
Abstract
Purpose of Review
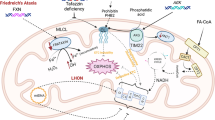
To elucidate the role metabolic pathways have on pathophysiological mechanisms involving the heart, it is essential to understand how biochemical pathways influence pathophysiology, clinical findings, and treatment. In this review, we focus on fatty acid oxidation disorders that affect the heart. We enumerate the defects that arise from each step of fatty acid oxidation leading to cardiomyopathy, conduction defects, arrhythmias, and sudden death.
Recent Findings
With the advent of newborn screening, careful interpretation of diagnostic findings and early treatment, morbidity, and mortality associated with these conditions have significantly decreased. However, treatment of the energy defect alone is not enough. New potential therapies and pathobiological mechanisms will be mentioned.
Summary
Mitochondrial fatty acid oxidation disorders associated with energy defects leading to cardiac disease are reviewed (clinical, biochemical findings and treatment); while highlighting the biochemical pathways involved in the pathophysiology of these disorders.

Similar content being viewed by others


References
Recently published papers of particular interest have been highlighted as: • of importance
Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi:10.1146/annurev.pathol.4.110807.092314.
Cox GF. Diagnostic approaches to pediatric cardiomyopathy of metabolic genetic etiologies and their relation to therapy. Prog Pediatr Cardiol. 2007;24:15–25. doi:10.1016/j.ppedcard.2007.08. 013.
Bing R. The metabolism of the heart. Harvey Lect. 1955;50:27–70.
Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi:10.1152/physrev.00006.2004.
Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–58. doi:10.1152/physrev.00015.2009.
Wanders RJA, Vreken P, den Boer MEJ, Wijburg FA, van Gennip AH, IJlst L. Disorders of mitochondrial fatty acyl-CoA β-oxidation. J Inherit Metab Dis. 1999;22:442–87. doi:10.1023/A:1005504223140.
Schulz H. Oxidation of fatty acids in eukaryotes. Amsterdam: Elsevier; 2007. p. 131–54. doi:10.1016/S0167-7306(02)36007-1.
Das AM, Steuerwald U, Illsinger S. Inborn errors of energy metabolism associated with myopathies. J Biomed Biotechnol. 2010; doi:10.1155/2010/340849.
Mitchell P. Chemiosmotic coupling in energy transduction: a logical development of biochemical knowledge. J Bioenerg. 1972;3:5–24. doi:10.1007/978-1-4684-2016-6_2.
Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy orally? J Physiol. 2006;574:95–112. doi:10.1113/jphysiol.2006.109389.
Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. BioEssays. 2001;23:1112–9. doi:10.1002/bies.10009.
Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi:10.1038/nrm2249.
Kudo N, Gillespie JG, Kung L, Witters LA, Schulz R, Clanachan AS, Lopaschuk GD. Characterization of 5′AMP-activated protein kinase activity in the heart and its role in inhibiting acetyl-CoA carboxylase during reperfusion following ischemia. Biochim Biophys Acta. 1996;1301:67–75. doi:10.1128/MCB.20.18.6704-6711.2000.
Lopaschuk GD, Witters LA, Itoi T, Barr R, Barr A. Acetyl-CoA carboxylase involvement in the rapid maturation of fatty acid oxidation in the newborn rabbit heart. J Biol Chem. 1994;269:25871–8.
Makinde AO, Kantor PF, Lopaschuk GD. Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Mol Cell Biochem. 1998;188:49–56. doi:10.1023/A:1006860104840.
Sakamoto J, Barr RL, Kavanagh KM, Lopaschuk GD. Contribution of malonyl-CoA decarboxylase to the high fatty acid oxidation rates seen in the diabetic heart. Am J Physiol Heart Circ Physiol. 2000;278:H1196–204.
Jaswal JS, Gandhi M, Finegan BA, Dyck JR, Clanachan AS. p38 mitogen-activated protein kinase mediates adenosine-induced alterations in myocardial glucose utilization via 5′-AMP-activated protein kinase. Am J Physiol Heart Circ Physiol. 2007;292:H1978–85. doi:10.1152/ajpheart.01121.2006.
Xi X, Han J, Zhang JZ. Stimulation of glucose transport by AMP-activated protein kinase via activation of p38 mitogen-activated protein kinase. J Biol Chem. 2001;276:41029–34. doi:10.1074/jbc.M102824200.
Bennett MJ. Pathophysiology of fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:533–7. doi:10.1007/s10545-010-9170-y.
Sewell AC, Bender SW, Wirth S, et al. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a severe fatty acid oxidation disorder. Eur J Pediatr. 1994;153:745–50. doi:10.1007/BF01954492.
