
Abstract
Background and Objective
As pazopanib plasma trough concentrations are correlated with treatment outcome, we explored whether single nucleotide polymorphisms in the elimination pathway of pazopanib affect systemic pazopanib concentrations.
Methods
The decreased function alleles CYP3A4 15389 C > T (*22), ABCB1 3435 C >T, ABCG2 421 C >A, and ABCG2 34G >A were analyzed within a recently developed population-pharmacokinetic model.
Results
Incorporation of CYP3A4*22 in the model resulted in a 35% lower clearance for variant carriers (0.18 vs. 0.27 L/h; difference in objective function value: − 9.7; p < 0.005). Simulated median trough concentrations of cancer patients with CYP3A4*22 with 600 mg once daily or 800 mg once daily were 31 and 35 mg/L, respectively. The simulated trough concentrations for the population excluding the CYP3A4*22 carriers after 600 mg once daily or 800 mg once daily were 18 and 20 mg/L, respectively.
Conclusion
This analysis shows that CYP3A4*22 heterozygotes have a substantial lower pazopanib clearance and that dose adjustments based on CYP3A4*22 status could be considered.
Similar content being viewed by others


Patients carrying CYP3A4*22 have a lower pazopanib clearance than CYP3A4 wild types. |
In our simulations, a pazopanib dose of 600 mg in these patients leads to equal pazopanib exposure as wild type patients dosed at 800 mg. |
This knowledge may potentially lead to alternative dosing of patients carrying CYP3A4*22. |
1 Introduction
Pazopanib is a tyrosine kinase inhibitor that is currently a first-line treatment option for renal cell carcinoma, and a second-line treatment or beyond for non-adipocytic soft-tissue sarcoma [3, 4]. Despite its proven clinical efficacy in these tumor types, dosing may be a challenge, as toxicity may become a serious limitation: 14–15% of patients discontinue pazopanib because of adverse events [3, 4]. Nausea, diarrhea, headache, hand-foot skin reaction, fatigue, and liver function disorders are among the most common side effects. From the outset, it is unknown which patients will develop side effects and which patients will not.
Likewise, it cannot be predicted which cancer patients will benefit from treatment with pazopanib. There are many factors that determine the outcome of an individual patient to a particular treatment; the pharmacology of an anti-tumor drug being one of these. As trough concentrations below 20.5 µg/mL have been associated with a worse clinical outcome [5, 6], it seems vital to dose patients up to this threshold value. Recently, we showed that this strategy is feasible and does not lead to increased toxicity in most patients [7]. However, toxicity is dose limiting in a substantial number of patients, and may be the result of high systemic concentrations [8].
Several factors have been described to influence pazopanib exposure. Its absorption has been shown to increase with concomitant food intake [9, 10] and to decrease when stomach pH is reduced [11], whereas its elimination is known to be increased as a result of cytochrome P450 (CYP)3A4 induction by ifosfamide or aprepitant [2, 12]. Although these drugs are generally not being used in combination with pazopanib, these drug–drug interactions stress the importance of the function of the enzyme CYP3A4. However, drug transporters such as ABCB1 (P-glycoprotein) and ABCG2 (breast cancer resistant protein), for which pazopanib is a substrate, may also be relevant according to the summary of product characteristics.
Transporter and enzyme function are not solely affected by co-medication, but also by single nucleotide polymorphisms (SNPs) in their encoding genes. Up till now, genetic polymorphisms have been associated only with pazopanib pharmacodynamics rather than with its pharmacokinetics [13, 14]. Therefore, in the current study, it was our goal to associate four potentially relevant SNPs in CYP3A4, ABCB1, and ABCG2 with pazopanib pharmacokinetics. These SNPs have earlier been shown to be associated with the exposure of other tyrosine kinase inhibitors such as sunitinib [15]. We did this by implementing SNP data into a recently developed population-pharmacokinetic model for pazopanib systemic exposure, describing the complex metabolism of pazopanib in cancer patients [16].
2 Methods
2.1 Study Population
Patients were selected from three separate studies in which pazopanib pharmacokinetics had been assessed: two studies assessed the pharmacokinetic and pharmacodynamic drug interactions between pazopanib and either docetaxel (NTR2309) [1] or ifosfamide (NTR2063) [2] and the third study investigated the feasibility of therapeutic drug monitoring in pazopanib-treated patients (NTR3293) [17]/ Respectively 40, 41, and 13 patients were included in these studies. All patients signed informed consent.
