
Abstract
Nosema ceranae infection induces energetic stress, malnutrition, and precocious foraging in Apis mellifera. This study investigated effects of N. ceranae infection on the gene expression, protein content, and enzyme activity in the hypopharyngeal gland of nurse bees. The 285 differentially expressed genes were found between N. ceranae-infected and control bees using RNA-Seq, and 279 were upregulated and 6 were downregulated in infected bees. The protein level was significantly lower in infected bees than in controls, implying protein dysmetabolism and energetic stress in infected bees. The ELISA test showed that the specific activity of amylase was significantly higher in infected bees than in controls. The elevated enzymic activities were accompanied by an increased expressed level of hemolymph juvenile hormone-binding protein-encoding gene in infected bees, suggesting the possibility of Nosema-infected nurse bees to engage in behaviors that are normally performed by foragers.
Similar content being viewed by others

1 Introduction
The European honeybee, Apis mellifera L., is the primary species of domestic bees worldwide and are subject to many biotic and abiotic stressors including parasites, pesticides, and habitat destruction (Vanengelsdorp and Meixner 2010; Goulson et al. 2015). The microsporidian parasite Nosema ceranae was originally found infecting the Asian honeybee Apis cerana (Fries et al. 1996), and this intracellular gut parasite has become a common pathogen of A. mellifera L. since it was presented in European bees about 20 years ago (Paxton et al. 2007). Honeybees infected with N. ceranae show higher levels of hunger and high mortality due to nutritional stress (Mayack and Naug 2009), and infected bees exhibit increased sucrose responsiveness and are less likely to share food with their fellow bees (Naug and Gibbs 2009). Precocious foraging is observed in nurse-aged bees after infection with N. ceranae (Goblirsch et al. 2013; Natsopoulou et al. 2016). Furthermore, N. ceranae infection can cause an immune dysregulation in bees, making the host highly susceptible to other pathogens (Antúnez et al. 2009; Doublet et al. 2017).
Polymerase chain reaction (PCR) results indicated the presence of N. ceranae nucleic acids in the midgut and hypopharyngeal glands (HPGs) of A. mellifera, suggesting that N. ceranae is not tissue specific (Chen et al. 2009; Copley and Jabaji 2012). The HPGs, located in the frontal area of the heads of worker bees, are responsible for secreting protein-rich food royal jelly which is used to feed all larvae and queen of colonies (Winston 1991; Hrassnigg and Crailsheim 1998). Protein synthesis in the HPGs of bees, 8–16 days old, exhibits the highest activity (Knecht and Kaatz 1990), and the HPGs reach their full development when bees are about 9 days old (Jaycox et al. 1974). In addition, the functional changes of the HPGs in worker bees are closely related to age and task (Winston 1991). The HPGs in nurse-aged bees produce royal jelly for brood rearing, and the HPGs in bees performing foraging activities secrete invertases, including amylase, glucose oxidase, and α-glucosidase for nectar processing (Winston 1991; Ueno et al. 2009).
The development of the gland is negatively affected by biotic and abiotic stresses (DeGrandi-Hoffman et al. 2010; Jack et al. 2016). The HPGs of Nosema-infected bees are smaller than those of uninfected bees (Alaux et al. 2010), and decreased protein content was observed in HPGs of N. ceranae-infected bees (Jack et al. 2016). Moreover, the gland malfunctions were observed in bees infected with Nosema apis (Wang and Moeller 1971). The protein content of the HPGs is lower in bees fed with sugar water alone than those in bees fed with sugar water plus pollen diets (DeGrandi-Hoffman et al. 2010), and transcriptome analysis of the HPGs revealed that a greater number of upregulated genes were observed in the pollen-deprived undernourished bees compared with those of the pollen-fed, well-nourished bees (Corby-Harris et al. 2016).
Although extensive studies have been conducted on nutritional, physiological, and behavioral changes in honeybees exposed to Nosema sp. infection, little is known about changes in transcriptional profiles and enzymatic activities in the HPGs of bees infected with N. ceranae. We conducted study to determine whether differences exist in enzymatic-specific activities between N. ceranae-infected nurse-aged bees and control bees by sandwich enzyme-linked immunosorbent assay (ELISA). RNA sequencing (RNA-Seq) was employed to analyze the transcriptional response of the HPGs to N. ceranae stress, and differentially expressed genes (DEGs) were used for gene ontology (GO) and KEGG pathway enrichment analysis.