Ventura FV, Ruiter JPN, Ijlst L, Tavares de Almeida I, Wanders RJA. Inhibitory effect of 3-hydroxyacyl-CoAs and other long-chain fatty acid b-oxidation intermediates on mitochondrial oxidative phosphorylation. J Inher Metab Dis. 1996;19:161–4.
Vockley J, Marsden D, McCracken E, DeWard S, Barone A, Hsu K, Kakkis E. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment a retrospective chart review. Mol Genet Metab. 2015;116:53–60. doi:10.1016/j.ymgme.2015.06.006.
• Vockley J, Charrow J, Ganesh J, Eswara M, et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016;119:223–31. doi:10.1016/j.ymgme.2016.08.008. This paper is a case series of patients who have LCFAODs and severe heart failure. It describes the course of the disease and the response of patients to the anaplerotic investigational agent used.
Bonnet D, Martin D, de Lonlay P, Villain E, Jouvet P, Rabier D, Brivet M, Saudubray JM. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100:2248–53. doi:10.1161/01.CIR.100.22.2248.
Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477–502. doi:10.1146/annurev.physiol.64.082201.154705.
• Byers SL, Ficicioglu C. The infant with cardiomyopathy: when to suspect inborn errors of metabolism? World J Cardiol. 2014;26:1149–55. doi:10.4330/wjc.v6.i11.1149. This paper provides a guide on how to consider the diagnosis of children with cardiomyopathy and when to consider inborn error of metabolism as a cause.
Smith EC, El-Gharbawy A, Koeberl D. Metabolic myopathies: clinical features and diagnostic approach. Rheum Dis Clin N Am. 2011;37:2201–17. doi:10.1016/j.rdc.2011.01.004.
Tein I, De Vivo DC, Bierman F, Pulver P, De Meirleir LJ, Cvitanovic-Sojat L, et al. Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr Res. 1990;28:247–55. doi:10.1203/00006450-199009000-00020.
Tang NLS, Ganapathy V, Wu X, et al. Mutations of OCTN2, an organic cation/carnitine transporter, lead to a deficient cellular carnitine uptake in primary carnitine deficiency. Hum Mol Genet. 1999;8:655–60. doi:10.1093/hmg/8.4.655.
Bremer J, Buist NRM. Carnitine—metabolism and functions. Physiol Rev. 1983;63:1420–80.
Tein I. Carnitine transport: pathophysiology and metabolism of known molecular defects. J Inherit Metab Dis. 2003;26:147–69. doi:10.1023/A:1024481016187.
Rinaldo P, Stanley CA, Hsu BYL, Sanchez LA, Stern HJ. Sudden neonatal death in carnitine transporter deficiency. J Pediatr. 1997;131:304–5. doi:10.1016/S0022-3476(97)70171-9.
Stanley CA, De Leeuw S, Coates PM, Vianey-Liaud C, Divry P, Bonnefont JP, Saudubray JM, et al. Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann Neurol. 1991;30:709–16. doi:10.1002/ana.410300512.
Pons R, De Vivo DC. Primary and secondary carnitine deficiency syndrome. J Child Neurol. 1995;10:S8–S24.
Lamhonwah AM, Olpin SE, Pollitt RJ, Vianey-Saban C, Divry P, Guffon N, Tein I. Novel OCTN2 mutations: no genotype–phenotype correlations: early carnitine therapy prevents cardiomyopathy. Am J Med Genet. 2002;111:271–84. doi:10.1002/ajmg.10585.
Rijlaarsdam RS, van Spronsen FJ, Bink-Boelkens MTH, et al. Ventricular fibrillation without overt cardiomyopathy as first presentation of organic cation transporter 2 deficiency in adolescence. Pacing Clin Electrophysiol. 2004;27:675–6. doi:10.1111/j.1540-8159.2004.00507.x.
Lund AM, Joensen F, Hougaard DM, Jensen LK, Christensen E, Christensen Norgaard-Petersen B, Schwartz M, Skovby F. Carnitine transporter and holocarboxylase synthetase deficiencies in the Faroe Islands. J Inherit Metab Dis. 2007;30:341–9. doi:10.1007/s10545-007-0527-9.
Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol Asp Med. 2004;25:521–32. doi:10.1016/j.mam.2004.06.007.
Brivet M, Slama A, Ogier H, Boutron A, Demaugre F, Saudubray J, Lemonnier A. Diagnosis of carnitine acylcarnitine translocase deficiency by complementation analysis. J Inherit Metab Dis. 1994;17:271–4.
Al Aqeel AI, Rashed MS, Wanders RJA. Carnitine-acylcarnitine translocase deficiency is a treatable disease. J Inherit Metab Dis. 1999;22:271–5.
Gellera C, Verderio E, Floridia G, et al. Assignment of the human carnitine palmitoyl transferase II gene (CPT1) to chromosome 1p32. Genomics. 1997;24:195–7.
