (standard error of the mean) ibuprofen plasma concentrations after single administration of FDC-IV (treatment A: 3 mg/mL ibuprofen + 10 mg/mL paracetamol, 100 mL IV), ibuprofen IV (treatment C: 3 mg/mL ibuprofen, 100 mL IV), FDC-IV half dose (treatment D: 1.5 mg/mL ibuprofen + 5 mg/mL paracetamol, 100 mL IV) and FDC-oral (treatment E: ibuprofen 300 mg + paracetamol 1000 mg, 2 tablets). FDC fixed-dose combination, IV intravenous
Intravenous infusions resulted in mean C max values of 26,709.6 ng/mL (FDC-IV) and 26,236 ng/mL (paracetamol IV) immediately after the 15-min infusion. A half dose of FDC-IV provided a mean C max of 12,880 ng/mL. The concentration of paracetamol from the FDC-oral tablets peaked at 0.73 h, with a mean C max of 14,907 ng/mL. The oral route provided paracetamol C max values 44 % lower than those observed after administration of FDC-IV or paracetamol IV.
Similarly, IV infusions resulted in C max values of 39,506.7 ng/mL (FDC-IV) and 40,292.97 ng/mL (ibuprofen IV) immediately after the 15-min infusion. A half dose of FDC-IV provided a mean C max of 20,352 ng/mL. The concentration of ibuprofen from the FDC-oral tablets peaked at 1.49 h, with a mean C max of 19,637 ng/mL. The oral route provided ibuprofen C max values 50 % lower than those observed after administration of FDC-IV or ibuprofen IV.
The corresponding AUC t and AUC∞ values were very similar across both the full-dose IV formulation and the FDC-oral tablets. The back-transformed 90 % CIs for FDC-IV in comparison with paracetamol IV and ibuprofen IV all fell within the 80–125 % acceptable bioequivalence range, thus confirming the lack of any pharmacokinetic interaction when both paracetamol and ibuprofen are given in combination as a parenteral preparation (Table 3).
Comparisons of the C max, AUC t and AUC∞ values for FDC-IV and a half dose of FDC-IV demonstrated consistent dose proportionality for both paracetamol and ibuprofen (Table 4). The t ½ values for paracetamol and ibuprofen were found to be comparable between all treatment groups (Table 2).
The relative bioavailability of paracetamol and ibuprofen from the FDC-oral tablets was calculated from the back-transformed ratios of the loge means of the AUC∞ values for the oral and IV formulations (AUCFDC-oral/AUCFDC-IV). For FDC-oral, the relative bioavailability of paracetamol was 93.78 % (90 % CI 90.98–96.67 %) and the absolute bioavailability of ibuprofen was 96.45 % (90 % CI 93.13–99.89 %), suggesting nearly complete absorption of both compounds from the gastrointestinal mucosa. These results are summarized in Table 5.
3.3 Safety Analysis
Twenty-five non-serious AEs were reported by 15 subjects (50 %). The majority of AEs were mild (80 %), and all other AEs were moderate (20 %). There were no severe AEs. Seventy-six percent of AEs were not related to the study medication in that a temporal association between the onsets of the events relative to the administration of the product was not reasonable. Twenty-four percent of AEs had a reasonable temporal relationship between their onset and administration of the study medication, and were deemed either ‘unlikely related’ or ‘possibly related’ on the basis of the subjective likelihood of the association. These data, and the distribution of AEs across the study periods (study drug or washout), are presented in Table 6. One subject did suffer a serious AE: bilateral ankle fractures due to a fall.
4 Discussion
For oral medications, increased absorption time and first-pass metabolism, or inconsistent absorption in hospitalized patients, due to delayed gastric emptying perioperatively, may result in a latency to or lack of clinical effect [15]. Indeed, in a study of 106 patients undergoing ear, nose and throat surgery, plasma concentrations of paracetamol 1000 mg sufficient to provide an analgesic effect (10 mg/L) were reached in 96 % of patients who received the drug parenterally and in only 67 % patients who received it orally [16]. FDC-IV has been developed for use in instances where oral therapy is limited by sedation, severe illness or debilitation, or in patients with difficulty in swallowing.