2 Materials and methods
2.1 N. ceranae spores
Nosema spores were purified in accordance with a previous study (Chen et al. 2009). Briefly, about 60 forager bees were caught at the entrance of a Nosema-infected colony. Then, the midguts were dissected and homogenized in a glass grinder containing 1 ml of sterile water. Afterward, the suspension was centrifuged at 3000 rpm, and the pellet was resuspended in 1 ml of sterile water. The resuspended solution was loaded on the discontinuous gradient consisting of 25%, 50%, 75%, and 100% Percoll solutions (1 ml each, v/v), and then centrifuged twice at 5000 rpm for 20 min at 4 °C. Subsequently, the supernatant was discarded, and the spore pellet was collected carefully. The spore pellet was then washed twice with the sterile water and further centrifuged at 8000 rpm for 10 min at 4 °C. The purified Nosema spore pellet was resuspended in sterile water and stored at 4 °C for later use. Finally, the spore concentration was counted using a hemocytometer (Cantwell 1970), and the species of Nosema spores was verified in accordance with a previously described method (Hamiduzzaman et al. 2010).
2.2 Experimental infection and spore counts
2.2.1 Experimental infection of bees with N. ceranae
Four colonies were used in the study, and one comb frame that had the most brood with emerging bees was collected from each colony. Each capped brood comb was placed into a single frame cage, and the four single frame cages were placed in an incubator at 34 °C with 65–70% relative humidity. After 24-h incubation, we used roughly equal numbers of newly emerged bees from each brood comb, so the newly emerged bees were therefore mixed approximately equally from four different colonies. The newly emerged bees were selected randomly for further treatment in the study, and the ubiquitous deformed wing virus (DWV) was regarded as the background pathogen of bees used in this study (Lanzi et al. 2006). Bees that were fed individually with 2 μl of 50% sugar water containing 105N. ceranae spores were assigned to the treatment group. Bees that were fed only with 2 μl of 50% sugar water were assigned to the control group.
Treated bees were randomly distributed into each of the plexiglass cages (9 cm × 7 cm × 8 cm) with multiple ventilation holes covering four sides of the cage. A 10-ml syringe containing 50% sugar water was then inserted into the cage through a hole of 1.7-cm diameter on the top side of the cage. Each cage of bees was provided with sugar water and a sufficient amount of pollen diet under ad libitum feeding conditions during the experimental period. To avoid possible N. ceranae spore contamination in pollen sources consumed by newly emerged bees, we used commercial pollen substitute diet composed of a mixture of 30% irradiated rape pollen and 70% soybean flour (Wangshi Animal Health Co., Ltd., Sichuan, China) as protein source for the bees. The pollen diet for each cage of bees was prepared by mixing the pollen substitute with 50% sugar water at a ratio of 5:1 (i.e., 5 g of the pollen substitute with 1 ml of 50% sugar water). The pollen diet was then distributed into two 1.5-ml centrifuge tubes, which were placed in the bottom of each cage.
The cages containing bees were then kept in an incubator set at 30 °C with 65–70% RH, and each cage contained 65 bees (nine cages per group). Of nine cages per group, three cages of bees were used for RNA-Seq analysis, three cages of bees were used for qRT-PCR confirmation, and three cages of bees were used for the quantification of total proteins and the enzyme activity in HPGs. The samples were collected for the experimental and control groups on day 9 post-infection and were stored at − 80 °C for later use. A table displaying the experimental design is shown in Online Resource 1.
2.2.2 Nosema spore counts
Before dissection of HPGs from the bees, the midguts of 30 bees from three control cages (10 bees each) and 30 bees from three treatment cages (10 bees each) were dissected and homogenized individually in 200 μl sterile water to assess the intensity of N. ceranae infections 9 days after infection. The homogenate was diluted 10 to 50 times based on the spore load present in the original homogenate, and a 10 μl sample of each diluted homogenate was used for counting the spore concentration using the method described above.
2.3 RNA-Seq analysis
For both treatment and control groups, 30 bees were collected from one cage, and 90 bees in total from three cages were used. The HPGs were dissected on dry ice under a stereomicroscope. A total of 30 pairs of HPGs were placed in a 1.5-ml centrifuge tube, and six tubes were used. Total RNA was extracted from HPGs in each tube using Trizol reagent (Invitrogen, CA, USA) in accordance with the manufacturer’s instructions. The mRNA was selected by oligo (dT) magnetic beads from a total of 3 μg of high-quality total RNA. Sequencing libraries were prepared by the NEBNext Ultra RNA library prep kit for Illumina (NEB, USA) according to the manufacturer’s recommendations. In brief, the mRNA was fragmented at an elevated temperature in NEBNext First-Strand Synthesis Reaction Buffer (5×). First-strand cDNA was prepared using random oligonucleotides and SuperScript II Reverse Transcriptase, followed by second-strand cDNA synthesis using DNA polymerase I and RNase H. DNA fragments were blunt ended, and the 3′ ends were adenylated. NEBNext adaptors with hairpin loop structure were then added for hybridization, and the resulting library fragments were purified using the Agencourt AMPure XP system (Beckman Coulter, Inc., CA, USA) to select 150–200 bp cDNA fragments. Exactly 3 μl of USER Enzyme (NEB, USA) was then used with the size-selected, adaptor-ligated cDNA at 37 °C for 15 min and then at 95 °C for 5 min prior to PCR. PCR was carried out using the Phusion High-Fidelity DNA Polymerase, universal PCR primers, and index (X) primer. Afterward, PCR products were then cleaned by AMPure XP system, and the quality assessment of the library was performed on a Bioanalyzer 2100 (Agilent). The index-coded samples were clustered using Illumina’s cBot cluster generation system with the TruSeq PE cluster kit version 3-cBot-HS following the manufacturer’s instructions.
