
Abstract
Neurodegenerative diseases are broadly characterized neuropathologically by the degeneration of vulnerable neuronal cell types in a specific brain region. The degeneration of specific cell types has informed on the various phenotypes/clinical presentations in someone suffering from these diseases. Prominent neurodegeneration of specific neurons is seen in polyglutamine expansion diseases including Huntington’s disease (HD) and spinocerebellar ataxias (SCA). The clinical manifestations observed in these diseases could be as varied as the abnormalities in motor function observed in those who have Huntington’s disease (HD) as demonstrated by a chorea with substantial degeneration of striatal medium spiny neurons (MSNs) or those with various forms of spinocerebellar ataxia (SCA) with an ataxic motor presentation primarily due to degeneration of cerebellar Purkinje cells. Due to the very significant nature of the degeneration of MSNs in HD and Purkinje cells in SCAs, much of the research has centered around understanding the cell autonomous mechanisms dysregulated in these neuronal cell types. However, an increasing number of studies have revealed that dysfunction in non-neuronal glial cell types contributes to the pathogenesis of these diseases. Here we explore these non-neuronal glial cell types with a focus on how each may contribute to the pathogenesis of HD and SCA and the tools used to evaluate glial cells in the context of these diseases. Understanding the regulation of supportive and harmful phenotypes of glia in disease could lead to development of novel glia-focused neurotherapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Polyglutamine Disease
Polyglutamine diseases including Huntington’s disease (HD) and spinocerebellar ataxias (SCA1-3, SCA6, SCA7, SCA17) (Table 1) are members of a group of diseases characterized by expansion of the trinucleotide cytosine-adenosine-guanine (CAG) repeat that translates into glutamine resulting in an expanded polyglutamine (polyQ) repeat in the proteins encoded by the disease-causing genes. These polyglutamine diseases present clinically with motor abnormalities including chorea, ataxia, dystonia, and sometimes parkinsonism [1,2,3,4]. In addition, patients with these disorders also present with other clinical symptoms including psychiatric, cognitive, sensory, and bulbar, muscle atrophy, and visual impairments. These diseases can lead to substantial brain atrophy and result in significant degeneration that is often limited or most severe in a specific subset of neurons in different brain regions. In HD, degeneration and atrophy are most prominent in the striatum and cortex. In the SCAs, there is significant atrophy and neurodegeneration of cerebellar Purkinje cells with some of the SCAs also showing degeneration of cerebellar nuclei and brainstem atrophy. The degeneration of these cells contributes to the clinical impairments observed in the various SCAs. While degeneration in these diseases is largely present in select brain regions, most of the polyglutamine proteins are expressed throughout the brain and found in non-neuronal glial cells.
Understanding polyQ diseases requires appreciation of the complexity of different brain cell types and how their intrinsic physiology and interactions contribute to disease progression. A significant body of work using constitutive and conditional mouse models, transcriptome, and pathological analyses provides evidence that both neurons and glia are altered in these diseases and actively contribute to their pathogenesis. Moreover, while mutant polyQ proteins are often present from developmental stages, polyQ diseases most often manifest in midlife. Manipulation of glial responses at different stages of disease indicates the complex glia contribution to disease that is in part enacted by early beneficial effects of glial cells that help maintain almost normal performance for decades. As neurodegeneration progresses over decades, these initial beneficial reactions of glia become chronic and promote an irreversible pathology of the brain.
Glial Cells
Glial cells constitute half of the cells in the human brain and are important for proper nervous system function and development. They are responsible for maintaining homeostasis in the brain. These homeostatic functions are carried out by different types of glial cells, including astrocytes and oligodendrocytes that originate from neural tissue and microglia that are of myeloid lineage originating from the mesoderm. Thus, this heterogenous population of cells support broad functions/activities in the nervous system, including but not limited to neurogenesis, blood–brain barrier maintenance, energy metabolism, ion homeostasis, immune defense, modulation of neuroinflammation, synapse formation, neurotransmission and maintenance of neurotransmitter levels, blood flow regulation, sleep, and provide metabolic support [5,6,7,8,9].
Astrocytes are the most numerous glial cell type and they tile the entire brain. While originally perceived as merely responsible for the maintenance of brain structure, astrocytes play critical roles during development including guiding neurons, promoting synapse formation and are key regulators of synaptic function and neuronal metabolism in the adult brain. For instance, fine astrocytic processes envelop many synapses where they maintain homeostasis of ions and neurotransmitters, thus regulating excitability and responsiveness of neurons [10,11,12]. Astrocytes respond to brain injury by undergoing reactive gliosis, a process of gene expression, morphological and functional changes. Morphological changes in reactive astrocytes have been described in the nineteenth century [13]. Recent transcriptomic studies demonstrated diversity of gene expression changes in reactive astrocytes across different diseases. Reactive astrocytes can be neuroprotective and ameliorate disease pathology or harmful and exacerbate neurodegeneration.
Microglia are the resident immune cell of the nervous system and can contribute to both protective and toxic mechanisms in the nervous system. These cells play roles in the development of proper connections in the nervous system. While microglia play essential roles as brain macrophages, they are derived from the primitive yolk sac and lymphatic-dependent precursors unlike neurons, astrocytes, and oligodendrocytes that are derived from neuroectoderm. However, the microglia are distinct from peripheral macrophages/monocytes as they arise from different precursors, and although originally thought to be maintained in the brain by resident progenitor cells [14, 15], it is now believed that these cells are self-renewing [16]. During nervous system development, microglia prune synapses to ensure proper development of neural circuits. In adulthood astrocytes and microglia contribute to the maintenance of brain homeostasis and are responsive to changes in neuronal activity under normal physiological conditions [17]. Microglia change and become more neuroinflammatory primed with age [18]. These cells respond to injuries to the brain, whether caused by chronic neurological conditions or acute insults, by altering their cellular morphology, proliferating, releasing inflammatory molecules, and engulfing cellular debris to maintain homeostasis. Consequently, the best described and most prominent role for microglia is in immune defense and their involvement in neuroinflammatory changes in neurodegenerative diseases is widely studied. However, like astrocytes, the way these cells contribute to the pathogenesis of disease is varied and can be influenced by age in disease; including their ability to respond to the presence of mutated disease-causing proteins.
Oligodendrocytes are glial cells that produce myelin and myelinate axons in the central nervous system. These cells develop from glial progenitor cells; specifically, oligodendrocyte precursor cells (OPCs) [19,20,21,22] originate in and migrate from the ventricular zone. The OPCs continue to persist in the adult central nervous system, largely expressing neuron-glial antigen 2 (NG2) and platelet-derived growth factor receptor alpha (PDGFR-α) and their numbers seem to remain constant in the mature nervous system [23,24,25]. The OPCs migrate from their sites of origin along blood vessels [26, 27]. A fully differentiated and mature oligodendrocyte can be identified by the production of myelin and expression of myelin basic protein (MBP), myelin-associated glycoprotein (MAG), and myelin-oligodendrocyte glycoprotein (MOG) [28, 29]. Myelin insulates axons and allows for rapid propagation of electrical signals allowing for rapid conduction of action potentials in the nervous system and provide trophic support for the axon. Deficits in myelination, either due to changes in oligodendrocytes or their precursors, can cause axonal changes which contribute to motor behavioral and cognitive deficits associated with neurological diseases. Oligodendrocytes and OPCs can also become reactive, although the role of their reactivity in neurodegenerative diseases is less understood. Glial cells exhibit significant heterogeneity across and within brain regions that at least in part originates from unique needs of neurons they interact with. As such, glia may contribute to brain region–specific vulnerability of neurons as well as provide a neuron specific support in disease that can be harnessed for therapies.
Huntington’s Disease
Observations from HD Patients
Huntington’s disease (HD) is a progressive, autosomal dominant, fatal neurodegenerative disorder characterized clinically by deficits in motor function, cognitive impairment, and psychiatric disturbances [30]. George Huntington published the first clinical description of HD in 1872 when he described those suffering from this disease as having a “hereditary chorea” where chorea is defined by “dancing propensities” [31]. Although the motor symptoms are the most visible manifestations of HD, the cognitive and psychiatric impairments can also manifest early in the disease process. The average age of onset for HD is the mid-40s with death usually occurring about 20–25 years after clinical onset [1, 30]. Huntington’s disease is a rare disorder with a prevalence of about 2–3 persons per 100,000 and is caused by a CAG repeat expansion of greater than 40 in the gene encoding the widely expressed protein Huntingtin (HTT) [32, 33]. A CAG repeat expansion in the 36–39 range is incompletely penetrant, and individuals with repeats in this range are at risk for developing Huntington’s disease [34, 35]. Clinically, HD is diagnosed based on the presence of motor abnormalities and these motor changes become more prominent and debilitating as the disease progresses [30, 36, 37].
The striatum is the most significantly atrophied brain region, with degeneration also prominent in the cortex [38, 39]. Other brain regions including hypothalamus, hippocampus, and thalamus are also affected in HD, mostly at later stages of disease [40, 41]. While the mutant protein is widely expressed, neuropathologically, the striatal GABAergic MSNs are the most degenerated neuron in HD patient brains [39]. The MSNs are central to the normal function of the basal ganglia, a critical brain region involved in motor and limbic functions. The function of these cells is modulated by input from the cortex, thalamus, and substantia nigra [42, 43]. Additional modulation of these MSNs is from intrastriatal interactions with interneurons and glial cells. The glial cells, including astrocytes, oligodendrocytes, and microglial cells, express HTT. Using RNA in situ hybridization to target human HTT revealed its expression in glial cells in normal and in HD patient postmortem tissue [44]. More recent studies of isolated astrocytes from human tissue also show HTT mRNA expression in astrocytes [45]. While the human HTT mRNA seems to be higher in neurons than in glial cells, the human HTT protein is present in astrocytes and is comparable to the level observed in neurons [46,47,48]. The expansion of the polyglutamine repeat in HTT results in an aggregation prone protein and mutant HTT (mHTT) aggregates are observed in astrocytes in the gray and white matters (region of myelinated axons) in HD patient postmortem brains [48,49,50].