Wieser T, Deschauer M, Olek K, Hermann T, Zierz S. Carnitine palmitoyltransferase II deficiency: molecular and biochemical analysis of 32 patients. Neurology. 2003;60:1351–3. doi:10.1006/geno.1994.1605.
Thuillier L, Rostane H, Droin V, Demaugre F, Brivet M, Kadhom N, Prip-Buus C, Gobin S, Saudubray JM, Bonnefont JP. Correlation between genotype, metabolic data and clinical presentation in carnitine palmitoyl transferase 2 (CPT2) deficiency. Hum Mutat. 2003;21:493–501. doi:10.1002/humu.10201.
Isackson PJ, Bennett MJ, Lichter-Konecki U, Willis M, Nyhan WL, Sutton VR, Tein I, Vladutiu GD. CPT2 gene mutations resulting in lethal neonatal or severe infantile carnitine palmitoyl transferase II deficiency. Mol Genet Metab. 2008;94:422–7. doi:10.1016/j.ymgme.2008.05.002.
Deschauer M, Wieser T, Zierz S. Muscle carnitine palmitoyl transferase II deficiency: clinical and molecular genetic features and diagnostic aspects. Arch Neurol. 2005;62:37–41. doi:10.1001/archneur.62.1.37.
Gillingham MB, Scott B, Elliott D, et al. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3 hydroxyl acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2006;89:58–63. doi:10.1016/j.ymgme.2006.06.004.
Uchida Y, Izai K, Orii T, Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. I. Purification and properties of very-long-chain acyl-coenzyme a dehydrogenase. J Biol Chem. 1992;267:1027–33.
Boneh A, Andresen BS, Gregersen N, Ibrahim M, Tzanakos N, Peters H, Yaplito-Lee J, Pitt JJ. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab. 2006;88:166–70. doi:10.1016/j.ymgme.2005.12.012.
McHugh DM, Cameron CA, Abdenur JE, Abdulrahman M, Adair O, Al Nuaimi SA, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011;13:230–54. doi:10.1097/GIM.0b013e31820d5e67.
Vianey-Saban C, Divry P, Brivet M, et al. Mitochondrial very-long-chain acyl-coenzymeA dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta. 1998;269:43–62. doi:10.1016/S0009-8981(97)00185-X.
Solis JO, Singh RH. Management of fatty acid oxidation disorders: a survey of current treatment strategies. J Am Diet Assoc. 2002;102:1800–3. doi:10.1016/S0002-8223(02)90386-X.
Behrend AM, Harding CO, Shoemaker JD, Martern D, Sahn DJ, Elliot DL, Gillingham MB. Substrate oxidation and cardiac performance during exercise in disorders of long chain fatty acid oxidation. Mol Genet Metab. 2012;105:110–5. doi:10.1016/j.ymgme.2011.09.030.
Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, et al. Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96:81–2. doi:10.1016/j.ymgme.2008.09.008.
Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, Strauss AW. General mitochondrial trifunctional protein (TFP) deficiency as a result of either α- or β-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr Res. 2004;55:190–6. doi:10.1203/01.PDR.0000103931.80055.06.
Das AM, Illsinger S, Lucke T, et al. Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. Clin Chem. 2006;52:530–4. doi:10.1373/clinchem.2005.062000.
Scheuerman O, Wanders RJA, Waterham HR, Dubnov-Raz G, Garty B-Z. Mitochondrial trifunctional protein deficiency with recurrent rhabdomyolysis. Pediatr Neurol. 2009;40:465–7. doi:10.1016/j.pediatrneurol.2008.12.017.
Den Boer MEJ, Dionisi-Vici C, Chakrapani A, Van Thuijl AOJ, Wanders RJA, Wijburg FA. Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003;142:684–9. doi:10.1067/mpd.2003.231.
Olsen RKJ, Pourfarzam M, Morris AAM, et al. Lipid-storage myopathy and respiratory insufficiency due to ETFQO mutations in a patient with late-onset multiple acyl-CoA dehydrogenation deficiency. J Inherit Metab Dis. 2004;27:671–8. doi:10.1023/B:BOLI.0000042986.10291.e9.
Frerman FE, Goodman SI. Defects of electron transfer flavoprotein and electron transfer flavoprotein ubiquinone oxidoreductase: glutaric acidaemia type II. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 2357–65. doi:10.1036/ommbid.131.
Christensen E, Kolvraa S, Gregersen N. Glutaric aciduria type II: evidence for a defect related to the electron transfer flavoprotein or its dehydrogenase. Pediatr Res. 1984;18:663–7. doi:10.1203/00006450-198407000-00020.
Olsen RKJ, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–54. doi:10.1093/brain/awm135.