The library preparations were sequenced using the Illumina HiSeq 2000 platform, which generated paired-end reads of 125 bp/150 bp. After quality filtering, the paired-end clean reads were mapped to genome sequences of three most common viruses infecting bees, black queen cell virus (BQCV), DWV, and Israeli acute paralysis virus (IAPV) using bowtie2 (Langmead and Salzberg 2012). The clean reads were then mapped to the A. mellifera reference genome (version 4.5) using TopHat v2.0.9 after filtering out viral reads (Kim et al. 2013). The number of reads mapped to each gene was counted using HTSeq v0.6.1 (Anders et al. 2015). DESeq R package (1.18.0) was used for differential expression analysis between the two groups. The GO terms and the KEGG pathway enrichment analysis of differentially expressed genes were performed by the GOseq R package and KOBAS software, respectively.
2.4 Validation of RNA-Seq data using quantitative RT-PCR
Quantitative RT-PCR was used to validate the ten selected DEGs from RNA-Seq data. Another set of 90 pairs of HPGs were used for RNA extraction for each of the two groups. For each group, 30 pairs of HPGs were put into a 1.5-ml centrifuge tube, and three tubes were used. Total RNA was extracted using TransZol Up reagent (TransGen Biotech, China) according to the manufacturer’s instructions. The concentration of total RNA was quantified by using a spectrophotometer (NanoDrop 2000, USA). The cDNA was synthesized using the One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen Biotech, China) following the manufacturer’s protocol. In brief, 500 ng of total RNA, 1 μl of oligo dT primer (0.5 μg/μl), 10 μl of 2× reaction mix, 1 μl of RT/RI enzyme mix, 1 μl of gDNA remover, and RNase-free dH2O were mixed to a final volume of 20 μl. The 20 μl reaction solution was incubated at 42 °C for 15 min, followed by 5 s at 85 °C. The cDNA was stored at − 20 °C. Real-time PCR was performed on a Bio-Rad CFX384 real-time system as follows: 1 μl of 1:3 diluted cDNA, 5 μl of 2× SYBR Premix Ex Taq (Tli RNaseH Plus, Takara, Japan), 0.4 μl of each specific primer (10 μM each) listed in Online Resource 2 (Huising and Flik 2005), and RNase-free dH2O were added to a final volume of 10 μl. The reactions were carried out in triplicates. The thermal profile consisted of initial denaturation at 94 °C for 3 s, 45 cycles of 94 °C for 5 s and 60 °C for 30 s, followed by melting curve analysis. Amplification efficiencies of the target and reference genes were determined as previously described (Chen et al. 2005).
2.5 Quantification of total proteins in HPGs
Twenty bees from each cage and 60 bees from three cages were used for the gland dissection from each group. Total proteins in the HPGs were extracted using a total protein extraction kit (TansGen Biotech, China). Then, 20 pairs of HPGs were transferred to each 1.5-ml centrifuge tubes and rinsed twice with 1 ml of ice-cold phosphate-buffered solution. Each tube was centrifuged at 500 rpm for 5 min at 4 °C. After discarding the supernatant, the gland pellet was resuspended in 1 ml of total protein extraction buffer and homogenized in a glass homogenizer. After homogenization, the suspension was transferred to a 1.5-ml centrifuge tube and incubated on ice for 30 min. The suspension was vibrated every 10 min during incubation. The tubes were centrifuged individually at 14,000 rpm for 5 min at 4 °C. The supernatants were collected carefully, and the total protein content was determined using a bicinchoninic acid (BCA) (Smith et al. 1985) protein assay kit (TansGen Biotech, China). Precisely 20 μl of each sample solution was added to the wells of a 96-well plate, and each sample was measured in four replicates. Subsequently, 200 μl of the BCA working solution was added to each sample well of the plate. The plate was incubated at 37 °C for 60 min. The absorbance value of each sample solution was recorded at 595 nm with a microplate reader (TECAN Infinite F50). The protein concentration of each sample was calculated according to a standard curve generated by a series of known concentrations of bovine serum albumin (BSA) solutions (25, 50, 100, 200, 300, 400, and 500 μg/ml).