2.2 Single Nucleotide Polymorphism Selection
As pazopanib is acknowledged as a substrate for CYP3A4, ABCB1, and ABCG2, four SNPs in the encoding genes for these proteins were selected, based on previous studies investigating other tyrosine kinase inhibitors [18,19,20,21]. Details of the investigated SNPs are depicted in Table 1.
2.3 DNA Isolation
If available, 2 mL of plasma or 3 mL of whole blood, stored at − 20 °C, was extracted on the MagNAPure Compact instrument (Roche Diagnostics GmbH, Mannheim, Germany) using the Nucleic Acid Isolation Kit I (Roche Diagnostics GmbH) and a final elution volume of 200 μL.
2.4 Taqman® Genotyping
Predesigned drug-metabolizing enzymes (Taqman® allelic discrimination assays) were used to perform genotyping on the Life Technologies Taqman® 7500 system (Applied Biosystems, Life Technologies Europe BV, Bleiswijk, The Netherlands). Each assay consisted of two allele-specific minor groove-binding probes, labeled with the fluorescent dyes VIC and FAM. Polymerase chain reactions were performed in a reaction volume of 10 μL, containing assay-specific primers, allele-specific Taqman® minor groove-binding probes, Abgene Absolute QPCR Rox Mix (Thermo Scientific, Life Technologies Europe BV, Bleiswijk, The Netherlands), and genomic DNA (20 ng). The thermal profile consisted of 40 cycles of denaturation at 95 °C for 20 s and annealing at 92 °C for 3 s and extension at 60 °C for 30 s. Allele-specific fluorescence was measured using the 7500 software Version 2.3 for allelic discrimination (Applied Biosystems) to score genotypes.
2.5 Population-Pharmacokinetic Model
A previously developed population-pharmacokinetic model with pharmacokinetic data from the same patients was used as a base model for the current analysis [16]. This model described the complex pazopanib pharmacokinetics including the saturable absorption process and the drug–drug interaction with ifosfamide [22]. The remaining unexplained variation in pazopanib pharmacokinetics was predominantly reflected in the between-subject variability of the fast absorption rate constant (coefficient of variation = 140%) and clearance (coefficient of variation = 30.9%).
2.6 Statistical Analysis
All SNPs were in Hardy–Weinberg equilibrium. The genetic data were added to the data used for developing the pharmacokinetic model. NONMEM (Version 7.2; ICON, Ellicott City, MD, USA) with the first-order conditional estimation method with interaction was used for parameter estimation. Piraña was used as the modeling environment [23] and R (Version 3.3.1) was used for graphical interpretation of the data.
Genotypes with a frequency of more than 5% were tested in the base model and genotypes with a frequency of less than 5% were ignored. Covariates were included as dichotomous covariates using the following equation:
where Ppop is the pharmacokinetic parameter of interest, pg was scored ‘1’ for patients of whom the genotype of interest was present and ‘0’ for patients of whom the genotype was absent, θx is the typical parameter value of the wild-type population, and θy is the covariate effect size estimate. The association of the SNPs with pazopanib clearance and bioavailability was investigated following a stepwise procedure. The potential association of all SNPs was univariately tested for both pharmacokinetic endpoints. Univariate forward inclusion was performed using a p value of < 0.05 as statistically significant (likelihood ratio test; 1 degree of freedom, difference in objective function value: 3.84). This was followed by a backward elimination procedure using a p value of < 0.005 (difference in objective function value: 7.88) as the significance threshold (likelihood ratio test).
Ultimately, the final population-pharmacokinetic model [see Electronic Supplementary Material (ESM)] was used for simulating median trough concentrations for patients with and without CYP3A4*22 dosed at 400, 600, and 800 mg once daily. For each combination of dose and genotype, 500 individuals were simulated. The simulated results were analyzed using R to present a concentration–time curve at steady state (6 weeks after start) and to calculate the percentage of simulated individuals reaching the target trough concentration of 20.5 ug/L. Goodness-of-fit plots are depicted in Fig. 1 of the ESM for all patients and in Fig. 2 of the ESM for the CYP3A4*22 carriers.