The change most observable in astrocytes that suggests altered function in neurological diseases, including polyglutamine diseases like HD, is the presence of reactive astrocytes [49, 51, 52]. Although the exact role of reactive astrocytes in neurological dysfunction is debatable, astrocytes in postmortem HD tissue have a reactive phenotype as demonstrated by changes in their morphology and an increase in the expression of GFAP with increasing disease grade from 0 to 4 (neuropathologically defined based on severity of striatal degeneration) [49, 51, 53, 54]. The location and pattern of reactive astrocytes seen in the HD post-mortem brains seem to follow the pattern of neurodegeneration observed in HD where degeneration is most prominent first in the dorsal striatum and then in the ventral striatum and observed earliest in the striosomal compartments of the striatum [55]. The presence of reactive astrocytes clearly denote alterations in astrocytes that include functional and molecular changes that depend on the specific insults or contexts to define whether those changes are protective or toxic [52, 56].
Studies using post-mortem tissue have identified significant gene expression changes in astrocytes that could contribute to altered astrocyte functions that will impinge on the proper function of the nervous system (Fig. 1). Multiple studies have identified a heterogenous population of astrocytes from mouse and human tissue. Astrocytes can be defined by a number of common genes; however, subtypes can be defined based on enrichment of additional genes [57,58,59,60,61]. Single nucleus RNA-Seq experiments using post-mortem tissue from the cingulate cortex of grade 3–4 HD tissue revealed the presence of a heterogenous population of astrocytes based on expression profiles [62]. The identified astrocytes could be subdivided into clusters that consisted of reactive astrocyte “states” as defined by the authors as having different patterns of expression of known astrocyte identity genes [62]. These gene expression changes include an elevation in the amount of GFAP expressed in the caudate putamen with increasing neuropathological grade, indicating a reactive astrocyte phenotype [62, 63]. Another study identified what have been called “A1” astrocytes in post-mortem HD tissue; those that significantly upregulate classical complement genes that have been shown to be destructive to synapses [64, 65]. Interestingly, when the genes defining this core A1 astrocyte signature were analyzed in additional studies of the cingulate cortex and striatum of post-mortem HD tissue, most of the genes comprising the A1 phenotype were not altered or the changes in expression of those genes were in the opposite direction indicative of an A1 phenotype [63]. Nonetheless, these studies have been very valuable in identifying astrocyte transcriptomic changes in specific brain regions at different neuropathological grades in HD, including at grade 0–1, suggesting a progressive loss of essential homeostatic astrocyte functions in the striatum prior to very significant neurodegeneration [63]. These data highlight the complexity of analyzing post-mortem tissue for transcriptional changes and the complexity in defining and attributing a single astrocyte gene signature to define a “toxic” cell type. These data also highlight differences in identifying gene expression signatures when analyzing bulk RNAseq as compared to sn-RNA seq. Nonetheless, these data point to specific pathway alterations that could be targeted for therapeutic intervention.

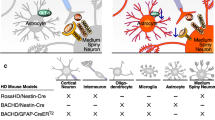
Huntington’s disease and glial cells. A Left—early-stage HD (H) and mutant Huntingtin (yellow) expressing mouse (M) brains. Reactive astrocytes (red) and reactive microglia (green) are present at early stages of HD, and myelin integrity (oligodendrocytes [purple]) is reduced. Gene expression changes are present at early disease stages. Top right—Reduced astrocytic homeostatic gene expression (red and blue) and myelin protein expression (green) is at early HD stages in cortex, striatum, and white matter. B Left—The number of reactive astrocytes (red) and reactive microglia (green) increases more at later HD stages, and myelin integrity (oligodendrocytes [purple]) is further reduced. Right—Astrocyte gene expression changes (red and blue) and myelin protein reduction (green) are exacerbated at later disease stages
Studying Mutant Huntingtin Expressing Astrocyte Contribution to HD in Mice
Many observations have been made about neuropathological changes in astrocytes in post-mortem tissue, in gene expression changes in astrocytes from bulk RNA and snRNA-Seq analyses, and even by using human iPSC-derived astrocytes. However, the ability to express mHTT in the mouse model selectively and widely allows for study and manipulation of astrocytes and their expression of mHTT to ascertain how the presence of mHTT affects these cells autonomously and how that may impact the neuronal circuit and behavior more broadly. Astrocytes in mice contain Htt mRNA [66] where the mRNA expression level is lower than what is observed in neuronal populations. However, the HTT protein in astrocytes [67, 68] is expressed at a similar level to what is observed in neurons [46, 50]. While the exact function of HTT in astrocytes remains an important area of study, there was a significant increase in the number of glial fibrillary acid protein (GFAP) positive cells and a decrease in the number of microtubule-associated protein 2 (MAP2) positive neurons when comparing cultures differentiated from mouse Hdh knock-out neural stem cells and control cultures. This finding suggests that wild-type HTT is involved in controlling the differentiation of neuronal and glial cells and that production of neurons from neural stem cells requires a normal level of wild-type HTT [69]. This data is in line with previous studies showing that wild-type HTT plays a role in central nervous system development and neuronal survival [70, 71]. However, the exact role of wild-type HTT in astrocytes in the nervous system will need to be further assessed in conditional knock-in mouse models, where one can specifically reduce the expression of endogenous HTT only in astrocytes.
Many different mouse models have been used to determine how astrocytes contribute to the pathogenesis of HD (Table 2). These mouse models express mHTT with varying CAG lengths, driven by different promoters and include knock-in mice with expanded CAG repeats in the endogenous mouse Hdh locus and transgenic mice overexpressing HTT in astrocytes alone or throughout the brain. Some of the mouse studies of mHTT in astrocytes have been performed using viral vectors designed to drive mHTT expression in astrocytes. Taking the approach of studying astrocytes in these various models has allowed the probing of cell-autonomous versus non-cell autonomous changes in astrocytes and neurons in vulnerable brain regions in HD. However, all results must be carefully considered, and the limitations acknowledged, given the differences in how the expression of mHTT is driven in these models, including genetically in transgenic N-terminal mHTT overexpressing mouse models driven by different promotors, transgenics overexpressing human full-length mHTT using the endogenous HTT promotor, and full-length mHTT knock-in models with expanded CAG repeats in the endogenous mouse Hdh gene. All these models express different levels of mHTT and have HD-like phenotypes of varying severity, timeframe of phenotypic onset, and lifespan of the model, each of which could impact the interpretation of the results obtained using a specific model [72].
The mHTT-expressing mouse models contain mHTT positive aggregates in astrocytes in the white matter as well as the gray matter [73,74,75]. Like the aggregation found in neurons in these mice, aggregation in the astrocytes also appears to be progressive, with the number of mHTT aggregates increasing as the animal ages and are found not only in the striatum and cortex of these mice but also in the corpus callosum [75, 76]. In addition, aggregates were found in astrocytes in the cortex, striatum, brainstem, and spinal cord in the GFAP-HD mouse model generated using the human GFAP promoter to drive the expression of a N-terminal fragment of mHTT with 160Q only in astrocytes [67]. Further analysis of various mouse models expressing mHTT using the S829 antibody reveals the presence of nuclear inclusions in astrocytes in those models [50]. A study using viral vectors to drive the N171 fragment of human mHTT in astrocytes or neurons separately revealed fewer mHTT aggregates in astrocytes as compared to neurons [77]. This study revealed that although both cell types can form aggregates with the same fragment, the propensity to do so is quite diminished in astrocytes, thus suggesting additional mechanisms in astrocytes to protect against aggregate accumulation.
Previous studies performed in culture demonstrated that mHTT-expressing astrocytes in the presence of neurons that did not contain mHTT resulted in increased neuronal cell death and cell autonomous astrocyte aggregation of mHTT [75]. This result suggested that cell autonomous changes in mHTT-expressing astrocytes were sufficient to cause neuronal cell death. Whether cell autonomous expression of mHTT in astrocytes causes HD-like phenotypes has been studied in vivo in the GFAP-HD mouse model with selective expression of an N-terminal fragment of mHTT in astrocytes driven by glial fibrillary acidic protein (GFAP) promoter, revealing motor dysfunction (rotarod) and a reduction in the expression of the glutamate transporter [67]. Another study used an AAV to drive expression of a fragment of mHTT (N-171-82Q) in astrocytes in the striatum of mice and compared this model with an AAV using the chicken β-actin promoter to dive the expression of the same fragment in neurons [77]. This study revealed mild deficits on the rotarod when N-terminal mHTT was expressed only in striatal astrocytes at a later timepoint after infection than what was observed when N-terminal mHTT was expressed only in striatal neurons [77]. Thus, the studies using N-terminal mHTT fragments revealed that mHTT expression only in astrocytes causes behavioral and cell-autonomous transcriptional changes (as well as exacerbated non-cell autonomous transcriptional changes) which was most evident as the animal aged after expression of the mHTT. However, many of these changes observed when mHTT was only expressed in astrocytes were significantly worsened by the concomitant expression of mHTT in neurons [76, 77].