2.6 Measurement of specific enzyme activities in HPGs
The activities of amylase, glucose oxidase, and α-glucosidase were determined using the total protein samples obtained above by sandwich ELISA kits according to instructions of the manufacturer (mlbio, Shanghai, China). Briefly, the three antibodies used were anti-amylase, anti-α-glucosidase, and anti-glucose oxidase, and the wells of a microtiter plate were precoated with the respective antibody. For determining the activity of each enzyme in each group, 10 μl of total protein from HPGs (1:5 dilution) was added to the well, and reactions were performed in triplicate wells. Then, 100 μl of each respective antibody labeled with horseradish peroxidase (HRP) was added to each reaction well. The microtiter plate was incubated at 37 °C for 60 min. The reaction solutions were discarded, and each reaction well was washed five times with wash buffer. A total of 100 μl of tetramethylbenzidine (TMB) was added to each reaction well, and the chromogenic reaction between TMB and HRP was performed at 37 °C for 60 min. The optical density of the reaction product was measured at 450 nm, and the activity of each enzyme was calculated on the basis of the standard curve generated by the serial dilutions of standard enzymes with known activities. The units of enzyme activity per gram of protein were used to express the specific activity of each enzyme (Habig et al. 1974).
2.7 Data analysis
The P values were set according to the Benjamini–Hochberg multiple comparison procedure for controlling the false discovery rate (FDR) (Benjamini and Hochberg 1995), and P values less than 0.05 were used to determine the significance of differences in differentially expressed genes and the GO and KEGG enrichment analysis of the differentially expressed genes based on RNA-Seq data. The differences in expression levels of the ten selected genes, the total protein content, and the specific enzyme activities in the HPGs of the two groups of bees were analyzed by Student’s t test using SPSS 18.0 (SPSS Inc., Chicago), and data are presented as mean ± SE.
3 Results
3.1 N. ceranae spore loads
All bees of the treatment group were confirmed to be infected with N. ceranae, and the average of the intensity of N. ceranae was 2.3 ± 0.16 × 107 spores/bee in the N. ceranae-infected bees (n = 30). N. ceranae spores were not observed in the midgut of control bees (n = 30).
3.2 Analysis of DEGs in HPGs
The percentage of clean reads in raw reads ranged from 92 to 96% across all six libraries (SRA accession number SRP133700), and approximately 53.7 and 54.3 million paired-end clean reads were obtained for the sequencing libraries of the N. ceranae-infected and control bees, respectively. Among these clean reads, the percentage of clean reads mapping to the DWV genome for the N. ceranae-infected and control bees was 3.1% and 3.4%, respectively, and no reads were mapped to the genome of BQCV and IAPV. The clean reads mapping to the DWV genome were filtered out, and we only used clean reads mapping to the A. mellifera reference genome for gene expression analysis. Approximately 38.3 and 36.7 million paired-end clean reads were mapped to the A. mellifera reference genome for the sequencing libraries of the N. ceranae-infected and control bees, respectively.
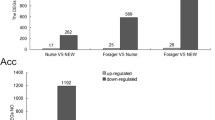
A total of 285 DEGs that met both P value and fold-change criteria were found in the HPGs between the N. ceranae-infected and control bees (P < 0.05; Figure 1). Among them, 279 and 6 genes were upregulated and downregulated in the HPGs of the N. ceranae-infected bees, respectively (Online Resource 3; Online Resource 4). Considerably more differential expressed genes were upregulated in the HPGs of the N. ceranae-infected bees compared with those of the control bees. N. ceranae infection in the HPGs upregulated the expression levels of genes, including neuropeptide genes (allatostatin A, FMRFamide, etc.) and genes involved in disease resistance and immune response in the N. ceranae-infected bees. In addition, genes downregulated in the HPGs of the N. ceranae-infected bees mainly participated in the modulation of protein-processing activities.

Differential expressed genes in HPGs between N. ceranae-infected bees and control bees (P < 0.05). Red and green represent the upregulated and downregulated genes in N. ceranae-infected bees, respectively; no differentially expressed genes are shown in blue.
3.3 GO and KEGG enrichment analysis
We identified the upregulated genes that were significantly enriched for 23 GO terms in the N. ceranae-infected bees (P < 0.05; Table I). The enriched GO terms were mainly involved in G protein-coupled receptor activity, single-organism localization and transport, transmembrane transporter activity, signal transduction, and neurotransmitter transport. However, no significant GO terms were found in the genes downregulated in N. ceranae-infected bees. In addition, six significantly enriched KEGG pathways associated with the nine upregulated genes were identified in the N. ceranae-infected bees (P < 0.05), and upregulated genes that participated in KEGG pathways included the metabolism of fatty acids, ether lipid, and glycerophospholipid; neuroactive ligand-receptor interaction; and ECM-receptor interaction (Table II). Three significantly enriched KEGG pathways associated with the three downregulated genes were identified in N. ceranae-infected bees (P < 0.05), and the pathways included protein processing in the endoplasmic reticulum, sphingolipid metabolism, and other glycan degradations (Table II). Notably, the two genes (GB45495, GB40976) involved in protein processing in endoplasmic reticulum were significantly downregulated in the N. ceranae-infected bees.