Schematic overview of the pazopanib (pazo) pharmacokinetic model [16]. ALAG lag time, CL clearance, fr fraction of pazopanib, KaF fast absorption rate constant, KaS slow absorption rate constant, Vc central volume of distribution, Vp peripheral volume of distribution

Median simulated pazopanib trough concentration after a single dose at steady state. The red line and the red dashed line represent the simulated concentration–time curves for CYP3A4*22 patients, treated with 800 mg and 600 mg of pazopanib daily, respectively. The black line and the black dashed line represent the simulated concentration–time curve for wild-type (WT) patients for CYP3A4, treated with 800 mg and 600 mg of pazopanib daily, respectively. 1dd once daily
3 Results
3.1 Patients
From the 96 patients in the original population-pharmacokinetic model by Yu et al. [16], germline DNA was genotyped successfully in 94 (98%) of them. From these 94 patients, 761 samples had been drawn for pharmacokinetic analysis. Because most patients had participated in phase I studies in which pazopanib was combined with either ifosfamide or docetaxel, tumor types varied widely between patients and were therefore not recorded in this study. Pazopanib starting dose ranged from 200 to 1000 mg daily. Most patients were male (71%) and Caucasian (97%; Table 2).
3.2 Effect of Single Nucleotide Polymorphisma on Clearance and Bioavailability
Genotyping details are shown in Table 1. A planned backwards elimination procedure was not performed because only a single association was found to be significantly associated with an endpoint in the primary univariate tests; incorporation of CYP3A4*22 in the model resulted in a 35% lower clearance for variant carriers (0.18 vs. 0.27 L/h; difference in objective function value: – 9.47; p = 0.002). Effects of the other four SNPs on clearance and bioavailability are presented in Table 3.
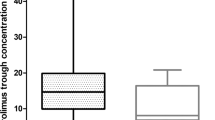
Incorporation of the CYP3A4*22 in the model (Fig. 1) slightly reduced the inter-individual variability in clearance from 31 to 27%. Parameter estimates and between-subject variability of other pharmacokinetic parameters remained largely unaffected (Table 4).
To translate our findings to clinical practice, the final model (including CYP3A4*22) was used for the simulation of several dosing regimens for CYP3A4*22 carriers. Next, we compared these with the 600- and 800-mg once-daily dose for non-carriers. The median trough concentrations for the CYP3A4 wild-type population after 400, 600, or 800 mg once daily were 16, 18 and 20 mg/L, respectively (Fig. 2; Table 1 of the ESM). The median trough concentrations for the CYP3A4*22 heterozygotes with 400, 600, or 800 mg once daily were 27, 31, and 35 mg/L, respectively.
4 Discussion
In the current analysis, we found that CYP3A4*22 is associated with a significantly lower pazopanib clearance. Our simulations estimate that trough concentrations at steady state are more than 50% higher for these patients, compared with patients who are wild type for this gene. This percentage is deemed clinically relevant, as this is not an uncommon mutation in the European population (minor allele frequency 5% [24]) and CYP3A4*22 carriers could be more prone to develop toxicity compared with wild-type patients.
Five years previously, it was shown that CYP3A4*22 is associated with a lower expression of the CYP3A4 enzyme, which resulted in decreased CYP3A4-dependent activity in the liver [25]. This reduced CYP3A4 activity has been shown to be associated with altered pharmacokinetics of several drugs, e.g., tacrolimus, midazolam, and erythromycin [26], and to be associated with an increased risk of side effects with for example, cyclosporine and paclitaxel [27, 28]. Furthermore, the clearance of sunitinib, another vascular endothelial growth factor receptor tyrosine kinase inhibitor, was found to be approximately 23% lower in CYP3A4*22 carriers [15]. In transplant patients, for whom it is important to quickly dose optimally to prevent organ rejection, this SNP has been used to predict tacrolimus exposure and to adjust its starting dose depending on the genotype [29, 30]. In contrast, in drugs that are also metabolized by CYP2D6, such as tamoxifen and several antipsychotics, CYP3A4*22 appears to be of less importance [31, 32].
