
Abstract
Recent genome-wide association studies of Huntington’s disease (HD) primarily highlighted genes involved in DNA damage repair mechanisms as modifiers of age at onset and disease severity, consistent with evidence that more DNA repair genes are being implicated in late age–onset neurodegenerative diseases. This provides an exciting opportunity to advance therapeutic development in HD, as these pathways have already been under intense investigation in cancer research. Also emerging are the roles of other polyglutamine disease proteins in DNA damage repair mechanisms. A potential universal trigger of oxidative DNA damage shared in these late age–onset diseases is the increase of reactive oxygen species (ROS) in human aging, defining an age-related mechanism that has defied other hypotheses of neurodegeneration. We discuss the potential commonality of DNA damage repair pathways in HD and other neurodegenerative diseases. Potential targets for therapy that may prove beneficial across many of these diseases are also identified, defining nodes in the ataxia telangiectasia-mutated (ATM) complex, mismatch repair, and poly ADP-ribose polymerases (PARPs).
Similar content being viewed by others


Huntington’s disease (HD) was precisely defined in 1993 when the huntingtin gene was cloned and the disease-causing CAG nucleotide expansion was defined, showing an inverse correlation between expansion length and age at onset [1]. The problem of HD was daunting, as the gene encoded one of the largest non-membrane proteins in the human proteome at 3144 amino acids or 350 kDa. Over the next 25 years, the normal function of huntingtin also eluded strict definition. Cell biology and biochemical studies revealed the protein to have no enzymatic activity and no conserved functional motifs, with the exception that proteins with polyglutamine tracts were often transcriptionally active and huntingtin contained HEAT repeats common to several protein scaffolds [2, 3]. Interactome studies showed huntingtin at the heart of several complexes [4] and it has been localized to the nucleus [5], endoplasmic reticulum [6], and early, recycling, and late endosomes [7], the neuronal postsynaptic density [8], mitotic spindle [9, 10], and the primary cilium [11, 12]. This ubiquitous location of huntingtin made it difficult to define one primary function of the protein, but rather suggested that this was a large, mobile scaffold with many dynamic complexes, with location and conformation sensitive to cell stress events.
From 1993, the predominant hypothesis of toxicity in HD was protein misfolding. The presence of > 37 continuous CAG repeats, hence, continuous glutamine residues, would lead to protein misfolding and accumulation of inclusions that triggered neuronal dysfunction and eventually neurodegeneration [13]. This hypothesis was heavily influenced by research in Alzheimer’s and Parkinson’s diseases, which were also associated with misfolded protein [14]. It was subsequently reinforced by studies in early mouse models in which a small fragment of huntingtin representing 3% of the total protein was overexpressed in trans [15]. The extreme polyglutamine length required to induce phenotypes in mice also caused abundant inclusions, and the protein misfolding hypothesis guided the field away from other viable avenues of research for many years.
Motivated by the need to find novel mechanisms and therapeutic targets in people, there has been a more recent return to unbiased statistical genetics. Although CAG expansion accurately predicts disease and correlates with age at onset, CAG length at typical clinical alleles, with a mean length of 43 repeats [16], is actually poor at predicting age at onset, with a variability of decades. Genome-wide association studies (GWAS) were therefore conducted to define modifiers of age at onset and disease severity.
A Return to an Unbiased Approach Nets New Leads in HD in DNA Damage Repair
A large global GWAS of 6000 to 9000 HD patients with broad genetic diversity and numbers for high statistical power primarily identified genes involved in DNA damage repair as significant modulators of age at onset and disease severity, with some pathways related to redox signaling and mitochondrial health [17,18,19]. There was a surprising lack of protein homeostasis regulators and the conclusion was that disease modifiers of HD are mainly involved in DNA repair mechanisms (Table 1). The most recent analyses make it clear that the length of uninterrupted CAG repeats, and not the length of the encoded polyglutamine tract, has the strongest association with age at onset [19, 23].
The single nucleotide polymorphisms (SNPs) in DNA repair genes significant to HD are also significant in some spinocerebellar ataxias (SCAs), a related group of CAG repeat diseases [20]. This suggests that DNA repair pathways underlie a common genetic mechanism of disease [20].