4.1 Interaction
The results of this study demonstrate that concurrent administration of ibuprofen and paracetamol in a novel IV fixed-dose combination did not significantly alter the extent of absorption of ibuprofen or paracetamol, in comparison with either agent alone. A lack of a pharmacokinetic interaction between ibuprofen and paracetamol has been documented for the following oral formulation, fixed-dose combinations: ibuprofen 300 mg + paracetamol 1000 mg [10], ibuprofen 400 mg + paracetamol 1000 mg [11] and ibuprofen 400 mg + paracetamol 650 mg [12]. In these products, after oral administration, the values of the pharmacokinetic parameters C max, AUC t and AUC∞ for both ibuprofen and paracetamol were similar with the fixed-dose combinations and monotherapies, and the back-transformed 90 % CI for each parameter fell within the acceptable bioequivalence range (80–125 %) [10, 11]. Similar results were obtained from comparison of pharmacokinetic parameters by ANOVA for the third fixed-dose combination [12]. Consequently, the results from the present study confirm and extend such results for an IV formulation.
4.2 Dose Proportionality
The dose proportionality of the FDC-IV formulation provided here is consistent with that reported for IV ibuprofen; a twofold lesser dose results in decreases of 44–51 % in C max, AUC t and AUC∞ [6, 14]. Absorption of paracetamol from a half dose of FDC-IV appeared to be slightly delayed; however, the longer mean T max in this group was due to a single patient whose paracetamol C max occurred 20 min after the start of the 15-min infusion (5 min after the end of the infusion). The clearance of both compounds remained constant (2.4 h for paracetamol and 1.8 h for ibuprofen), which is consistent with previous findings for ibuprofen [6]. The clear dose proportionality of absorption of both active ingredients in FDC-IV could provide practitioners with the ability to easily titrate analgesia according to their patient’s pain.
4.3 Relative Bioavailability
This study determined that both active ingredients in the FDC-oral formulation have very high bioavailability. In comparison with FDC-IV, the relative bioavailability values for ibuprofen and paracetamol in the FDC-oral tablets are 93.59 and 96.34 % respectively, confirming the assertion that both compounds are not subject to significant pre-systemic metabolism. These results are congruent with previously published data [5–9].
Absorption via the gastrointestinal mucosa results in delayed absorption and decreased C max values. In comparison with FDC-IV, the C max values for ibuprofen and paracetamol were reduced by 50 and 44 %, respectively, after administration of FDC-oral. For ibuprofen, the results of this study are in line with other published data; an equivalent oral dose of ibuprofen results in a C max value that is 50 % of the value observed after an IV dose (63 versus 120 µg/mL) [5]. For paracetamol, oral dosing has been shown to reduce C max of paracetamol 1000 mg by 41–43 % in healthy volunteers [9, 13].
5 Conclusion
This study confirmed that a combination of 3 mg/mL ibuprofen and 10 mg/mL paracetamol (FDC-IV) produces ibuprofen and paracetamol plasma concentration–time profiles that are similar to those observed after IV administration of each component on its own. The pharmacokinetic profile of the IV formulation has been demonstrated to be dose proportional following administration of single doses of 1.5 mg/mL ibuprofen and 5 mg/mL paracetamol (FDC-IV half dose) and 3 mg/mL ibuprofen and 10 mg/mL paracetamol (FDC-IV).
The pharmacokinetic parameters were observed to be similar when a single dose of FDC 300/1000 was administered in either an IV formulation (FDC-IV) or an oral formulation (FDC-oral), except for C max of the IV formulation, which was twice that of the oral formulation, and, as expected, T max was much shorter than with the oral dose. The relative bioavailability of paracetamol (93.78 %) and ibuprofen (96.45 %) confirmed the pharmacokinetic equivalence of the oral and IV FDC 300/1000 formulations.
Consequently, in instances where delayed gastric emptying is expected, an IV formulation of FDC 300/1000 may provide more efficient analgesic delivery.
References
Bailey E, Worthington HV, van Wijk A, Yates JM, Coulthard P, Afzal Z. Ibuprofen and/or paracetamol (acetaminophen) for pain relief after surgical removal of lower wisdom teeth. Cochrane Database Syst Rev. 2013;12:CD004624. doi:10.1002/14651858.CD004624.pub2.
Malmberg AB, Yaksh TL. Cyclooxygenase inhibition and the spinal release of prostglandin E2 and amino acids evoked by paw formalin injection: a microdialysis study in unanesthetized rats. J Neurosci. 1995;15:2768–78.
Anderson BJ. Paracetamol (acetaminophen): mechanisms of action. Paediatr Anaesth. 2008;18:915–21.
Merry AF, Gibbs RD, Edwards J, Ting GS, Frampton C, Davies E, Anderson BJ. Combined acetaminophen and ibuprofen for pain relief after oral surgery in adults: a randomized controlled trial. Br J Anaesth. 2010;104:80–8.
Pavliv L, Voss B, Rock A. Pharmacokinetics, safety, and tolerability of a rapid infusion of i.v. ibuprofen in healthy adults. Am J Health Syst Pharm. 2011;68:47–51.