3.4 qRT-PCR analysis
The relative expression levels of genes encoding zinc finger (GB40145), hemolymph juvenile hormone binding (GB43129), carboxylesterase type B (GB43549), CHK kinase-like (GB47148), allatostatin (GB47928), amine oxidases (GB50076), fatty acid desaturase type 1 (GB51236), neuropeptide-like 1 (GB43772), heat shock protein 90 (GB40976), and glutamine synthetase 2 (GB51371) were selected at random and validated by qRT-PCR assay. Given that similar amplification efficiencies were observed between the target and reference genes (actin), the comparative Ct method 2−△△Ct method was used to analyze the relative expression levels of the genes (Livak and Schmittgen 2001). Consistent with the gene expression in RNA-Seq data, all ten genes exhibited a higher level of expression in the HPGs of the N. ceranae-infected bees, and eight of the ten genes were significantly upregulated in the HPGs of the N. ceranae-infected bees in comparison with those of the control bees (*P ≤ 0.05, **P < 0.01; Figure 2).

The relative expression levels (qRT-PCR) and the corresponding RPKM (reads per kilobase of exon model per million mapped reads) values of ten DEGs in HPGs of N. ceranae-infected bees relative to controls. The error bar indicates SE; asterisk represents significant differences (Student’s t test, *P ≤ 0.05; **P < 0.01).
3.5 Analysis of total protein and specific enzyme activities in HPGs
A significantly lower total protein concentration of the HPGs was found in the N. ceranae-infected bees (569 ± 61 μg/ml) than in the control bees (897 ± 105 μg/ml) at day 9 post-infection (P ≤ 0.05; Figure 3a). In addition, the specific activity of amylase was significantly higher in the HPGs of the N. ceranae-infected bees than those of the controls (P < 0.01; Figure 3b). The specific activities of glucose oxidase and α-glucosidase were higher in the HPGs of the N. ceranae-infected bees than in those of the controls, but no significant difference was found (Figure 3c, d).

Comparative analysis of total protein contents (a), specific activities of amylase (b), glucose oxidase (c), and α-glucosidase (d) in HPGs between N. ceranae-infected bees and controls. The error bar indicates SE; asterisk represents significant differences (Student’s t test, *P ≤ 0.05; **P < 0.01).
4 Discussion
Honeybee HPGs which are involved with age polyethism play a fundamental role in the development of colonies (Winston 1991). N. ceranae infection negatively affects many aspects of bee health including causing poor development of the HPGs in infected bees (Alaux et al. 2010; Jack et al. 2016). In the present study, the gene expression profiles revealed that a large number of genes were upregulated in the HPGs of the N. ceranae-infected bees compared with those of controls after 9 days of infection. After comparing differentially expressed genes to the data of the previous study (Corby-Harris et al. 2016) by the hypergeometric test (http://nemates.org/MA/progs/overlap_stats.html), a total of 46 upregulated genes in the HPGs of the N. ceranae-infected bees significantly overlapped with those upregulated genes in the HPGs of pollen-deprived bees (P < 0.0001; Online Resource 5), suggesting that these genes may be involved in the mechanism underlying nutrient and energy imbalance in bees induced by N. ceranae infection.
N. ceranae-infected nurse bees exhibited relatively elevated levels of juvenile hormone (JH) at 8 days after infection and were more likely to engage in precocious foraging compared to non-infected bees (Goblirsch et al. 2013). Our data revealed that the N. ceranae-infected bees exhibited significantly higher levels of the gene encoding hemolymph juvenile hormone-binding protein (JHBP) than control bees. The binding protein plays key roles in protecting hemolymph juvenile hormone against hydrolysis by hemolymph esterases in insects and in distributing the JH more quickly throughout the body of the insect (Hammock et al. 1975; Kołodziejczyk et al. 2003). The upregulation of the gene encoding JHBP in our observation may be related to the elevated JH titers in N. ceranae-infected bees. Our results suggest that the HPGs of N. ceranae-infected nurse bees exhibited increased specific activities of amylase which may well correspond to precocious foraging induced by Nosema infection in nurse-aged bees (Goblirsch et al. 2013).