In retrospect, the GWAS results should have been anticipated. Many age-onset neurodegenerative diseases are known to be caused by mutations in bona fide DNA repair factors. Tyrosyl DNA-phosphodiesterase 1 (TDP1), aprataxin (APTX), and polynucleotide kinase/phosphatase (PNKP) are three factors involved in repairing broken DNA strands [24]. A series of neurodegenerative and neurodevelopmental disorders have been associated with mutations affecting the functions of these proteins. Mutations in TDP1 and APTX cause spinocerebellar ataxia with axonal neuropathy (SCAN1) [25] and ataxia-oculomotor apraxia 1 (AOA1) [26], respectively. Mutations in PNKP cause microcephaly with seizures (MCSZ) [27] and ataxia-oculomotor apraxia 4 (AOA4) [28], while decreased PNKP activity is associated with spinocerebellar ataxia type 3 (SCA3 or Machado-Joseph disease) [29]. Mutations in the X-ray repair cross-complementing protein, XRCC1, have been linked to ataxia-ocular motor apraxia-XRCC1 (AOA-XRCC1) [30]. XRCC1 interacts with DNA ligase III, polymerase beta, and poly ADP-ribose polymerase 1 (PARP1) to participate in base excision repair (BER) to correct either mismatched or damaged bases. This repair pathway is critical in neurons that are postmitotic, as they cannot rely on DNA replication proofreading to repair the mismatched or damaged adducts.
One of the first identified genetic ataxias was ataxia telangiectasia, or Louis–Bar Syndrome. This is a pediatric-onset ataxia, with high risk of cancer later in life due to defective DNA repair induced by loss of function mutations in ataxia telangiectasia mutated (ATM) [31]. Similar to huntingtin, ATM is a large 350-kDa scaffolding protein, rich in HEAT repeats, and ubiquitous in location, found in the nucleus, cytoplasm, vesicles, primary cilium, and mitotic spindle [32]. However, unlike huntingtin, it has a known catalytic activity as a serine-threonine kinase [32]. As a signaling factor, ATM modifies DNA repair factors, as well as genomic integrity regulators tumor protein p53 (TP53) [33] and mouse double minute 2 homolog (MDM2) [34]. Understanding the broader landscape of DNA repair defects in neurodegenerative diseases will shed light on GWAS leads and why they affect HD age at onset and severity, and more importantly, where there could be targets for disease modification.
The Huntingtin Protein and DNA Damage Repair
In 2014, ATM was seen as a modifier of HD phenotypes in a mouse model, as crosses with heterozygous null ATM mice resulted in a milder phenotype [35]. What drove that study was the observation that there is elevated DNA damage in HD and SCA mouse models [36], and this damage preceded protein aggregation [37]. Indeed, even in human HD fibroblasts with only 43 CAG repeats, a phenotype of elevated DNA damage can be detected [38]. In an HD clinical study in 2018, elevated damage was seen longitudinally in HD patient peripheral blood mononuclear cells, and this damage preceded mitochondrial markers of dysfunction [39]. Similarly in 2019, another clinical study measured increased DNA damage in prodromal HD in the blood cell population [40]. Thus, from cells to mouse models, and most importantly to patient clinical samples, there is a clear progressive level of DNA damage in HD. This DNA damage starts in the prodromal phase of disease and is present with typical clinical alleles.
Although these observations and GWAS results provide hints of DNA damage mechanisms in HD, one question is the exact cause of the damage. Inhibition of DNA repair in postmitotic neurons would be sufficient to cause an accumulation of DNA lesions that occur through normal metabolic processes. Furthermore, during human aging, decreased mitochondrial efficiency leads to increased reactive oxygen and sulfur species, and this decreased efficiency is amplified in neurodegeneration [41]. In HD, these problems may be caused by impaired function of the huntingtin protein.
Huntingtin protein localization and conformation are stress dependent. Huntingtin is a ROS sensor and will relocate from the outer leaf of the pervasive endoplasmic reticulum bilayer to the nucleus upon oxidation of a single methionine at position 8 [42]. Reactive oxygen species (ROS) also increases the number of nuclear speckles: liquid–liquid phase separated droplets at which huntingtin colocalizes with DNA repair factors including ATM [38, 42,43,44]. Huntingtin acts as a scaffold that can localize to DNA damage, and modulates its associated complex in the presence of ROS stress [38]. Importantly, localization to damage is dependent on ATM kinase activity [38]. Huntingtin was identified in the transcription-coupled repair (TCR) complex that finds lesions and mediates repair during transcription. Mutant huntingtin impairs the function of the TCR complex components PNKP and ataxin-3, leading to elevated DNA damage and ATM hyperactivation [45].