Experiments to determine whether mHTT expression in astrocytes is necessary to cause HD-like phenotypes in vivo has been performed using the conditional full-length human mHTT (fl-mHTT)-expressing BACHD mouse model. This model contains a human bacterial artificial chromosome carrying a modified full-length HTT gene with a floxed exon 1 with a mixed CAA-CAG repeat encoding 97 glutamines driven by the human promotor [78]. The expression of mHTT is found in neuronal and non-neuronal cells throughout the brain [68, 79] and results in phenotypes reminiscent of what is observed in HD patients including behavioral abnormalities and neuropathological changes. The BACHD model allows for cell-type-specific assessment of the contribution of mHTT from one cell type to the overall phenotypes observed in the BACHD model. Therefore, in the presence of Cre recombinase, one can specifically reduce the level of mutant HTT in specific cell types and explore the resulting phenotypes. Studies to explore the necessity of fl-mHTT in astrocytes to the phenotypes observed in this model reveals that decreasing fl-mHTT expression in GFAP positive astrocytes results in a slowing of the progressive HD-like phenotypes in this model. The mice with reduced expression in astrocytes showed significant improvements in motor and psychiatric-like behaviors. There was also a significant improvement in striatal atrophy after reduction of mHTT in astrocytes in this model. Furthermore, the abnormal evoked NMDAR currents in the MSNs were normalized after reducing mHTT in astrocytes, revealing a non-cell autonomous contribution of mHTT to this phenotype. Most of the behavioral improvements in this model after reduction of mHTT in astrocytes appeared more than 6 months after the manifestations of the disease-like phenotypes in this model. Thus, demonstrating that the presence of mHTT in astrocytes is likely not the initiator of disease in HD, but likely contributes to worsening of disease phenotypes and disease progression. This result is like what was observed using the lentiviral expression of mHTT in striatal astrocytes where mild rotarod impairment was observed at a later timepoint than a more significant deficit observed when mHTT was expressed only in neurons. However, the expression of mHTT in both cell types caused rotarod impairment at an even earlier age than what was observed in neurons alone [77].
Striatal astrocytes in HD display a reactive phenotype that increases in severity in post-mortem tissue of increasing neuropathological grade [49, 51]. Reactive astrocytes were observed in the striatum of some mouse models expressing mHTT specifically in astrocytes [49, 76, 77]. Reactive astrocytes are also observed in the knock-in Q175 (homozygous) mouse model [80]. Furthermore, reactive astrocytes are observed in the cortex and striatum when N-terminal mHTT is expressed throughout the brain in both neurons and glia [81]; however, when the N-terminal mHTT expression was restricted only to neurons in the striatum, there was no significant number of reactive astrocytes observed [82]. Nonetheless, the severity of this phenotype varies in the mHTT-expressing mice, where reactive astrocytes were observed and appeared most prominently at timepoints/ages where significant behavioral abnormalities and neuronal dysfunction are present. No overt reactive astrocyte phenotype was observed in R6/2 mice [83], a rapidly progressing transgenic mouse model (death at about 15 weeks of age) that contains an N-terminal fragment of human mHTT [84, 85]. The BACHD mice do not recapitulate the significant increase in reactive astrocytes in the striatum, as demonstrated by an increase in the number of GFAP positive astrocytes with altered morphology that is observed in HD patient post-mortem tissue (Gray unpublished). However, in mouse models, including the BACHD model, behavioral and neuropathological abnormalities can be elicited without a significant increase in the number of reactive astrocytes.
Another morphological phenotype observed in the striatum of the R6/2 mouse models expressing N-terminal fragment of mHTT is astrocyte shrinkage. This observation was made using injection of lucifer yellow into astrocytes and with viral approaches to label astrocytes allowing for more thorough labeling of cell bodies and processes that are unobservable using standard immunolabeling approaches [84,85,86]. In these studies in the R6/2 mice, astrocyte territory was decreased in size at 6–8 weeks of age [84,85,86] with no change present at an earlier age (2 weeks) [86]. Furthermore, these astrocytes had reduced proximity to striatal excitatory synapses with a specific reduction at the cortical-striatal synapse [85]. Interestingly, human mHTT-expressing astrocytes derived from glial progenitor cells transplanted into the corpus callosum of R6/2 mice showed less fiber network complexity and occupied a smaller volume of its immediate surroundings [87]. These data, though intriguing, have yet to be recapitulated in other mHTT-expressing models especially the full-length mHTT-expressing models which show slower progression of HD-like phenotypes. Studies using a fluorescence resonance energy transfer (FRET)-based assay in the R6/2 mice also revealed a decrease in the proximity of striatal astrocytes to the cortical-striatal synapse, but an increased proximity of striatal astrocytes to the thalamo-striatal synapse in R6/2 mice. Together this data demonstrates abnormal morphology, resulting in changes in association of astrocytes with specific synapses that could ultimately contribute to neuronal dysfunction in the striatum and thus alterations in the basal ganglia circuit that is disrupted in HD.
Molecular and Functional Changes in Astrocytes in HD Mice
Analyses of gene expression profiles of astrocytes from mHTT-expressing models, including the rapidly progressing R6/2 transgenic mouse model that contains an N-terminal fragment of human mHTT and the Q175 knock-in model that expresses endogenous full-length mHTT, reveal progressive transcriptional changes in genes responsible for normal astrocyte function [63, 86, 88] (Fig. 1). The extent to which the changes observed in the R6/2 model and the Q175 knock-in model was concordant with what was observed in HD-post-mortem tissue varied with the Q175 model displaying a higher concordance in down-regulated genes than the R6/2 model demonstrating differences in transcriptional alterations due to the presence of full-length or N-terminal fragments of mHTT [86]. Given this data, it is likely that the molecular landscape of a given astrocyte based on the degree of mHTT fragmentation could have different non-cell autonomous effects on neurons in the vicinity of specific astrocytes. Nonetheless, the common changes between the mHTT-expressing mouse models (both fragment and knock-in) reveals signatures that are likely exploitable for therapeutic intervention.

A Early stages of SCA1. Brain regions indicated in blue exhibit increased (cortex) or trending (cerebellum) expression of astrocyte (blue cells) homeostatic genes likely providing increased support to neurons (gray), whereas regions in red exhibit loss of astrocyte (red astrocytes) homeostatic function (hippocampus and medulla oblongata). B Late, terminal stages of SCA1. Astrocytes in all brain regions exhibit reduced expression of genes necessary for astrocyte homeostatic function (cortex, cerebellum, hippocampus, and medulla). Microglia activation (pink) in different brain regions does not correlate with astrocyte reactivity or homeostatic gene expression. Yellow triangles represent expression of Atxn1 gene in all these cell types in mice and ATXN1 humans
The A1 astrocyte gene expression signature has been suggested to identify a neurotoxic reactive astrocyte phenotype (A1 phenotype) in disease including in HD that drive disease pathogenesis [65]. Analyses of some of the genes that identify the A1 signature by quantitative PCR in striatal tissue from R6/2 and the Q175 knock-in model did not reveal significant gene expression alterations (only Serping1g) that would clearly indicate the A1 signature in striatal astrocytes in these models [63]. While there are significant changes in gene expression and a likely disease associated gene expression signature in mouse models expressing mHTT, the presence of astrocytes with the specific neurotoxic A1 gene expression signature is questionable. Nonetheless, there are astrocytic molecular changes observed in these models with alterations in the expression of amino acid transporters, potassium channels, and other genes required for homeostatic functions of astrocytes. Furthermore, these changes seem largely due to the presence of mHTT within astrocytes as they are normalized when mHTT was reduced in the R6/2 model [63].
Using a viral approach to express N-terminal fragment of human mHTT (N-171) in striatal astrocytes revealed changes in the glutamine-glutamate cycle as also revealed in the R6/2 and Q175 knock-in studies [77]. This data agrees with changes observed in HD patient tissue which formed part of the basis for the excitotoxicity hypothesis in HD, where glutamate toxicity could be due to alterations in glutamate transporter levels critically important for the clearance of glutamate by astrocytes [89, 90]. This analysis revealed decreased expression of Glul which is responsible for the conversion of glutamate to glutamine as well as decreased expression of Slc1a2 (Glt1) and Slc1a3 (Glast) which are glutamate transporters responsible for the uptake of glutamate. Interestingly when N-terminal mHTT was also expressed in neurons, the expression of Glul, Slc1a2, and Slc1a3 was further decreased, revealing cell autonomous and non-cell autonomous contributions of mHTT to the regulation of the expression of these critical components of the glutamate-glutamine cycle [77]. These same gene expression changes were observed when N-terminal N171-82 fragment was expressed using a lentivirus [49]. Thus, in vivo genetic expression of N-terminal mHTT or fl-mHTT and viral approaches to express N-terminal fragments of mHTT in mice provides largely overlapping and consistent data about astrocytic gene expression changes in striatum, although analyses of astrocytic expression changes are not possible with the specific viral approaches targeting only one brain region.
Analyses of molecular changes in the various mouse models have been very informative and suggest significant alterations in normal astrocyte function. We focus here on alterations in calcium signals in astrocytes in mHTT-expressing mouse models. Astrocytes play many important roles in the nervous system including neurotransmitter homeostasis and release of gliotransmitters [7, 91]. Properly maintaining many of the critical astrocyte functions needed for proper nervous system development and function is regulated by astrocyte Ca2+ signals; whether they be those observed in microdomains of astrocytes [92] or encompassing more of the astrocyte cell body [93]. Studies in R6/2 mice at P50–P70 days of age using an AAV vector with the gfaABC1D (GFAP) promoter to drive expression of the calcium indicator GCaMP3 in striatal astrocytes revealed a reduction in the frequency, amplitude, and duration of spontaneous Ca2+ signals [94, 95]. An interesting result from this study found that normal striatal astrocytes do not typically respond to cortico-striatal stimulation; however, mHTT-expressing striatal astrocytes responded by significantly increasing Ca2+ signals in the astrocytic cell bodies and processes [94]. Therefore, mHTT-expressing striatal astrocytes can respond to signals at the cortico-striatal synapse likely reflecting changes at this stage of disease progression in the R6/2 model due to alterations in the expression of the glutamate transporter Glt-1. In the R6/2 mice, the increased evoked Ca2+ signals observed in striatal astrocytes were dependent on the expression of Glt-1 that is reduced at this stage in R6/2 mice [94, 96]. These data are quite intriguing; however, most of the studies to date on striatal astrocyte Ca2+ calcium signals have only been performed in this rapidly progressing N-terminal fragment mHTT-expressing mouse model. Whether these alterations in Ca2+ signals in striatal astrocytes are also reflected in models that express full-length mHTT remain to be seen.