N. ceranae infection can cause malnutrition in honeybees through absorption of nutrients from the intestines of honeybees; thus, it may impair the development of the HPGs of infected bees (Martín-Hernández et al. 2011). Consistent with previous studies (Jack et al. 2016), the total protein contents of the HPGs were significantly lower in N. ceranae-infected bees than in controls. Previous studies showed that N. ceranae spores proliferated slowly before day 7 post-infection in N. ceranae-infected bees (105 spores/bee), and N. ceranae infection exerted mild physiological effects on bees (Li et al. 2018). In addition, bees consumed massive amounts of pollen diet at around 2–5 days old (Haydak 1970). In other words, it is therefore possible that N. ceranae-infected bees can be normally nourished before N. ceranae produces a higher number of spores and exerts its severe effects in bees at day 7 post-infection. Therefore, the decreased protein content in HPGs of N. ceranae-infected bees at day 9 post-infection was possibly because of a great increase in N. ceranae infestation levels. The two genes (GB45495, GB40976) involved in protein processing in the endoplasmic reticulum pathway were significantly downregulated in the N. ceranae-infected bees (Kanehisa and Goto 2000), and the HPGs of infected bees may produce royal jelly with significantly lower concentrations of proteins compared with those of the non-infected controls. Given that approximately one half of the dry mass of royal jelly are proteins (Bıliková et al. 2002), N. ceranae infection adversely affects the function of the HPGs in nurse-aged bees, and the nutritionally imbalanced food fed to the queen and larvae may ultimately negatively affect the development and survival of honeybee colonies. The HPGs of infected bees exhibited a lower protein content and a higher amylase activity, indicating that Nosema-infected nurse bees underwent pronounced changes in the hypopharyngeal gland function, and these physiological alterations may be closely related to those upregulated genes in HPGs of infected bees. In addition, N. ceranae infection poses nutritional and energetic stress to bees (Martín-Hernández et al. 2018), and therefore, HPG degradation is probably more active in Nosema-infected bees with smaller HPGs (Alaux et al. 2010), resulting in the large number of upregulated genes in Nosema-infected undernourished bees (Corby-Harris et al. 2016).
Additionally, our data revealed that four neuropeptide-encoding genes, namely, allatostatin A (GB47928), FMRFamide (GB41296), short neuropeptide F-like (GB42980), and neuropeptide-like 1 (GB43772), were significantly upregulated in the N. ceranae-infected bees compared with controls. Increased expression of allatostatin A was found in Drosophila preferring to consume protein-rich food strongly (Hentze et al. 2015). N. ceranae-infected bees may exhibit more pronounced protein hunger behaviors than controls because of impairment of protein synthesis in the HPGs, and an increased protein intake ultimately promotes the proliferation of N. ceranae in N. ceranae-infected bees (Jack et al. 2016). In addition, short neuropeptide F enhances feeding in Drosophila (Lee et al. 2004), and N. ceranae-infected bees eat significantly more than controls (Mayack and Naug 2009). Neuropeptides act as signaling molecules that play important roles in regulating behaviors, stress physiology, and immune response in insects (Gäde and Goldsworthy 2003; Schoofs et al. 2017), and changes in feeding behavioral response of N. ceranae-infected bees may be regulated in part via upregulation of neuropeptide-encoding genes in bees.
The findings provide further evidence demonstrating that N. ceranae can cause energetic stress in infected bees, resulting in gene expression changes associated with physiological homeostasis. Further work is needed to investigate the changing patterns of gene expression in the HPGs at different time points during the infection process of N. ceranae and to understand the direct or indirect alteration in HPG development caused by N. ceranae infection.