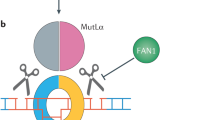
Thus, we have a new role of huntingtin scaffolding activity in DNA repair, in which dysregulation may explain the predominance of DNA repair pathways identified by GWAS. However, within the genes uncovered by GWAS and subsequent SNP genotyping, there are very specific, seemingly unrelated repair pathways that have appeared. Mismatch repair (MMR) is represented by SNPs in several genes, whereas one of the most significant hits, FAN1, is associated with interstrand cross-link (ICL) repair.
Mismatch Repair and Somatic CAG Expansion
The mismatch repair (MMR) pathway is of particular interest because of its influence on somatic expansion of CAG repeats, which can undergo progressive length increases over time, particularly in the brain [46]. Somatic expansion of huntingtin is associated with earlier age of HD onset [47] and more severe symptoms [48]. In mouse models, expansion is prevented upon deletion of MMR genes, mutS homologs 2 and 3 (msh2 and msh3), mutL homolog 1 (mlh1), or PMS1 homolog 2 (pms2) [49,50,51,52]. These proteins normally work in concert to repair insertion-deletion loops in the DNA, but they also drive disease-associated repeat expansions [53].
The initial evidence from mouse models has since been bolstered by human data, clearly implicating the MMR pathway in disease progression. Although the chromosomal region bearing MLH1 was identified by GWAS, but did not initially reach genome-wide significance [17], a subsequent SNP genotyping study confirmed a modifier haplotype at the MLH1 locus [21]. The genomic region bearing MSH3 was identified by a second GWAS with more in-depth measures of disease progression [18], and PMS2 was among the SNP locations independently confirmed by genotyping [20]. Disease-associated SNPs near the MLH1, MSH3, PMS2, and PMS1 loci have recently been confirmed and the importance of uninterrupted CAG repeats within the huntingtin gene itself, which also impacts somatic instability, is now clear [19, 23]. PMS2 and MSH3 have also been implicated in SCA1 and myotonic dystrophy, respectively [20, 22]. Modifier genes are summarized in Table 1.
Although it is likely that MMR proteins influence pathology via somatic expansion, this may not be the sole mechanism, as tissue specificity of somatic expansion correlates with striatal neurodegeneration in HD, but not in SCAs [54], despite ubiquitous transcription of the CAG-containing proteins. It is possible that MMR proteins also contribute to disease modification through other DNA repair factors, including TP53 [55], breast cancer associated-1 (BRCA1) [56], and ATM [57]. The MMR machinery has also been implicated in the repair of several types of DNA damage [58,59,60], including ICLs [61].
In the patient population, pathology is likely to be caused by a combination of these mechanisms. The fact that MMR proteins, and scaffolds such as huntingtin and ATM are common components of many DNA repair complexes [38, 56, 57, 60], offers the intriguing possibility that mutant huntingtin protein dysfunction may affect CAG expansion within its own gene. DNA repair mechanisms are paradoxical in that the pathways are very specific to types of DNA damage, yet many players operate in more than one pathway [62]. Most DNA damage repair mechanisms have been studied through the disease lens of cancer or universal base mechanisms from prokaryotes, which may explain why huntingtin was never identified previously in DNA damage repair complexes: there appear to be some very basic differences critical to neuronal populations. It is well established that the bulk of neuronal DNA damage is acquired by oxidative damage rather than replication errors [63], and novel mechanisms such as the generation of double-strand breaks in targeted promoters of active neurons have more recently been uncovered [64].
FAN1 and Interstrand Cross-Link Repair
Another lead gene identified in HD GWAS is the Fanconi anemia FANCD2- and FANCI-associated nuclease 1 (FAN1) [17, 20, 21]. FAN1 is well defined in the repair of interstrand cross-links (ICLs) as a nuclease inducing strand cleavage around ICL adducts [65]. These covalent structures, if not repaired, distort DNA and suppress transcription by preventing strand separation [66]. Like other forms of DNA damage, ICLs can occur as a result of cellular metabolism, and can form between strands at abasic sites produced by oxidative damage and base excision repair [67].