Microglia in Huntington’s Disease
Morphological changes in microglia in the striatum, cortex, and white matter are observed in HD post-mortem patient tissue where they display an ameboid shape demonstrating a reactive phenotype [97, 98]. Positron emission tomography (PET) imaging studies using [11C]PK11195 BPND that targets TSPO in presymptomatic subjects carrying the HD mutation without overt motor phenotypes revealed activated microglia in the cortex, basal ganglia, and thalamus [99]. PET imaging also revealed that microglial activation seems to increase in HD patients with increased striatal neuron loss and as disease progresses [100, 101]. Although the specificity of this PET ligand to microglia is in question [102], these studies together with those in post-mortem tissue do implicate changes in microglia in HD patients.
Analysis of inflammatory markers from HD patient samples including plasma, CSF, and postmortem brain tissue revealed significant elevations in several inflammatory cytokines including IL-1β, IL-6, IL-8, and TNF-α [99, 103,104,105,106]. Interestingly, there was regional specificity to some of these changes with IL-1β and TNF-α being increased only in the striatum, whereas IL-6, IL-8, and MMP-9 were also upregulated in the cortex and cerebellum [99, 103, 104]. Some of the changes in the inflammatory cytokines from the CSF and plasma are observed prior to the onset of overt clinical manifestations of HD [103, 105, 107].
Studies in mouse models expressing mHTT including N-terminal mHTT-expressing R6/2 and full-length human mHTT YAC128 have revealed microglia with altered morphology adopting an ameboid shape and a decrease in processes [98, 108, 109]. Functionally, microglia isolated from BACHD and YAC128 mice exposed to chemoattractants displayed impaired migration and a reduction in their response to injury [110]. The levels of inflammatory cytokines including IL-6, IL-1β, and TNFα increase in the striatum of R6/2, YAC128, BACHD, and zQ175 mice at an advanced disease stage specific to each mHTT-expressing mouse model as well as in primary microglia isolated from zQ175 and R6/2mice [103, 108, 111,112,113]. Abnormal activation of NFκB in HD can cause an increase in pro-inflammatory cytokines [114, 115]. This increased activation of NFκB in R6/2 mice has been proposed to lead to increased levels of galectin-3 (Gal3) in microglia [108], a lectin whose decrease has been shown to reduce inflammation [116]. When Gal3 is decreased in the R6/2 mice, it dramatically decreased the inflammatory signature observed in R6/2 mice [108]. Furthermore, depletion of microglia rescued many of the deficits observed in R6/2 mice including behavioral and neuropathological changes [117].
Genetic expression of N-terminal mHTT in myeloid lineage cells, including microglia, in the RosaHD knock-in mouse model [81] bred to Cx3cr1-Cre mice resulted in an increase in the number of activated microglia and an increase in proinflammatory cytokines [112]. In addition, studies using cultured microglia from R6/2 mice also demonstrated significant increase of IL-6 secreted when compared to microglia cultured from nontransgenic mice and RNA-seq analysis of microglia from adult Q175 homozygous mice revealed a proinflammatory gene signature [112]. Taken together, this study indicates that mHTT within microglia alone is sufficient to cause a proinflammatory state. Additional studies using the conditional BACHD mouse bred to a Lys2-Cre (LysMCre) mouse model to reduce mHTT expression in myeloid lineage cells including microglia (BACHD/LysMCre) demonstrated no significant rescue of the motor abnormalities (coordination or locomotion) or the neuropathological phenotype (brain weight and volume) previously observed in the BACHD model. However, the significant secretion of IL-6 from cultured stimulated microglia from the BACHD mice was significantly reduced when mHTT was reduced in BACHD/LysMCre mice [113]. Although, in these studies, microglia expression or decrease of mHTT was verified, care must be taken in the interpretation of these data given that these studies targeted all myeloid cells and not microglia alone and the LysMCre does not completely reduce mHTT expression in microglia. Together these studies suggest cell-autonomous phenotypes exist in mHTT expressing microglia; however, the degree to which mHTT expression in microglia contributes to the overall phenotypes in HD is still debatable. Interestingly, data obtained from the reduction of mHTT from astrocytes in the BACHD mouse model suggested that mHTT in astrocytes did not contribute to the onset of phenotypes in the model, but contributed to the progression of those phenotypes in the model. Thus, given the slow phenotypic progression of the BACHD mouse model, it is possible that the timepoints assessed in the model did not allow for rescue of the behavioral and neuropathological changes observed in the BACHD mice.
Oligodendrocytes in Huntington’s Disease
Several studies have described early white matter (WM) loss in HD. Post-mortem brain tissues of HD patients exhibit a loss of myelin and WM volume, and significant changes in the numbers of oligodendrocytes. Moreover, non-invasive imaging demonstrated that white matter atrophy is present in premanifest carriers of expanded HTT, preceding the onset of the symptoms [118,119,120]. The integrity of the white matter, as demonstrated by fractional anisotropy in HD patients at premanifest and early stages, is correlated with the degree of caudate atrophy [121]. White matter defects in HD patients were also found to be associated with motor and cognitive deficits [122]. Although another study revealed abnormalities in myelin integrity in premanifest carriers that correlated with cognitive changes, they found no correlation with atrophy, the correlation was only observed in those at the early stages of HD [123]. Interestingly, an increase in oligodendrocyte numbers in HD patient striatal tissue has been observed [53]. Mouse models of HD, including the full length mHTT expressing models YAC128, BACHD, zQ175 HD, and the N-terminal mHTT expressing R6/2 mice, exhibit changes in white matter, including decreased volume of myelin-rich corpus callosum, thinner myelin sheaths, and reduced expression of myelin-related genes. These WM abnormalities in HD mouse models allowed for investigation of the etiology and molecular mechanisms of WM changes in HD.
To ask whether expression of mutant HTT in oligodendrocytes is sufficient to cause HD-like oligodendrocyte pathology and behavioral deficits, Huang et al. established a transgenic mouse model that selectively expresses N-terminal mHtt in oligodendrocytes, the PLP-150Q mice. These mice showed axonal degeneration and an early onset polyQ disease phenotype that includes impaired rotarod performance, body weight loss, and early death. Huang et al. also showed that mHTT binding to myelin regulatory factor (MYRF) affects MYRF’s transcription activity and consequently reduces myelin gene expression in mature oligodendrocytes [124]. This study provides strong evidence that mHTT in oligodendrocytes is sufficient to cause neuronal and oligodendrocyte pathology. In another study using a knock-in HD mouse model (~ 250 CAG), myelin deficits were observed, including thinner myelin sheaths in the corpus callosum and reduced level of MYRF and myelin basic protein, MBP [125]. Recently Ferrari Bardile et al. demonstrated that intrinsic mutant huntingtin (mHTT)-mediated deficits in oligodendroglia contribute to myelination abnormalities and behavioral manifestations in HD [126]. To do so, they crossed BACHD mice with the NG2-Cre mice that express the Cre recombinase in NG2 + OPCs. As such, BACHD;NG2-Cre mice had reduced mHTT expression specifically in oligodendrocytes and oligodendrocyte progenitor cells. Selective inactivation of mutant huntingtin (mHTT) in the NG2 + oligodendrocyte progenitor cell population prevented myelin abnormalities and certain behavioral deficits, such as anxiety and depression in HD mice, despite the continued expression of mHTT in neurons, astrocytes, and microglia. Using RNA-seq and ChIP-seq, Ferrari Bardile et al. showed enhanced activity of polycomb repressive complex 2 (PRC2), proposing that mHTT increases PRC2 activity in OPCs, resulting in a delay in their maturation and myelination defects in HD [126]. This intrinsic effect of mutant HTT was further supported by studies showing decreased expression of myelin genes in OPCs isolated from zQ175 and R6/2 mouse models [127] or HD patient-derived glial progenitor cells (hGPCs, the human homologs of rodent OPCs), produced from human embryonic stem cells (hESCs) derived from HD patients [87].
Finally, therapeutic approaches that target myelin were found to be effective in HD mice. Laquinimod (LAQ) is an immunomodulatory agent used to alleviate demyelination in multiple sclerosis [128]. LAQ treatment rescued the expression of myelin genes and improved white matter pathology and behavioral phenotypes in the YAC128 and the PLP-150Q mice [129]. Together these results suggest that further investigations of OPC and OL pathologies and their contribution to HD are needed for a better understanding and consequently better treatments for HD.
Spinocerebellar Ataxias
Observations from SCA Patients
Spinocerebellar ataxias (SCAs) are a group of dominantly inherited and progressive neurodegenerative diseases [130]. Most common SCAs, including SCA1-3, SCA6, SCA7, SCA17, and DRPLA, are caused by the expansion of CAG repeats that encode polyglutamine (polyQ) tract in the respective disease proteins [2]. SCAs are characterized by gait ataxia and limb incoordination likely arising from the pathology in the cerebellum [131]. Additional symptoms of cognitive decline and premature death are found in some patients with SCAs [132, 133]. Pathologically, patients display cerebellar and brainstem degeneration, with profound loss of Purkinje cells in the cerebellum [134]. In addition, loss of neurons in cortical, subcortical, and spinal regions have also been reported [135].
Spinocerebellar ataxia Glial pathology was described in patients with SCAs using postmortem pathological analysis and magnetic resonance spectroscopy (MRS) [136,137,138,139]. Myelin pallor and/or atrophy were observed in the cerebellar and brainstem white matter, including cerebellar peduncles, cranial nerves, somatosensory, auditory, and precerebellar fiber tracts in SCA1 patients. Importantly, gliosis was observed throughout the brain regardless of neuronal loss or sparing [135]. Several MRS studies detected neurochemical abnormalities, reflective of gliosis (increased myo-inositol) in SCA1 patients not only at early disease stage but also in premanifest stage [139]. Diffusion MRI (dMRI) identified cross-sectional degeneration in the cerebellum, corpus callosum, and internal capsule in SCA patients at symptomatic as well as premanifest stages of disease [137]. These studies showing pathological changes in glia even before disease onset indicate a likely contribution of glia to SCA disease pathogenesis, yet the causes and consequences of glial pathology in SCAs remain largely unexplored (Fig. 2) [140, 141].