The investigated SNPs in ABCB1 and ABCG2 were not significantly associated with pazopanib bioavailability or clearance, although both proteins encoded by these genes are located on intestinal enterocytes and on hepatocytes. Possibly, the full contribution of these transporters to pazopanib pharmacokinetics is not covered by the selected polymorphisms or is not reflected by bioavailability or clearance. However, given the fact that little to no literature on these transporters and pazopanib pharmacokinetics is available, apart from the summary of product characteristics, it is likely that their contribution to pazopanib pharmacokinetics is rather small.
Given the limited number of investigated patients in our study, only a few SNPs could be selected to have sufficient statistical power for the analyses. Although the analyses might therefore have not been comprehensive, we deemed that the investigated SNPs were the most likely candidates to cause relevant pharmacokinetic alterations, based on the currently available literature. As reported previously [16], bootstrapping is not a suitable method for validating this model given its complexity and the different sampling schedules used.
Furthermore, clinical outcome (i.e., toxicity and survival) was not assessed in this analysis. As the pharmacokinetic data were collected from different phase I and pharmacokinetic studies, the study population was largely heterogeneous regarding confounding factors such as pazopanib dose, tumor type, disease stage, and concomitant (anti-cancer) medication. These factors should be taken into account to determine true effects on clinical outcome. The effect of CYP3A4*22 on the effect of pazopanib therefore remains to be investigated further in prospective clinical trials, but our simulations show that a dose reduction to 600 mg daily in CYP3A4*22 carriers provided even higher pazopanib trough concentrations than the approved 800-mg dose in wild-type patients, which potentially makes them more susceptible to toxicity.
5 Conclusion
CYP3A4*22 carriers have a significantly reduced pazopanib clearance, which potentially makes them more susceptible to (severe) toxicity. Simulations suggest that dosing these patients at pazopanib 600 mg daily leads to adequate pazopanib trough concentrations, higher than those in wild-type patients dosed at 800 mg daily. Future studies should investigate whether dose reductions also lead to less toxicity, without compromising the anti-cancer effect in this specific group of patients.
References
Hamberg P, Mathijssen RH, de Bruijn P, Leonowens C, van der Biessen D, Eskens FA, et al. Impact of pazopanib on docetaxel exposure: results of a phase I combination study with two different docetaxel schedules. Cancer Chemother Pharmacol. 2015;75(2):365–71.
Hamberg P, Boers-Sonderen MJ, van der Graaf WT, de Bruijn P, Suttle AB, Eskens FA, et al. Pazopanib exposure decreases as a result of an ifosfamide-dependent drug-drug interaction: results of a phase I study. Br J Cancer. 2014;110(4):888–93.
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28(6):1061–8. https://doi.org/10.1200/jco.2009.23.9764.
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–86. https://doi.org/10.1016/s0140-6736(12)60651-5.
Suttle AB, Ball HA, Molimard M, Hutson TE, Carpenter C, Rajagopalan D, et al. Relationships between pazopanib exposure and clinical safety and efficacy in patients with advanced renal cell carcinoma. Br J Cancer. 2014;111(10):1909–16.
Verheijen RB, Swart LE, Beijnen JH, Schellens JHM, Huitema ADR, Steeghs N. Exposure-survival analyses of pazopanib in renal cell carcinoma and soft tissue sarcoma patients: opportunities for dose optimization. Cancer Chemother Pharmacol. 2017;80(6):1171–8. https://doi.org/10.1007/s00280-017-3463-x.
Verheijen RB, Bins S, Mathijssen RH, Lolkema MP, van Doorn L, Schellens JH, et al. Individualized pazopanib dosing: a prospective feasibility study in cancer patients. Clin Cancer Res. 2016;22(23):5738–46. https://doi.org/10.1158/1078-0432.ccr-16-1255.
Soomers V, Desar IME, van Erp NP, Verwiel J, Kaal SEJ, van der Graaf WTA. Fatal heart failure in a young adult female sarcoma patient treated with pazopanib. Acta Oncol. 2017;56(9):1233–4. https://doi.org/10.1080/0284186x.2017.1296582.
Heath EI, Chiorean EG, Sweeney CJ, Hodge JP, Lager JJ, Forman K, et al. A phase I study of the pharmacokinetic and safety profiles of oral pazopanib with a high-fat or low-fat meal in patients with advanced solid tumors. Clin Pharmacol Ther. 2010;88(6):818–23.