Importantly, FAN1 has also been linked to somatic expansion in fragile X-related disorders [68] and HD [69]. In contrast to MMR proteins, FAN1 expression is associated with reduced expansion and later age at onset, in a mechanism that surprisingly does not require nuclease activity [69]. As with many other factors in DNA damage repair, such as XRCC1 and ATM, FAN1 is both an enzyme and a scaffold, which is a critical consideration when interpreting genetic data and assuming the target is enzymatic inhibition alone.
The relative contributions of somatic expansion suppression and ICL repair to pathology have yet to be elucidated. These seemingly disparate pathways may be connected by products of oxidative DNA damage, such as N6-furfuryladenine (N6FFA), which has been defined as a beneficial compound in HD models and a potential drug lead as described below [43, 70].
DNA Repair Mechanisms in HD May Uncover New Targets and Drug Leads
N6FFA was identified in a screen for compounds affecting the phosphorylation of serines 13 and 16 within the N17 domain of huntingtin. N17 comprises the first 17 amino acids that modulate the location of the massive 3144 amino acid protein as a “tail that wags the dog” [6, 71]. This modification is deficient in HD [9, 44] and its restoration is beneficial in HD model systems [9, 43, 72, 73] making it an attractive therapeutic target.
N6FFA is a naturally occurring product of oxidative DNA damage and an excretory metabolite [74, 75]. Furan moieties are formed in the presence of reactive oxygen species and further react by addition to adenosine. This modified base forms bulky lesions in DNA and causes mismatches at adjacent bases, requiring BER and MMR mechanisms for repair [76, 77]. The excised product, N6FFA, is salvaged to the ATP analog, KTP, via the promiscuous activity of adenine phosphoribosyltransferase (APRT) [78]. KTP can be used as a “neo-substrate” by kinases with large enzymatic pockets, including CK2, the kinase responsible for phosphorylating the huntingtin N17 domain [9, 43]. This would be especially important under conditions of DNA damage, which are associated with low levels of ATP [79]. CK2 is an important player in the DNA damage response, having numerous substrates and activating ATM at the very first steps of DNA mismatch recognition [80].
Huntingtin found at sites of DNA damage is phosphorylated at N17 [38], and N6FFA, APRT, and CK2 colocalize with huntingtin at damage sites [43]. We therefore proposed that in proximity to DNA damage, N6FFA is salvaged to KTP by APRT, and CK2 uses locally generated KTP to phosphorylate huntingtin and its other substrates. In this way, KTP would enhance the ability of CK2 to phosphorylate its targets, in proximity to DNA damage sites, under conditions of depleted ATP. By this mechanism, a product of DNA damage signals the activation of repair proteins, resulting in the production of additional signaling molecules in a feed-forward loop that dampens naturally once N6FFA adducts are corrected (Fig. 1, left panel).

Proposed mechanism of N6FFA action. Left panel: Products of oxidative DNA damage are excised by the repair machinery yielding N6FFA, which is salvaged to KTP by APRT. KTP is used by CK2 to phosphorylate its targets, including normal huntingtin. The signal dampens naturally as adducts are repaired. Middle panel: Expanded huntingtin is inefficiently phosphorylated and impaired in its scaffolding function, resulting in the accumulation of adducts. Without N6FFA excision and conversion to KTP, the signal is stifled. Right panel: Exogenous N6FFA is salvaged by APRT, providing a source of KTP to activate CK2 signaling and restore repair
We hypothesize that polyglutamine expansion inhibits the phosphorylation of N17 by CK2 [9, 44] and impairs the scaffolding function of huntingtin in the process of DNA repair [38]. Excision of damaged bases would therefore be diminished, leaving N6FFA trapped in DNA and shutting down the critical signaling by CK2 via KTP in a negative cascade (Fig. 1, middle panel). This mechanism provides an opportunity for therapeutic intervention: the feed-forward repair signaling would be restored by adding free N6FFA in trans, which is salvaged to KTP [78] for use by CK2 on huntingtin and its other targets (Fig. 1, right panel). Indeed, N6FFA was beneficial in mouse cortical neuron assays as well as mouse models to correct HD phenotypes, with an intriguing loss of huntingtin inclusions in mouse brains [43] that could be attributed to the restoration of energy levels as described in the next section.