Approaches Used to Understand How Glia Contribute to SCAs
Animal models and more recently induced pluripotent stem cells (iPSCs) have been very helpful in understanding pathological mechanisms of SCAs [142]. Being inherited in a dominant manner, SCAs are well modeled in mice with each polyQ SCA having different mouse models that recapitulate many aspects of human disease [2, 143,144,145,146,147,148,149,150]. With its stereotypical anatomical organization, well-understood neuronal circuitry, and intimate interaction of Purkinje cells with Bergmann glia, cerebellum offers a unique opportunity to study the role of glial cells in neurodegeneration [151, 152]; yet we still have limited understanding of the glial involvement in SCAs [140, 141].
Bergmann glia are a special type of astrocytes with radial fibers that are intimately associated with Purkinje cell dendrites where they both regulate and are regulated by Purkinje cell synaptic activity [153, 154], making Bergmann glia a prime candidate cell type for involvement in SCA cerebellar degeneration. One of the first studies indicating the active role of astrocytes in SCA was in SCA7. Dr. Albert La Spada’s group used mouse genetic approach to show that expressing mutant ATXN7 under the control of the Gfa2 promoter, a version of the GFAP promoter typically used for restricted expression in Bergmann glia of the cerebellum, is sufficient to cause ataxia and Purkinje cell pathology [155]. Moreover, authors demonstrated that expression of mutant ATXN7 in glia causes reactive gliosis and reduces expression of glutamate transporters in Bergmann glia. By showing that mutant ATXN7 affects glutamate clearance by Bergmann glia, Custer et al. linked mutant ATXN7-induced glial dysfunction with excitotoxicity of Purkinje cells [155], elegantly providing a mechanism by which Bergmann glia can contribute to disease pathogenesis in SCA7. Recent study showed that 4 days of optogenetic activation of Bergmann glia induces reactive gliosis with reduced expression of glutamate transporters that causes Purkinje cell dysfunction, atrophy, and death [156], further supporting the importance of Bergmann glia glutamate transport for Purkinje cell function and viability [157].
In addition to dysfunction directly caused by mutant protein expression in glial cells, cerebellar glia can be altered in response to neuronal dysfunction. Reactive astrogliosis is a term used to describe the process of astrocyte changes in gene expression, morphology, and function in response to brain insult and neurodegeneration [52]. Recent single-nuclei RNA sequencing study by Borgenheimer et al. provided insight into the reactive astrogliosis gene expressing changes in SCA1 [158]. Authors used the transgenic SCA1 mouse model, Pcp2- ATXN1[82Q] line in which mutant ATXN1 is expressed only the cerebellar Purkinje cells to identify the gene expression changes in cerebellar glia solely in response to Purkinje cell pathology. Despite mutant ATXN1 being expressed only in Purkinje cells, authors identified similar number of gene expression changes in Bergmann glia and Purkinje cells. Authors also provided insight to the reactive astrogliosis of velate astrocytes, little understood type of cerebellar astrocytes. As some of the altered glial genes play key roles in regulating firing rate of Purkinje cells, these complex molecular changes are likely to impact on the accuracy of cerebellar encoding and can thereby contribute to motor deficits in SCA1.
Previous studies demonstrated that expression of mutant ATXN1 in Purkinje cells is sufficient to induce reactive astrogliosis via NF-kB signaling in Bergmann glia [159], implicating NF-kB signaling as a pathway by which Purkinje dysfunction can contribute to reactive Bergman astrogliosis in SCA1. A key characteristic of SCAs is their relentless progression. Thus, it is important to investigate whether glial contribution to pathogenesis is dynamic and changes with disease progression. This was examined by Kim et al. using mouse Cre-lox genetic approach to modulate reactive gliosis at different stages of SCA1 progression [160]. In particular, Kim et al. used the TMX-dependent Cre genetic approach to inhibit NF-kB signaling selectively in astrocytes during early and late stages of SCA1 disease progression [160]. This study demonstrated that reactive Bergmann glia had a beneficial effect on Purkinje cells early in SCA1 disease, delaying onset of motor symptoms and ameliorating cerebellar pathology [160], but later in disease, reactive Bergman glia become harmful. Understanding regulation and mechanism underlying beneficial and harmful effects of Bergmann glia is key for developing glia-focused therapies. Two recent studies implicate JNK and Wnt as additional signaling pathways that can regulate Bergmann glia phenotypes in SCA1 [161, 162]. Further investigation by Mellesmoen et al. identified increased neurotrophic support and increased expression of homeostatic genes in Bergmann glia as factors that contribute to the early protective role of astrocytes [163]. Later in disease, reactive Bergmann glia were found to be harmful [160], in part due to the reduced expression of glutamate transporters [164], echoing previous studies from La Spada group in SCA7 and studies in HD astrocytes. What remains unknown is the extent to which intrinsic and extrinsic factors contribute to Bergmann gliosis. For instance to which extent is reduced expression of glutamate transporters caused cell-autonomously by mutant ATXN1 expression in Bergmann glia and to which extent it is caused in non-cell autonomous manner by Purkinje cell dysfunction. Conditional mouse models in which mutant SCA genes can be selectively deleted in neurons or glia will allow us to directly address this question.
Transcriptome analysis revealed large numbers of differentially expressed genes in SCA1 microglia in patients and knock-in mouse cerebella [165]. Microglia show signs of reactivity in the cerebellum of SCA mice as measured as increased density of Iba1 + microglia. Moreover, using mouse genetic approach with LysM-Cre mice to inhibit inflammatory NF-kB signaling selectively in microglia or eliminating microglia with PLX reduced cerebellar neuroinflammation and ameliorated disease pathogenesis in SCA1 mice, implicating microglial contribution to SCA1 disease pathogenesis [141, 166, 167]. These results implicate the active role of microglia in SCA1 with further studies needed to investigate underlying mechanisms.
Recent studies implicated changes in oligodendrocytes (OL) and oligodendrocyte precursor cells (OPCs) in SCA1 and SCA3 [149, 165, 168]. Single-cell RNA sequencing of SCA1 patients revealed large numbers of gene expression changes in OL and OPCs and protein analysis of OL and OPC proteins via western blotting, and immunohistochemistry is consistent with the reduction in OL numbers and myelination in the cerebellar cortex of SCA1 patients. Transmission electron microscopy confirmed reduced myelination in the cerebellum of SCA1 knock-in mice. Using a dimensionality-reduction method named potential of heat diffusion for affinity-based transition embedding (PHATE) analysis to capture differentiation trajectories of OPCs, Tejwani et al. suggested that deficit in transition from OPCs to OLs may underlie OL deficiency in SCA1 [165, 169]. Impaired OL maturation from OPCs was also identified in the SCA3 transgenic mouse model expressing human mutant ATXN3. Authors found decreased expression of OL genes, reduced numbers of Ols, and abnormalities in axonal myelination in the vulnerable brain regions in SCA3 mice [149]. Thus, changes in oligodendrocytes and OPCs have been described in at least two different mouse models of SCA, but whether and how they are regulated and contribute to disease pathogenesis remains to be determined.
Although these studies demonstrated that glia could contribute to SCA pathogenesis in the cerebellum, it is important to understand how glial cells contribute to pathogenesis in other brain regions that undergo neurodegeneration and could contribute to the clinical phenotypes. For instance, significant pathology is described in the brainstem, while motor cortex, spinal cord, and hippocampus undergo milder degeneration in SCA1 patients. Pathology in these regions could contribute to SCA1 symptoms such as cognitive deficits, mood disorders, difficulties in respiration and swallowing, and premature lethality.
To understand how glia contribute to SCA1 pathogenesis in these regions and compare it to changes in the cerebellum, Rosa et al. recently characterized glial and neuronal changes in brainstem, motor cortex, hippocampus, and cerebellum of Atxn1154Q/2Q mice, a knock-in mouse model of SCA1 [170]. They found that morphological and molecular changes in glia correspond to reduced neuronal activity and synaptic loss in a spatial and temporal manner. For instance, at the early disease stage, expression of core astrocytic homeostatic genes (including Slc1a2 and Kcnj10) was reduced in the hippocampus, while their expression was increased in the cortex of Atxn1154Q/2Q mice. Kcnj10 encodes a potassium rectifier, Kir4.1, that is involved in maintaining potassium homeostasis and solute carrier (Slc1a2) encodes a glutamate transporter responsible for removing glutamate from synaptic space. These astrocytic genes are critical for neuronal activity and have been implicated in the pathogenesis of several neurodegenerative diseases. Reduced expression of astrocyte neuro-supportive genes in the hippocampus correlated with reduced neuronal activity, while neuronal activity was preserved in the cortex of SCA1 mice [158]. Thus, it is possible that increased expression of these neuro-supportive astrocyte genes in the cortex indicates compensatory roles of astrocytes with role in delaying neuronal dysfunction in the SCA1 affected cortex. The spectrum of reactive glial phenotypes may indicate brain-region-specific glial dysfunctions and consequently brain-region-specific contributions of glia to SCA1 disease pathogenesis. Moreover, as observed glial changes precede neuronal loss, promoting protective glial phenotypes and preserving glial functionality may provide therapeutic benefits in SCA1.
Molecular Mechanisms of Pathogenesis in SCAs
Mouse models and more recently induced pluripotent stem cells (iPSCs) have been very fruitful in increasing our understanding of pathological mechanisms of SCAs. Similar to HD, it is thought that expanded polyQ causes toxicity in SCAs by affecting normal functions, causing novel toxic gain of functions and increasing protein misfolding and aggregation of the respective mutated proteins [3, 130, 168, 171, 172]. In addition, expanded CAG repeats can cause protein-mediated effects through repeat-associated non-ATG-initiated (RAN) translation [2, 173]. Transcriptional dysregulation, mitochondrial dysfunction, perturbed calcium, channelopathy, and autophagy have been implicated in SCA pathogenesis [2, 168, 174,175,176]. As mutant genes, such as ATXN1, are also expressed in glial cells, it is reasonable to assume that similar dysregulation occurs in glia as it does in neurons.