References
Alaux, C., Brunet, J.L., Dussaubat, C., Mondet, F., Tchamitchan, S., et al. (2010) Interactions between Nosema microspores and a neonicotinoid weaken honeybees (Apis mellifera). Environ. Microbiol. 12(3), 774–782
Anders, S., Pyl, P.T., Huber, W. (2015) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31(2), 166–169
Antúnez, K., Martín-Hernández, R., Prieto, L., Meana, A., Zunino, P., et al. (2009) Immune suppression in the honey bee (Apis mellifera) following infection by Nosema ceranae (Microsporidia). Environ. Microbiol. 11(9), 2284–2290
Benjamini, Y., Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 289–300
Bıliková, K., Hanes, J., Nordhoff, E., Saenger, W., Klaudiny, J., et al. (2002) Apisimin, a new serine–valine-rich peptide from honeybee (Apis mellifera L.) royal jelly: purification and molecular characterization 1. FEBS Lett. 528(1–3), 125–129
Cantwell, G. (1970) Standard methods for counting Nosema spores. Amer. Bee J. 110, 222
Chen, Y., Higgins, J., Feldlaufer, M. (2005) Quantitative real-time reverse transcription-PCR analysis of deformed wing virus infection in the honeybee (Apis mellifera L.). Appl. Environ. Microbiol. 71(1), 436–441
Chen, Y.P., Evans, J.D., Murphy, C., Gutell, R., Zuker, M., et al. (2009) Morphological, molecular, and phylogenetic characterization of Nosema ceranae, a microsporidian parasite isolated from the European honey bee, Apis mellifera. J. Eukaryot. Microbiol. 56(2), 142–147
Copley, T., Jabaji, S. (2012) Honeybee glands as possible infection reservoirs of Nosema ceranae and Nosema apis in naturally infected forager bees. J. Appl. Microbiol. 112(1), 15–24
Corby-Harris, V., Meador, C.A., Snyder, L.A., Schwan, M.R., Maes, P., et al. (2016) Transcriptional, translational, and physiological signatures of undernourished honey bees (Apis mellifera) suggest a role for hormonal factors in hypopharyngeal gland degradation. J. Insect Physiol. 85, 65–75
DeGrandi-Hoffman, G., Chen, Y., Huang, E., Huang, M.H. (2010) The effect of diet on protein concentration, hypopharyngeal gland development and virus load in worker honey bees (Apis mellifera L.). J. Insect Physiol. 56(9), 1184–1191
Doublet, V., Poeschl, Y., Gogol-Döring, A., Alaux, C., Annoscia, D., et al. (2017) Unity in defence: honeybee workers exhibit conserved molecular responses to diverse pathogens. BMC genomics 18(1), 207
Fries, I., Feng, F., da Silva, A., Slemenda, S.B., Pieniazek, N.J. (1996) Nosema ceranae n. sp.(Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana (Hymenoptera, Apidae). Eur. J. Protistol. 32(3), 356–365
Gäde, G., Goldsworthy, G.J. (2003) Insect peptide hormones: a selective review of their physiology and potential application for pest control. Pest Manag. Sci. 59(10), 1063–1075
Goblirsch, M., Huang, Z.Y., Spivak, M. (2013) Physiological and behavioral changes in honey bees (Apis mellifera) induced by Nosema ceranae infection. PLoS One 8(3), e58165
Goulson, D., Nicholls, E., Botías, C., Rotheray, E.L. (2015) Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science 347(6229), 2
Habig, W.H., Pabst, M.J., Jakoby, W.B. (1974) Glutathione S-transferases the first enzymatic step in mercapturic acid formation. J. Biol. Chem. 249(22), 7130–7139
Hamiduzzaman, M.M., Guzman-Novoa, E., Goodwin, P.H. (2010) A multiplex PCR assay to diagnose and quantify Nosema infections in honey bees (Apis mellifera). J. Invertebr. Pathol. 105(2), 151–155
Hammock, B., Nowock, J., Gooddman, W., Stamoudis, V., Gilbert, L.I. (1975) The influence of hemolymph-binding protein on juvenile hormone stability and distribution in Manduca sexta fat body and imaginal discs in vitro. Mol. Cell. Endocrinol. 3(3), 167–184
Haydak, M.H. (1970) Honey bee nutrition. Annu. Rev. Entomol. 15(1), 143–156
Hentze, J.L., Carlsson, M.A., Kondo, S., Nässel, D.R., Rewitz, K.F. (2015) The neuropeptide allatostatin A regulates metabolism and feeding decisions in Drosophila. Sci. Rep. 5, 11680
Hrassnigg, N., Crailsheim, K. (1998) Adaptation of hypopharyngeal gland development to the brood status of honeybee (Apis mellifera L.) colonies. J. Insect Physiol. 44(10), 929–939
Huising, M.O., Flik, G. (2005) The remarkable conservation of corticotropin-releasing hormone (CRH)-binding protein in the honeybee (Apis mellifera) dates the CRH system to a common ancestor of insects and vertebrates. Endocrinol. 146(5), 2165–2170
Jack, C.J., Uppala, S.S., Lucas, H.M., Sagili, R.R. (2016) Effects of pollen dilution on infection of Nosema ceranae in honey bees. J. Insect Physiol. 87, 12–19
Jaycox, E.R., Skowronek, W., Guynn, G. (1974) Behavioral changes in worker honey bees (Apis mellifera) induced by injections of a juvenile hormone mimic. Ann. Entomol. Soc. Am. 67(4), 529–534
Kanehisa, M., Goto, S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28(1), 27–30