Martin W, Koselowske G, Töberich H, Kerkmann T, Mangold B, Augustin J. Pharmacokinetics and absolute bioavailability of ibuprofen after oral administration of ibuprofen lysine in man. Biopharm Drug Dispos. 1990;11:265–78.
Perucca E, Richens A. Paracetamol disposition in normal subjects and in patients treated with antiepileptic drugs. Br J Clin Pharmacol. 1979;7:201–6.
Rawlins MD, Henderson DB, Hijab AR. Pharmacokinetics of paracetamol (acetaminophen) after intravenous and oral administration. Eur J Clin Pharmacol. 1977;11:283–6.
Singla NK, Parulan C, Samson R, Hutchinson J, Bushnell R, Beja EG, et al. Plasma and cerebrospinal fluid pharmacokinetic parameters after single-dose administration of intravenous, oral, or rectal acetaminophen. Pain Pract. 2012;12:523–32.
Atkinson HC, Stanescu I, Beasley CPH, Salem II, Frampton C. A pharmacokinetic analysis of a novel fixed dose oral combination of paracetamol and ibuprofen, with emphasis on food effect. J Bioequiv Availab. 2015;7:150–4.
Tanner T, Aspley S, Munn A, Thomas T. The pharmacokinetic profile of a novel fixed-dose combination tablet of ibuprofen and paracetamol. BMC Pharmacol Toxicol. 2010;10:10.
Wright CE, Antal EJ, Gillespie WR, Albert KS. Ibuprofen and acetaminophen kinetics when taken concurrently. Clin Pharmacol Ther. 1983;34:707–10.
Cadence Pharmaceuticals, Inc. Product monograph: Ofirmev (acetaminophen) injection. 2010. http://c.ymcdn.com/sites/www.npamonline.org/resource/resmgr/imported/OFIRMEV%20Monograph_FINAL.pdf. Accessed 12 Jan 2015.
Cumberland Pharmaceuticals. Product monograph: Caldolor (ibuprofen for intravenous injection). Nashville: Cumberland Pharmaceuticals Inc; 2009.
Power DBM, Forbes AM, van Heerden PV, Ilett APKF. Pharmacokinetics of drugs used in critically ill adults. Clin Pharmacokinet. 1998;34:25–56.
Van Der Westhuizen J, Kuo PY, Reed EW, Holder K. Randomised controlled trial comparing oral and intravenous paracetamol (acetaminophen) plasma levels when given as preoperative analgesia. Anaesth Intensiv Care. 2011;39:242–6.
Acknowledgments
We thank the staff at Christchurch Clinical Studies Trust Ltd (CCST; Christchurch, New Zealand) for the administration of the study protocol and data collection. We thank the staff at the International Pharmaceutical Research Centre (IPRC; Amman, Jordan) for the data analysis. We are grateful to the volunteers who participated in this study. This study was funded by AFT Pharmaceuticals Ltd (Auckland, New Zealand).
Author Contributions
H. Atkinson supervised the project and was involved in the design of the study, development of the protocol, interpretation of the data and writing of the manuscript. I. Stanescu was involved in the study design, protocol development, data analysis, data interpretation and writing of the manuscript. C. Frampton conducted the statistical analysis of the data and reviewed the manuscript. I. Salem performed the plasma drug assays and reviewed the manuscript. C. Beasley contributed to the statistical data interpretation and assisted in drafting the manuscript. R. Robson was involved in the data interpretation and critically reviewed the manuscript. All authors had full access to all of the data.
The authors have full control of all primary data and agree to allow the journal to review their data if requested.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study protocol received approval by the Health and Disability Ethics Committee, Ministry of Health, New Zealand, and was conducted in accordance with Good Clinical Practice (GCP), including the Declaration of Helsinki. Informed consent was obtained from all individual participants prior to their inclusion in the study.
This study was funded by AFT Pharmaceuticals Ltd.
H. Atkinson is a shareholder and Managing Director of AFT Pharmaceuticals Ltd, and I. Stanescu and C. Beasley are employees of AFT Pharmaceuticals Ltd. I. Salem is an employee of the IPRC. C. Frampton provides consultancy services to AFT Pharmaceuticals Ltd. R. Robson is the Director of CCST.
Additional information
This study was registered on the Australian New Zealand Clinical Trials Registry (trial ID: ACTRN12614000809639).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Atkinson, H.C., Stanescu, I., Frampton, C. et al. Pharmacokinetics and Bioavailability of a Fixed-Dose Combination of Ibuprofen and Paracetamol after Intravenous and Oral Administration. Clin Drug Investig 35, 625–632 (2015). https://doi.org/10.1007/s40261-015-0320-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-015-0320-8