Two recent studies reported on the gene expression changes in cerebellar SCA1 glia using single-nuclei RNA sequencing [158]. These studies indicated significant changes in glial gene expression and signaling pathways, including perturbed calcium signaling. One study used SCA1 knock-in mice and the other SCA1 Purkinje cell–specific transgenic mice providing an initial insight into which gene expression changes are in response to PC dysfunction (i.e., seen in PC-specific transgenic mice) and which ones are due to combination of direct effect of mutant ATXN1 in glia and response to neuronal dysfunction [158, 165].
Future studies, using conditional mouse models to selectively express or delete mutant proteins from glial cells, will distinguish between these two ways by which glial cells are affected in SCAs.
Human Glia and Aging
Most of the research on the role of glia in neurodegenerative diseases utilizes mouse models. While many aspects of glial biology are conserved across species, there are significant morphological, gene expression, and functional differences between human and murine glia [140, 177, 178]. For instance, unbiased genome-wide comparison of the human and mouse astrocytes revealed over 600 genes enriched in human but not in mouse astrocytes [179]. These differences between human and mouse astrocytes indicate a need to study disease pathology in human astrocytes for translational research. In addition, mouse models of polyQ diseases require additional manipulations not seen in patients; knock-in mice require longer polyQ extensions (146Q or more) to model disease than patients with SCA1 (from 39 to 82Q) [180]. While these significant species differences indicate the importance of investigating human SCA glia, there is little work published so far that examines human SCA iPSC–derived glial cells.
Neurodegenerative diseases are often characterized by a brain region–specific pathology and aging is an important contributor. Several studies indicate that regional heterogeneity of glial cells and effects of aging on glia may underlie these two important hallmarks of neurodegeneration. Grabert et al. performed genome-wide analysis of microglia isolated from four different brain regions 4, 12, and 22 months of age [181]. They found that microglia have distinct region- and age-dependent transcriptomes. For instance, in the young adult mice (4 months), differences in bioenergetic and immunoregulatory pathways suggested that microglia in the cerebellum and hippocampus are more immune vigilant compared to other brain regions. However, aging enhanced the cerebellar microglial immunophenotype, but aged hippocampal microglia became more like microglia in other brain regions.
In humans there are morphological changes that ensue in astrocytes (substantia nigra), where they start to show short and stubby processes in post-mortem tissue from older subjects, whereas in younger subjects the processes are long and slender [182, 183]. Interestingly, there is also a change in the gene expression profile. Boisvert et al. and Clarke et al. used the Ribotag technique to genetically label ribosomes in astrocytes from different brain regions through the lifespan of the mouse [184, 185]. They identified brain region–specific astrocyte transcriptomes and increased expression of reactive genes in astrocytes with aging. Recent study by Lee et al. used single-cell RNA seq to identify a unique subtype in aged hippocampus named autophagy-dysregulated astrocytes (APDAs) characterized by abnormal accumulation of autophagosomes in swollen processes, impairing protein trafficking and secretion. Moreover, APDA had impaired secretion of synaptogenic molecules and astrocytic synapse elimination, suggesting that aging leads to a loss of astrocyte’s ability to control synapses and homeostasis, in addition to more reactive astrocytes previously described [186].
The white matter, a region of myelin coated axons, constitutes more than half of the human brain. In the CNS, myelin is primarily made of oligodendrocytes (OL) and its primary function is to increase the conduction speed of axon potentials. OL are derived from oligodendrocyte progenitor cells (OPCs) that proliferate to self-renew and differentiate to provide new mature OL. Aging leads to white matter degeneration, evidenced by change in diffusor tensor index (DTI), reduced volume, and the accumulation of myelin abnormalities, such as myelin balloons and piling of paranodal loops, suggesting that OPCs and OL within the aging brain have a reduced capacity for producing and maintaining healthy myelin [187]. Recently, Spitzer et al. demonstrated using single-cell electrophysiological recordings that OPCs become functionally heterogeneous both within and between brain regions with age. This age-induced changes in OPCs correlate with their differentiation potential [188].
Similar brain region and age-induced changes in glial cells were found in humans. Soreq et al. performed a comprehensive analysis of transcriptomes across ten human brain regions from 480 individuals ranging in age from 16 to 106 years, which showed that cerebellar microglia and astrocytes are distinct from other brain regions, as well as that aging leads to an increase in immune MG-specific genes across all brain regions. On the other hand, neuron-specific transcriptomes seem to be more defined by brain region than by age, and both neuronal and oligodendrocyte numbers seem to decrease with age [18].
Conclusion
A recent study by Yu et al. demonstrated that how astrocyte respond to pathology is context dependent [84]. Combined with evidence of spatial and temporal heterogeneity of glia, it is reasonable to propose that glial response and contribution to neurodegeneration may also be brain-region and disease-stage specific. This level of complexity merits a deeper investigation into the glial contributions and may also provide a richness of potential therapeutic targets. We reviewed studies using different approaches to investigate glial contribution to polyglutamine diseases HD and SCAs. HD and SCA are characterized by increased vulnerability of specific neurons and brain regions despite wide-spread expression of mutant proteins. In case of HD, striatal medium spiny neurons are most affected, while in SCA1 cerebellar Purkinje cells are most vulnerable. Studies have convincingly shown that glial cells contribute to pathogenesis of HD and SCAs.
Together these studies demonstrate that glial cells are malleable, and that in-depth understanding of glial molecular changes can be used for identifying potential therapeutic targets. Validating these molecular changes in human iPSC-derived glia will be an important steppingstone before moving to clinical studies.
References
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005.
Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci. 2017;18(10):613–26.
Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621.
Bunting EL, Hamilton J, Tabrizi SJ. Polyglutamine diseases. Curr Opin Neurobiol. 2022;72:39–47.
Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129(4):877–96.
Pérez-Alvarez A, Araque A. Astrocyte-neuron interaction at tripartite synapses. Curr Drug Targets. 2013;14(11):1220–4.
Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35.
MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol. 2015;7(5).
Verkhratsky A, Nedergaard M. Physiology of Astroglia. Physiol Rev. 2018;98(1):239–389.
Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27(24):6473–7.
Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, et al. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 2015;18(2):210–8.
Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, et al. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol. 2013;141(5):633–47.
Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–67.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–43.
Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21(4):530–40.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225–42.
Soreq L, UK Brain Expression Consortium, North American Brain Expression Consortium, Rose J, Soreq E, Hardy J, et al. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 2017;18(2):557–70.
Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303(5916):390–6.
Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):37–53.
Hardy R, Reynolds R. Proliferation and differentiation potential of rat forebrain oligodendroglial progenitors both in vitro and in vivo. Development. 1991;111(4):1061–80.
Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9(2):173–9.
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11(12):1392–401.
Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3(6):191–7.
Dawson MR, Levine JM, Reynolds R. NG2-expressing cells in the central nervous system: are they oligodendroglial progenitors? J Neurosci Res. 2000;61(5):471–9.
Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H, et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 2016;351(6271):379–84.
Lachapelle F, Gumpel M, Baulac M, Jacque C, Duc P, Baumann N. Transplantation of CNS fragments into the brain of shiverer mutant mice: extensive myelination by implanted oligodendrocytes. I. Immunohistochemical studies. Dev Neurosci. 1983;6(6):325–34.
Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2’,3’-cyclic nucleotide 3’-phosphodiesterase in the CNS of adult rats. J Neurochem. 1989;52(1):296–304.
Trapp BD. Myelin-associated glycoprotein. Location and potential functions. Ann N Y Acad Sci. 1990;605:29–43.
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98.
Huntington G. On chorea. J Neuropsychiatry Clin Neurosci. 2003;15(1):109–12.
The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–83.
Dayalu P, Albin RL. Huntington disease: pathogenesis and treatment. Neurol Clin. 2015;33(1):101–14.
Ha AD, Beck CA, Jankovic J. Intermediate CAG Repeats in Huntington’s disease: analysis of COHORT. Tremor Other Hyperkinet Mov (N Y). 2012;2.
Nance MA. Genetics of Huntington disease. Handb Clin Neurol. 2017;144:3–14.
Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–16.
McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol. 2018;25(1):24–34.
Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–84.
Vonsattel JP, Keller C, Pilar Amaya MD. Neuropathology of Huntington’s disease. Handb Clin Neurol. 2008;89:599–618.
Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, et al. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8(9):791–801.
Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10(1):31–42.
Wall NR, De La Parra M, Callaway EM, Kreitzer AC. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron. 2013;79(2):347–60.
Haber SN. Corticostriatal circuitry. Dialogues Clin Neurosci. 2016;18(1):7–21.
Landwehrmeyer GB, McNeil SM, Dure LS, Ge P, Aizawa H, Huang Q, et al. Huntington’s disease gene: regional and cellular expression in brain of normal and affected individuals. Ann Neurol. 1995;37(2):218–30.
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89(1):37–53.
Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18(12):1819–31.
Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995;14(5):1065–74.
Singhrao SK, Thomas P, Wood JD, MacMillan JC, Neal JW, Harper PS, et al. Huntingtin protein colocalizes with lesions of neurodegenerative diseases: an investigation in Huntington’s, Alzheimer’s, and Pick’s diseases. Exp Neurol. 1998;150(2):213–22.
Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet. 2010;19(15):3053–67.
Jansen AH, van Hal M, Op den Kelder IC, Meier RT, de Ruiter AA, Schut MH, et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia. 2017;65(1):50–61.
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44(6):559–77.
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24(3):312–25.
Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, et al. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J Neuropathol Exp Neurol. 1991;50(6):729–42.
Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. 1998;55(3):215–24.
Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington’s disease. J Neuropathol Exp Neurol. 1995;54(1):105–20.
Escartin C, Guillemaud O, Carrillo-de Sauvage MA. Questions and (some) answers on reactive astrocytes. Glia. 2019;67(12):2221–47.
McKenzie AT, Wang M, Hauberg ME, Fullard JF, Kozlenkov A, Keenan A, et al. Brain cell type specific gene expression and co-expression network architectures. Sci Rep. 2018;8(1):8868.
Zhang Y, Sloan Steven A, Clarke Laura E, Caneda C, Plaza Colton A, Blumenthal Paul D, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89(1):37–53.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–47.
Bayraktar OA, Bartels T, Holmqvist S, Kleshchevnikov V, Martirosyan A, Polioudakis D, et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci. 2020;23(4):500–9.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14.
Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun. 2020;8(1):19.
Diaz-Castro B, Gangwani MR, Yu X, Coppola G, Khakh BS. Astrocyte molecular signatures in Huntington’s disease. Sci Transl Med. 2019;11(514).
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391–410.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I, et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging. 2014;35(1):1–14.
Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106(52):22480–5.
Lee W, Reyes RC, Gottipati MK, Lewis K, Lesort M, Parpura V, et al. Enhanced Ca-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol Dis. 2013;58C:192–9.
Conforti P, Camnasio S, Mutti C, Valenza M, Thompson M, Fossale E, et al. Lack of huntingtin promotes neural stem cells differentiation into glial cells while neurons expressing huntingtin with expanded polyglutamine tracts undergo cell death. Neurobiol Dis. 2013;50:160–70.
Reiner A, Dragatsis I, Zeitlin S, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003;28(3):259–76.
Lo Sardo V, Zuccato C, Gaudenzi G, Vitali B, Ramos C, Tartari M, et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci. 2012;15(5):713–21.
Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington’s disease. Nat Rev Neurosci. 2013;14(10):708–21.
Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO Jr, et al. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20(2):198–202.
Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington’s disease. J Neurosci. 2003;23(6):2193–202.
Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171(6):1001–12.
Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, Li S, et al. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem. 2010;285(14):10653–61.
Meunier C, Merienne N, Jolle C, Deglon N, Pellerin L. Astrocytes are key but indirect contributors to the development of the symptomatology and pathophysiology of Huntington’s disease. Glia. 2016;64(11):1841–56.
Gray M, Gu X, Shirasaki D, Cepeda C, Yamazaki I, Levine M, et al., editors. Cortical Control of striatal pathogenesis in the Cre/LoxP conditional BAC transgenic mouse model of Huntington’s disease (BACHD). Soc Neurosci. Washington, DC. 2008.
Wood TE, Barry J, Yang Z, Cepeda C, Levine MS, Gray M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum Mol Genet. 2019;28(3):487–500.
Vagner T, Dvorzhak A, Wojtowicz AM, Harms C, Grantyn R. Systemic application of AAV vectors targeting GFAP-expressing astrocytes in Z-Q175-KI Huntington’s disease mice. Mol Cell Neurosci. 2016;77:76–86.
Gu X, Li C, Wei W, Lo V, Gong S, Li SH, et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46(3):433–44.
Gu X, Andre VM, Cepeda C, Li SH, Li XJ, Levine MS, et al. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener. 2007;2:8.
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506.
Yu X, Nagai J, Marti-Solano M, Soto JS, Coppola G, Babu MM, et al. Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron. 2020;108(6):1146–62 e10.
Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, Khakh BS. An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron. 2018;98(1):49–66 e9.
Benraiss A, Mariani JN, Osipovitch M, Cornwell A, Windrem MS, Villanueva CB, et al. Cell-intrinsic glial pathology is conserved across human and murine models of Huntington’s disease. Cell Rep. 2021;36(1):109308.
Osipovitch M, Asenjo Martinez A, Mariani JN, Cornwell A, Dhaliwal S, Zou L, et al. Human ESC-Derived Chimeric Mouse Models of Huntington’s Disease Reveal Cell-Intrinsic Defects in Glial Progenitor Cell Differentiation. Cell Stem Cell. 2019;24(1):107–22 e7.
Malaiya S, Cortes-Gutierrez M, Herb BR, Coffey SR, Legg SRW, Cantle JP, et al. Single-nucleus RNA-Seq reveals dysregulation of striatal cell identity due to Huntington’s disease mutations. J Neurosci. 2021;41(25):5534–52.
Lievens JC, Salin P, Had-Aissouni L, Mahy N, Kerkerian-Le GL. Differential effects of corticostriatal and thalamostriatal deafferentation on expression of the glutamate transporter GLT1 in the rat striatum. J Neurochem. 2000;74(3):909–19.
Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105.
Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A. Gliotransmitters travel in time and space. Neuron. 2014;81(4):728–39.
Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, et al. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci. 2011;14(10):1276–84.
Haustein MD, Kracun S, Lu XH, Shih T, Jackson-Weaver O, Tong X, et al. Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron. 2014;82(2):413–29.
Jiang R, Diaz-Castro B, Looger LL, Khakh BS. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J Neurosci. 2016;36(12):3453–70.
Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, et al. Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron. 2018;99(6):1170–87 e9.
Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 2017;40(7):422–37.
Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60(2):161–72.
Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55(10):1074–84.
Politis M, Lahiri N, Niccolini F, Su P, Wu K, Giannetti P, et al. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol Dis. 2015;83:115–21.
Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66(11):1638–43.
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Imaging microglial activation in Huntington’s disease. Brain Res Bull. 2007;72(2–3):148–51.
Rashid K, Geissl L, Wolf A, Karlstetter M, Langmann T. Transcriptional regulation of translocator protein (18 kDa) (TSPO) in microglia requires Pu.1, Ap1 and Sp factors. Biochim Biophys Acta Gene Regul Mech. 2018;1861(12):1119–33.
Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205(8):1869–77.
Silvestroni A, Faull RL, Strand AD, Moller T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. NeuroReport. 2009;20(12):1098–103.
Chang KH, Wu YR, Chen YC, Chen CM. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav Immun. 2015;44:121–7.
Palpagama TH, Waldvogel HJ, Faull RLM, Kwakowsky A. The role of microglia and astrocytes in Huntington’s disease. Front Mol Neurosci. 2019;12:258.
Rodrigues FB, Byrne LM, McColgan P, Robertson N, Tabrizi SJ, Zetterberg H, et al. Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington’s disease. PLoS ONE. 2016;11(9):e0163479.
Siew JJ, Chen HM, Chen HY, Chen HL, Chen CM, Soong BW, et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat Commun. 2019;10(1):3473.
Franciosi S, Ryu JK, Shim Y, Hill A, Connolly C, Hayden MR, et al. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2012;45(1):438–49.
Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest. 2012;122(12):4737–47.
Kwan W, Magnusson A, Chou A, Adame A, Carson MJ, Kohsaka S, et al. Bone marrow transplantation confers modest benefits in mouse models of Huntington’s disease. J Neurosci. 2012;32(1):133–42.
Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17(4):513–21.
Petkau TL, Hill A, Connolly C, Lu G, Wagner P, Kosior N, et al. Mutant huntingtin expression in microglia is neither required nor sufficient to cause the Huntington’s disease-like phenotype in BACHD mice. Hum Mol Genet. 2019;28(10):1661–70.
Khoshnan A, Patterson PH. The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol Dis. 2011;43(2):305–11.
Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum Mol Genet. 2013;22(9):1826–42.
Jiang HR, Al Rasebi Z, Mensah-Brown E, Shahin A, Xu D, Goodyear CS, et al. Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J Immunol. 2009;182(2):1167–73.
Crapser JD, Ochaba J, Soni N, Reidling JC, Thompson LM, Green KN. Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington’s disease. Brain. 2020;143(1):266–88.
Johnson EB, Ziegler G, Penny W, Rees G, Tabrizi SJ, Scahill RI, et al. Dynamics of cortical degeneration over a decade in Huntington’s disease. Biol Psychiatry. 2021;89(8):807–16.
Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, et al. Myelin breakdown and iron changes in Huntington’s disease: pathogenesis and treatment implications. Neurochem Res. 2007;32(10):1655–64.
Phillips O, Squitieri F, Sanchez-Castaneda C, Elifani F, Caltagirone C, Sabatini U, et al. Deep white matter in Huntington’s disease. PLoS ONE. 2014;9(10):e109676.
Novak MJ, Seunarine KK, Gibbard CR, Hobbs NZ, Scahill RI, Clark CA, et al. White matter integrity in premanifest and early Huntington’s disease is related to caudate loss and disease progression. Cortex. 2014;52:98–112.
Bohanna I, Georgiou-Karistianis N, Sritharan A, Asadi H, Johnston L, Churchyard A, et al. Diffusion tensor imaging in Huntington’s disease reveals distinct patterns of white matter degeneration associated with motor and cognitive deficits. Brain Imaging Behav. 2011;5(3):171–80.
Rosas HD, Wilkens P, Salat DH, Mercaldo ND, Vangel M, Yendiki AY, et al. Complex spatial and temporally defined myelin and axonal degeneration in Huntington disease. NeuroImage Clin. 2018;20:236–42.
Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, et al. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85(6):1212–26.
Jin J, Peng Q, Hou Z, Jiang M, Wang X, Langseth AJ, et al. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2015;24(9):2508–27.
Ferrari Bardile C, Garcia-Miralles M, Caron NS, Rayan NA, Langley SR, Harmston N, et al. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc Natl Acad Sci U S A. 2019;116(19):9622–7.
Benraiss A, Mariani JN, Tate A, Madsen PM, Clark KM, Welle KA, et al. A TCF7L2-responsive suppression of both homeostatic and compensatory remyelination in Huntington disease mice. Cell Rep. 2022;40(9):111291.