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4), R36
Knecht, D., Kaatz, H. (1990) Patterns of larval food production by hypopharyngeal glands in adult worker honey bees. Apidologie 21(5), 457–468
Kołodziejczyk, R., Kochman, M., Bujacz, G., Dobryszycki, P., Ożyhar, A., et al. (2003) Crystallization and preliminary crystallographic studies of juvenile hormone-binding protein from Galleria mellonella haemolymph. Acta Crystallogr. D Biol. Crystallogr. 59(3), 519–521
Langmead, B., Salzberg, S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9(4), 357
Lanzi, G., de Miranda, J.R., Boniotti, M.B., Cameron, C.E., Lavazza, A., et al. (2006) Molecular and biological characterization of deformed wing virus of honeybees (Apis mellifera L.). J. Virol. 80(10), 4998–5009
Lee, K.-S., You, K.-H., Choo, J.-K., Han, Y.-M., Yu, K. (2004) Drosophila short neuropeptide F regulates food intake and body size. J. Biol. Chem. 279(49), 50781–50789
Li, W., Chen, Y., Cook, S.C. (2018) Chronic Nosema ceranae infection inflicts comprehensive and persistent immunosuppression and accelerated lipid loss in host Apis mellifera honey bees. Int. J. Parasitol. Parasites 48(6), 433–444
Livak, K.J., Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4), 402–408
Martín-Hernández, R., Botías, C., Barrios, L., Martínez-Salvador, A., Meana, A., et al. (2011) Comparison of the energetic stress associated with experimental Nosema ceranae and Nosema apis infection of honeybees (Apis mellifera). Parasitol. Res. 109(3), 605–612
Martín-Hernández, R., Bartolomé, C., Chejanovsky, N., Le Conte, Y., Dalmon, A., et al. (2018) Nosema ceranae in Apis mellifera: a 12 years postdetection perspective. Environ. Microbiol. 20(4), 1302–1329
Mayack, C., Naug, D. (2009) Energetic stress in the honeybee Apis mellifera from Nosema ceranae infection. J. Invertebr. Pathol. 100(3), 185–188
Natsopoulou, M.E., McMahon, D.P., Paxton, R.J. (2016) Parasites modulate within-colony activity and accelerate the temporal polyethism schedule of a social insect, the honey bee. Behav. Ecol. Sociobiol. 70(7), 1019–1031
Naug, D., Gibbs, A. (2009) Behavioral changes mediated by hunger in honeybees infected with Nosema ceranae. Apidologie 40(6), 595–599
Paxton, R.J., Klee, J., Korpela, S., Fries, I. (2007) Nosema ceranae has infected Apis mellifera in Europe since at least 1998 and may be more virulent than Nosema apis. Apidologie 38(6), 558–565
Schoofs, L., De Loof, A., Van Hiel, M.B. (2017) Neuropeptides as regulators of behavior in insects. Annu. Rev. Entomol. 62, 35–52
Smith, P.E., Krohn, R.I., Hermanson, G., Mallia, A., Gartner, F., et al. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150(1), 76–85
Ueno, T., Nakaoka, T., Takeuchi, H., Kubo, T. (2009) Differential gene expression in the hypopharyngeal glands of worker honeybees (Apis mellifera L.) associated with an age-dependent role change. Zool. Sci. 26(8), 557–563
Vanengelsdorp, D., Meixner, M. (2010) A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Invertebr. Pathol. 103, S80-S95
Wang, D.-I., Moeller, F. (1971) Ultrastructural changes in the hypopharyngeal glands of worker honey bees infected by Nosema apis. J. Invertebr. Pathol. 17(3), 308–320
Winston, M.L. (1991) The biology of the honey bee. Harvard University Press, Cambridge.
Funding
This research was supported by the earmarked fund for Modern Agro-Industry Technology Research System (No. CARS-45-KXJ3) and Fujian Agriculture and Forestry University Foundation for Outstanding Youth (XJQ201606).
Author information
Authors and Affiliations
Contributions
ZGL, JFH, and SKS conceived this research and designed experiments; ZGL, WFH, and HYN participated in the design and interpretation of the data; JFH, TTY, and JNH performed experiments and analysis; ZGL, YPC, SKS, and YZZ wrote the paper and participated in the revisions of it. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Manuscript editor: Marina Meixner
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Réponses transcriptionnelles et physiologiques des glandes hypopharyngées chez l’abeille ( Apis mellifera L.) infectées par Nosema ceranae
abeilles domestiques / Nosema ceranae / glande hypopharyngée / gènes exprimés de manière différentielle / réponses physiologiques
Transkriptionelle und physiologische Antwort der Hypopharynxdrüsen der Honigbiene ( Apis mellifera ) bei Infektion mit Nosema ceranae
Honigbienen / Nosema ceranae / Hypopharynxdrüsen / differentielle Genexpression / physiologische Antwort
Rights and permissions
About this article

Cite this article
Li, Z., He, J., Yu, T. et al. Transcriptional and physiological responses of hypopharyngeal glands in honeybees (Apis mellifera L.) infected by Nosema ceranae. Apidologie 50, 51–62 (2019). https://doi.org/10.1007/s13592-018-0617-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-018-0617-8