Interestingly, the N6FFA trapped in DNA due to poor BER and MMR due to mutant huntingtin can further oxidize to generate an ICL, in addition to DNA–RNA and DNA–protein cross-links [77, 81]. Thus, N6FFA can be considered the focal point of: an age-onset mechanism of increased ROS stress; the generation of DNA mismatches implicating MMR; and a maturation of the adduct to an ICL, which highlights FAN1.
Understanding the different mechanisms by which GWAS hits and expanded huntingtin affect DNA repair will be indispensable in the development of therapeutic strategies going forward. Regardless of mechanism, however, the consequences of DNA damage accumulation may be similar, including changes in transcription and epigenetic signatures, as well as important effects on energy metabolism.
DNA Repair Is Intimately Linked to Energy Defects in Neurodegeneration
The development of cerebellar ataxia in AOA-XRCC1 is associated with the elevation of poly ADP-ribose (PAR) levels [30]. The production of PAR chains by PARPs is one of the first steps in the process of DNA damage repair. These PAR chains transiently recruit DNA repair factors to adducts, including XRCC1, but the fast polymerization of ADP comes at the great metabolic cost of draining nicotinamide adenine dinucleotide (NAD+) levels [82] and inhibiting glycolysis [83]. Transient energy drops are relieved quickly by the breakdown of PAR by PAR glycohydrolase (PARG) [84] and nucleotide-salvaging enzymes are critical to restore intraneuronal nucleotide levels [85]. However, in AOA-XRCC1, the production of PAR persists due to low XRCC1 levels, leading to toxicity. Excessive and persistent PAR production can lead to neuronal death by the unique mechanism of parthanatos, which involves the nuclear translocation of apoptosis-inducing factor (AIF) from mitochondria [86].
Hyper-PARylation can also lead to protein aggregation. In 2018, this was highlighted elegantly in the Parkinson’s disease context showing PAR chains could nucleate alpha-synuclein aggregation [87]. Aggregation is also a downstream consequence of ROS-mediated energy depletion, as ATP-dependent chaperones struggle to maintain protein quality under conditions of oxidative stress [88, 89]. There is recent interest in repurposing PARP inhibitors aside from cancer treatment [90], but the caveat is that most PARP inhibitors are designed to be toxic to transformed cells by trapping PARP enzymes on DNA. One solution may be PARP expression knockdown with new generations of anti-sense oligonucleotides [91].
The relevance of PARylation to HD has yet to be determined, but from GWAS we may have a hint that ties hyper-PARylation to the ATM/huntingtin complex. Another major lead in HD GWAS is the ribonucleoside-diphosphate reductase subunit M2B, or RRM2B, also known as P53R2. RRM2B is a critical ribonucleotide salvager which nets a severe fatal pediatric disorder called mitochondrial DNA depletion syndrome (MDDS) that affects muscle, the brain, and the respiratory tract when null [92]. Like huntingtin, RRM2B is activated by the TP53 tumor suppressor [93], and is a known ATM interactor [94]. The ribonucleoside-diphosphate reductase activity could be critical downstream of PARG to salvage ADP back from hydrolyzed PAR chains during neuronal energy crisis. Thus, we can hypothesize a mechanism of RRM2B disease modification by catalyzing the conversion of poly ADP-ribose chains back to critical adenosine ribonucleosides.
HD GWAS in the Bigger Picture of Polyglutamine Diseases
Given the significance of HD genetic modifier SNPs to some spinocerebellar ataxias, we may find an intersection of molecular mechanisms of disease between HD and other CAG repeat disorders with respect to DNA repair. One node is the ATM complex, but huntingtin has also been defined at the TCR complex [45] along with ataxin-3, the affected protein in SCA3, and PKNP, the protein mutated in MCSZ [27]. Ataxin-3 has also been implicated by others in the double-strand break response [95]. CAG expansion in the androgen receptor, a transcription factor involved in DNA damage repair signaling, leads to spinal and bulbar muscle atrophy, SBMA, or Kennedy’s disease [96, 97]. CAG expansion in the TATA box-binding protein (TBP) causes SCA17, and TBP localizes to damaged DNA [98]. DNA repair has also been implicated in SCA1, as overexpression of DNA repair factors replication protein A1 and high mobility group box 1 in mouse and Drosophila models corrects motor phenotypes [99, 100]. Thus, we may anticipate increased relevance of DNA repair in many late age–onset neurodegenerative diseases, as the natural increased ROS stress during human aging and effects on DNA/RNA oxidation are tempting mechanisms to explain why these diseases typically occur later in life.