Comi G, Jeffery D, Kappos L, Montalban X, Boyko A, Rocca MA, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366(11):1000–9.
Sun Y, Tong H, Yang T, Liu L, Li XJ, Li S. Insights into White Matter Defect in Huntington's Disease. Cells. 2022;11(21).
Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res. 2018;7.
Diallo A, Jacobi H, Tezenas du Montcel S, Klockgether T. Natural history of most common spinocerebellar ataxia: a systematic review and meta-analysis. J Neurol. 2021;268(8):2749–56.
Buijsen RAM, Toonen LJA, Gardiner SL, van Roon-Mom WMC. Genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias. Neurotherapeutics. 2019;16(2):263–86.
Schmahmann JD. The cerebellum and cognition. Neurosci Lett. 2019;688:62–75.
Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124(1):1–21.
Rub U, Burk K, Timmann D, den Dunnen W, Seidel K, Farrag K, et al. Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights. Neuropathol Appl Neurobiol. 2012;38(7):665–80.
Oz G, Nelson CD, Koski DM, Henry PG, Marjanska M, Deelchand DK, et al. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2010;30(10):3831–8.
Park YW, Joers JM, Guo B, Hutter D, Bushara K, Adanyeguh IM, et al. Assessment of cerebral and cerebellar white matter microstructure in spinocerebellar ataxias 1, 2, 3, and 6 using diffusion MRI. Front Neurol. 2020;11:411.
Koscik TR, Sloat L, van der Plas E, Joers JM, Deelchand DK, Lenglet C, et al. Brainstem and striatal volume changes are detectable in under 1 year and predict motor decline in spinocerebellar ataxia type 1. Brain Commun. 2020;2(2):fcaa184.
Joers JM, Deelchand DK, Lyu T, Emir UE, Hutter D, Gomez CM, et al. Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann Neurol. 2018;83(4):816–29.
Sheeler C, Rosa JG, Ferro A, McAdams B, Borgenheimer E, Cvetanovic M. Glia in neurodegeneration: the housekeeper, the defender and the perpetrator. Int J Mol Sci. 2020;21(23).
Ferro A, Sheeler C, Rosa JG, Cvetanovic M. Role of microglia in ataxias. J Mol Biol. 2019;431(9):1792–804.
Cendelin J, Cvetanovic M, Gandelman M, Hirai H, Orr HT, Pulst SM, et al. Consensus paper: strengths and weaknesses of animal models of spinocerebellar ataxias and their clinical implications. Cerebellum. 2022;21(3):452–81.
Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34(6):905–19.
Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544(7650):362–6.
Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17(19):7385–95.
Cui Y, Yang S, Li XJ, Li S. Genetically modified rodent models of SCA17. J Neurosci Res. 2017;95(8):1540–7.
McLoughlin HS, Moore LR, Chopra R, Komlo R, McKenzie M, Blumenstein KG, et al. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann Neurol. 2018;84(1):64–77.
Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell. 2013;154(1):118–33.
Schuster KH, Zalon AJ, Zhang H, DiFranco DM, Stec NR, Haque Z, et al. Impaired oligodendrocyte maturation is an early feature in SCA3 disease pathogenesis. J Neurosci. 2022;42(8):1604–17.
Cendelin J. From mice to men: lessons from mutant ataxic mice. Cerebellum Ataxias. 2014;1:4.
Diedrichsen J, King M, Hernandez-Castillo C, Sereno M, Ivry RB. Universal transform or multiple functionality? Understanding the contribution of the human cerebellum across task domains. Neuron. 2019;102(5):918–28.
Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292(5518):926–9.
Wang F, Xu Q, Wang W, Takano T, Nedergaard M. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc Natl Acad Sci U S A. 2012;109(20):7911–6.
Bellamy TC. Interactions between Purkinje neurones and Bergmann glia. Cerebellum. 2006;5(2):116–26.
Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9(10):1302–11.
Shuvaev AN, Belozor OS, Mozhei OI, Khilazheva ED, Shuvaev AN, Fritsler YV, et al. Protective effect of memantine on Bergmann glia and purkinje cells morphology in optogenetic model of neurodegeneration in mice. Int J Mol Sci. 2021;22(15).
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–86.
Borgenheimer E, Hamel K, Sheeler C, Moncada FL, Sbrocco K, Zhang Y, et al. Single nuclei RNA sequencing investigation of the Purkinje cell and glial changes in the cerebellum of transgenic spinocerebellar ataxia type 1 mice. Front Cell Neurosci. 2022;16:998408.
Cvetanovic M, Ingram M, Orr H, Opal P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience. 2015;289:289–99.
Kim JH, Lukowicz A, Qu W, Johnson A, Cvetanovic M. Astroglia contribute to the pathogenesis of spinocerebellar ataxia type 1 (SCA1) in a biphasic, stage-of-disease specific manner. Glia. 2018;66(9):1972–87.
Luttik K, Tejwani L, Ju H, Driessen T, Smeets C, Lim J. Differential effects of Wnt-β-catenin signaling in Purkinje cells and Bergmann glia in SCA1. bioRxiv. 2021. https://doi.org/10.1101/2021.10.21.465333.
Edamakanti CR, Mohan V, Opal P. Reactive Bergmann glia play a central role in Spinocerebellar ataxia inflammation via the JNK pathway. bioRxiv. 2022. https://doi.org/10.1101/2022.06.29.498121.
Mellesmoen A, Sheeler C, Ferro A, Rainwater O, Cvetanovic M. Brain derived neurotrophic factor (BDNF) delays onset of pathogenesis in transgenic mouse model of spinocerebellar ataxia type 1 (SCA1). Front Cell Neurosci. 2018;12:509.
Cvetanovic M. Decreased expression of glutamate transporter GLAST in Bergmann glia is associated with the loss of Purkinje neurons in the spinocerebellar ataxia type 1. Cerebellum. 2015;14(1):8–11.
Tejwani L, Ravindra NG, Nguyen B, Luttik K, Lee C, Gionco J, et al. Longitudinal single-cell transcriptional dynamics throughout neurodegeneration in SCA1. bioRxiv. 2021. https://doi.org/10.1101/2021.10.22.465444.
Qu W, Johnson A, Kim JH, Lukowicz A, Svedberg D, Cvetanovic M. Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J Neuroinflammation. 2017;14(1):107.
Ferro A, Qu W, Lukowicz A, Svedberg D, Johnson A, Cvetanovic M. Inhibition of NF-kappaB signaling in IKKbetaF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of Spinocerebellar ataxia type 1. PLoS ONE. 2018;13(7):e0200013.
Tejwani L, Lim J. Pathogenic mechanisms underlying spinocerebellar ataxia type 1. Cell Mol Life Sci. 2020;77(20):4015–29.
Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, et al. Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol. 2019;37(12):1482–92.
Rosa J-G, Hamel K, Sheeler C, Borgenheimer E, Gilliat S, Soles A, et al. Spatial and temporal diversity of astrocyte phenotypes in Spinocerebellar ataxia type 1 mice. bioRxiv. 2022. https://doi.org/10.1101/2021.09.13.460129.
Muller U. Spinocerebellar ataxias (SCAs) caused by common mutations. Neurogenetics. 2021;22(4):235–50.
Sullivan R, Yau WY, O’Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol. 2019;266(2):533–44.
Banez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, et al. RAN translation in Huntington disease. Neuron. 2015;88(4):667–77.
Meera P, Pulst SM, Otis TS. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J Physiol. 2016;594(16):4653–60.
Bushart DD, Shakkottai VG. Ion channel dysfunction in cerebellar ataxia. Neurosci Lett. 2019;688:41–8.
Egorova P, Popugaeva E, Bezprozvanny I. Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin Cell Dev Biol. 2015;40:127–33.
Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006;29(10):547–53.
Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29(10):3276–87.
Li J, Pan L, Pembroke WG, Rexach JE, Godoy MI, Condro MC, et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun. 2021;12(1):3958.
Lorenzetti D, Watase K, Xu B, Matzuk MM, Orr HT, Zoghbi HY. Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus. Hum Mol Genet. 2000;9(5):779–85.
Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19(3):504–16.
Jyothi HJ, Vidyadhara DJ, Mahadevan A, Philip M, Parmar SK, Manohari SG, et al. Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol Aging. 2015;36(12):3321–33.
Cerbai F, Lana D, Nosi D, Petkova-Kirova P, Zecchi S, Brothers HM, et al. The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS ONE. 2012;7(9):e45250.
Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018;22(1):269–85.
Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 2018;115(8):E1896–905.
Lee E, Jung Y-J, Park YR, Lim S, Choi Y-J, Lee SY, et al. A distinct astrocyte subtype in the aging mouse brain characterized by impaired protein homeostasis. Nature Aging. 2022;2(8):726–41.
Sams EC. Oligodendrocytes in the aging brain. Neuronal Signal. 2021;5(3):NS20210008.
Spitzer SO, Sitnikov S, Kamen Y, Evans KA, Kronenberg-Versteeg D, Dietmann S, et al. Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron. 2019;101(3):459–71 e5.
Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995 Sep 22;82(6):937–48. https://doi.org/10.1016/0092-8674(95)90273-2. PMID: 7553854.
Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999 May;23(1):181–92. https://doi.org/10.1016/s0896-6273(00)80764-3. PMID: 10402204.
Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, . . . Howland D (2012) Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One, 7(12): e49838. https://doi.org/10.1371/journal.pone.0049838.
Acknowledgements
MG is supported by the Dixon Family Foundation, Hereditary Disease Foundation, and National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS089750 and NS089750-01A1 (MG)). This work was supported by National Institute of Health NINDS awards (R01 NS197387 and R01 NS109077) to M.C. We thank Juao-Guilerme Rosa for help with figures.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cvetanovic, M., Gray, M. Contribution of Glial Cells to Polyglutamine Diseases: Observations from Patients and Mouse Models. Neurotherapeutics 20, 48–66 (2023). https://doi.org/10.1007/s13311-023-01357-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-023-01357-5