Lubberman F, Gelderblom H, Jansman F, Colbers A, Van der Graaf WTA, Mulders P, et al. Food intervention to make therapy with pazopanib more patient-friendly and affordable. J Clin Oncol. 2016;34(15_suppl):11040. https://doi.org/10.1200/jco.2016.34.15_suppl.11040.
Tan AR, Gibbon DG, Stein MN, Lindquist D, Edenfield JW, Martin JC, et al. Effects of ketoconazole and esomeprazole on the pharmacokinetics of pazopanib in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71(6):1635–43.
Imbs DC, Dieras V, Bachelot T, Campone M, Isambert N, Joly F, et al. Pharmacokinetic interaction between pazopanib and cisplatin regimen. Cancer Chemother Pharmacol. 2016;77(2):385–92.
Maitland ML, Xu CF, Cheng YC, Kistner-Griffin E, Ryan KA, Karrison TG, et al. Identification of a variant in KDR associated with serum VEGFR2 and pharmacodynamics of pazopanib. Clin Cancer Res. 2015;21(2):365–72.
Xu CF, Johnson T, Wang X, Carpenter C, Graves AP, Warren L, et al. HLA-B*57:01 confers susceptibility to pazopanib-associated liver injury in patients with cancer. Clin Cancer Res. 2016;22(6):1371–7.
Diekstra MH, Klumpen HJ, Lolkema MP, Yu H, Kloth JS, Gelderblom H, et al. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin Pharmacol Ther. 2014;96(1):81–9. https://doi.org/10.1038/clpt.2014.47.
Yu H, van Erp N, Bins S, Mathijssen RH, Schellens JH, Beijnen JH, et al. Development of a pharmacokinetic model to describe the complex pharmacokinetics of pazopanib in cancer patients. Clin Pharmacokinet. 2017;56(3):293–303. https://doi.org/10.1007/s40262-016-0443-y.
de Wit D, van Erp NP, den Hartigh J, Wolterbeek R, den Hollander-van Deursen M, Labots M, et al. Therapeutic drug monitoring to individualize the dosing of pazopanib: a pharmacokinetic feasibility study. Ther Drug Monit. 2015;37(3):331–8. https://doi.org/10.1097/ftd.0000000000000141.
Elens L, van Gelder T, Hesselink DA, Haufroid V, van Schaik RH. CYP3A4*22: promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics. 2013;14(1):47–62.
Qin C, Cao Q, Li P, Wang S, Wang J, Wang M, et al. The influence of genetic variants of sorafenib on clinical outcomes and toxic effects in patients with advanced renal cell carcinoma. Sci Rep. 2016;6:20089. https://doi.org/10.1038/srep20089.
Kim DH, Sriharsha L, Xu W, Kamel-Reid S, Liu X, Siminovitch K, et al. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin Cancer Res. 2009;15(14):4750–8. https://doi.org/10.1158/1078-0432.ccr-09-0145.
Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H, et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet. 2010;55(11):731–7. https://doi.org/10.1038/jhg.2010.98.
Kerbusch T, Huitema AD, Ouwerkerk J, Keizer HJ, Mathot RA, Schellens JH, et al. Evaluation of the autoinduction of ifosfamide metabolism by a population pharmacokinetic approach using NONMEM. Br J Clin Pharmacol. 2000;49(6):555–61.
Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72–9. https://doi.org/10.1016/j.cmpb.2010.04.018.
Zhou Y, Ingelman-Sundberg M, Lauschke VM. Worldwide distribution of cytochrome P450 alleles: a meta-analysis of population-scale sequencing projects. Clin Pharmacol Ther. 2017;102(4):688–700. https://doi.org/10.1002/cpt.690.
Okubo M, Murayama N, Shimizu M, Shimada T, Guengerich FP, Yamazaki H. CYP3A4 intron 6 C > T polymorphism (CYP3A4*22) is associated with reduced CYP3A4 protein level and function in human liver microsomes. J Toxicol Sci. 2013;38(3):349–54.