Wen B, Dai T, Li W, et al. Riboflavin-responsive lipid storage myopathy caused by ETFDH gene mutations. Journal of Neurology Neurosurgery and Psychiatry. 2010;81:231–6. doi:10.1136/jnnp.2009.176404.
Gregersen N, Andresen BS, Pedersen CB, Olsen RKJ, Corydon TJ, Bross P. Mitochondrial fatty acid oxidation defects remaining challenges. J Inherit Metab Dis. 2008;31:643–57. doi:10.1007/s10545-008-0990-y.
Van Hove JLK, Grunewald S, Jaeken J, et al. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). Lancet. 2003;361:1433–5. doi:10.1016/S0140-6736(03)13105-4.
Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron- transferring -flavoprotein dehydrogenase (ETF-DH) gene. Brain. 2007;130:2037–44. doi:10.1093/brain/awm054.
Derks TG, Reijngoud DJ, Waterham HR, Gerver WJ, Van Den Berg MP, Sauer PJ, Smit GP. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr. 2006;148:665–70. doi:10.1016/j.jpeds.2005.12.028.
Iafolla AK, Thompson RJ Jr, Roe CR. Medium-chain acyl-coenzyme a dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994;124:409–15. doi:10.1016/S0022-3476(94)70363-9.
Ruitenbeek W, Poels PJE, Tumbull DM, Garavaglia B, Chalmers RA, Taylor RW, Gabreels FJM. Rhabdomyolysis and acute encephalopathy in late onset medium chain acyl-CoA dehydrogenase. Deficiency Journal of Neurology, Neurosurgery, and Psychiatry. 1995;58:209–14. PMC1073319
Lang TF. Adult presentations of medium-chain acyl-CoA dehydrogenase deficiency (MCADD). J Inherit Metab Dis. 2009;32:675–83. doi:10.1007/s10545-009-1202-0.
Rinaldo P, O'Shea JJ, Coates PM, Hale DE, Stanley CA, Tanaka K. Medium-chain acyl-CoA dehydrogenase deficiency. Diagnosis by stable-isotope dilution measurement of urinary n-hexanoylglycine and 3-phenylpropionylglycine. N Engl J Med. 1988;319:1308–13. doi:10.1056/NEJM198811173192003.
Saudubray JM, Martin D, De Lonlay P, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inher Metab Dis. 1999;22:488–502. doi:10.1023/A:1005556207210.
• Schiff M, Haberberger B, Xia C, Mohsen AW, Goetzman ES, Wang Y, Uppala R, Zhang Y, Karunanidhi A, Prabhu D, Alharbi H, Prochownik EV, Haack T, Haberle J, Munnich A, Rotig A, Taylor RW, Nicholls RD, Kim JJ, Prokisch H, Vockley J. Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum Mol Genet. 2015;24:3238–47. doi:10.1093/hmg/ddv074. This publication contains multiple functional studies on cell lines and in animal models to elucidate the effect and function of ACAD 9 in cells.
• Collet M, Assouline Z, Bonnet D, Rio M, Iserin F, Sidi D, et al. High incidence and variable clinical outcome of cardiac hypertrophy due to ACAD9 mutations in childhood. Eur J Hum Genet. 2016;24:1112–6. doi:10.1038/ejhg.2015.264. This study specifically addresses the cardiac manifestations associated with ACAD 9 deficiency and the outcomes in response to transplant and available therapies.
Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110:259–69. doi:10.1172/JCI15311.
Djouadi F, Aubrey F, Schlemmer D, Ruiter JP, Wanders RJ, Strauss AW, Bastin J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet. 2005;14:2695–703. doi:10.1093/hmg/ddi303.
Gobin-Limballe, S., Djouadi, F., Aubey, F., Olpin, S., Andersen, B.S., et.al. Genetic basis for correction of very-long-chain acyl-coenzyme a dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy. Am J Hum Genet 2007:81:1133–1143 DOI: 10.1086/522375.
• Orngreen MC, Madsen KL, Preisler N, Andersen G, Vissing J, Laforet P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. 2014;82:607–13. doi:10.1212/WNL.0000000000000118. This clinical study is important because it is a randomized human clinical trial reflecting lack of in vivo response to bezafibrate, in opposition to the beneficial results reported in vitro studies.
Wang Y, Mohsen A-W, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem. 2010;285:29834–41. doi:10.1074/jbc.M110.139493.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pathobiology of Orphan Diseases
Rights and permissions
About this article

Cite this article
El-Gharbawy, A., Goldstein, A. Mitochondrial Fatty Acid Oxidation Disorders Associated with Cardiac Disease. Curr Pathobiol Rep 5, 259–270 (2017). https://doi.org/10.1007/s40139-017-0148-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-017-0148-4