Elens L, Nieuweboer A, Clarke SJ, Charles KA, de Graan AJ, Haufroid V, et al. CYP3A4 intron 6 C > T SNP (CYP3A4*22) encodes lower CYP3A4 activity in cancer patients, as measured with probes midazolam and erythromycin. Pharmacogenomics. 2013;14(2):137–49.
Elens L, Bouamar R, Hesselink DA, Haufroid V, van Gelder T, van Schaik RH. The new CYP3A4 intron 6 C > T polymorphism (CYP3A4*22) is associated with an increased risk of delayed graft function and worse renal function in cyclosporine-treated kidney transplant patients. Pharmacogenet Genom. 2012;22(5):373–80.
de Graan AJ, Elens L, Sprowl JA, Sparreboom A, Friberg LE, van der Holt B, et al. CYP3A4*22 genotype and systemic exposure affect paclitaxel-induced neurotoxicity. Clin Cancer Res. 2013;19(12):3316–24. https://doi.org/10.1158/1078-0432.ccr-12-3786.
Elens L, Capron A, van Schaik RH, De Meyer M, De Pauw L, Eddour DC, et al. Impact of CYP3A4*22 allele on tacrolimus pharmacokinetics in early period after renal transplantation: toward updated genotype-based dosage guidelines. Ther Drug Monit. 2013;35(5):608–16. https://doi.org/10.1097/FTD.0b013e318296045b.
Woillard JB, Mourad M, Neely M, Capron A, van Schaik RH, van Gelder T, et al. Tacrolimus updated guidelines through popPK modeling: how to benefit more from CYP3A pre-emptive genotyping prior to kidney transplantation. Front Pharmacol. 2017;8:358. https://doi.org/10.3389/fphar.2017.00358.
van der Weide K, van der Weide J. The influence of the CYP3A4*22 polymorphism and CYP2D6 polymorphisms on serum concentrations of aripiprazole, haloperidol, pimozide, and risperidone in psychiatric patients. J Clin Psychopharmacol. 2015;35(3):228–36. https://doi.org/10.1097/jcp.0000000000000319.
Sanchez Spitman AB, Moes D, Gelderblom H, Dezentje VO, Swen JJ, Guchelaar HJ. Effect of CYP3A4*22, CYP3A5*3, and CYP3A combined genotypes on tamoxifen metabolism. Eur J Clin Pharmacol. 2017;73(12):1589–98. https://doi.org/10.1007/s00228-017-2323-2.
Acknowledgements
The authors thank Djoeke de Wit for her assistance in collecting clinical data, and Daan Hurkmans for designing Fig. 1. The authors also thank Roxanne C. Jewell (GSK/Novartis) and Gaaled Haj Mohammad (GSK/Novartis) for providing raw pazopanib data from two clinical trials (VEG109601 [1] and VEG110725 [2]).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflict of interest
Huixin Yu is currently employed by Novartis. All her contributions to this article were made before that employment. Nielka van Erp received grants from Astellas, Janssen-Cilag BV, Novartis, GSK, Boehringer-Ingelheim, Ipsen, Roche, Pfizer, Gilead, and Sanofi and payment for lectures by Novartis, Bayer, and Sanofi. Ron H.J. Mathijssen received grants and payment for lectures by Novartis. Stijn L.W. Koolen received payment for lectures by Novartis. Sander Bins, Alwin D.R. Huitema, Pim Laven, Samira el Bouazzaoui, Carla van Herpen, Paul Hamberg, Hans Gelderblom, Neeltje Steeghs, Stefan Sleijfer, and Ron H.N. van Schaik have no conflicts of interest that are directly relevant to the contents of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40262_2018_719_MOESM1_ESM.tiff
Supplementary Fig. 1 Goodness-of-fit plots for the base model and the final model including the CYP3A*22 covariate effect on clearance. Concentrations are given in mg/L. The smooth red line indicates the general data trend. The black line represents the line y = 0 in the population conditional weighted residuals (CWRES) plot or the identity line in the dependent variable (DV) vs. population-predicted concentration (PRED) plots
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bins, S., Huitema, A.D.R., Laven, P. et al. Impact of CYP3A4*22 on Pazopanib Pharmacokinetics in Cancer Patients. Clin Pharmacokinet 58, 651–658 (2019). https://doi.org/10.1007/s40262-018-0719-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0719-5