Increased DNA oxidation and hyper-PARylation appeared in neurodegenerative disease studies in the late 1990s [101, 102], but the early study of PAR chains and relevance of oxidation to disease mechanism were not further explored in favor of various amyloid hypotheses of late age–onset neurodegeneration. Although guanine oxidation products were the focus of these studies as a biomarker of DNA oxidation, it is not clear that all DNA bases are equally modified under oxidative stress nor processed in a similar manner. Adenosine bases are subject to nucleotide salvage in neurons and have unique utility after base excision repair to be salvaged back to adenosine nucleosides for energy production [85]. Neurons are highly metabolically active and thus generate high ROS loads, yet oxidative base damage to DNA cannot be repaired by DNA replication as in mitotic cell types. Brain subregions can also transiently flip to aerobic glycolysis or Warburg metabolism to generate ATP at times of energy stress [103], but this energy supply comes at a high cost of increased ROS stress. The burden of reactive oxygen levels on mitochondria, which have a diminished efficiency with human aging, might explain how mitochondrial dysfunction is a common aspect of all neurodegenerative diseases [104].
To fully understand the implications of defective DNA repair in neurodegeneration, it is important to understand how DNA repair imparts a severe energy stress on neurons. High rates of neuronal metabolism mean that even as energy is depleted, ROS by-products cause damage that further drains the energy supply. In this way, neurons must struggle to maintain energy levels and even a minor deficiency in a DNA repair protein would amount to undue neuronal death over time (Fig. 2).

DNA repair and neuronal energy homeostasis. The combined energetic costs of high metabolic rate, and repair of metabolism by-product–mediated damage, make neurons vulnerable to minor deficiencies in DNA repair proteins with age
The involvement of DNA repair pathways in neurodegenerative diseases presents a number of opportunities for therapeutic intervention. Although drugs against key potential targets have been developed for cancer, preclinical data on their applicability for neurodegenerative diseases is needed as the goal of anticancer therapy is to kill affected cells, whereas the goal of antineurodegeneration therapy is to save affected cells. The immediate goals in researching DNA repair in neurodegeneration are to define the roles of DNA repair factors, in terms of their scaffolding versus enzymatic functions, to precisely define therapeutic mechanisms of either inhibiting enzymatic activity or modulating protein levels, or both. Testing in the most clinically relevant models and special attention to measures of genotoxicity will help pave the way to the clinic.
References
Macdonald M (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983
Andrade MA, Bork P (1995) HEAT repeats in the Huntington’s disease protein. Nat Genet 11:115–116
Takano H, Gusella JF (2002) The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci 3:15
Brandi V, Di Lella V, Marino M, Ascenzi P, Polticelli F (2018) A comprehensive in silico analysis of huntingtin and its interactome. J Biomol Struct Dyn 36:3155–3171
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548
Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet 16:2600–2615
Kim M, Velier J, Chase K, Laforet G, Kalchman MA, Hayden MR, Won L, Heller A, Aronin N, Difiglia M (1999) Forskolin and dopamine D1 receptor activation increase huntingtin’s association with endosomes in immortalized neuronal cells of striatal origin. Neuroscience 89:1159–1167
Marcora E, Gowan K, Lee JE (2003) Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. Proc Natl Acad Sci U S A 100:9578–9583
Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, Truant R (2011) Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol 7:453–460
Godin JD, Colombo K, Molina-Calavita M, et al. (2010) Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 67:392–406
Maiuri T, Woloshansky T, Xia J, Truant R (2013) The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum Mol Genet 22:1383–1394
Keryer G, Pineda JR, Liot G, et al. (2011) Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest 121:4372–4382
Truant R, Atwal RS, Desmond C, Munsie L, Tran T (2008) Huntington’s disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. FEBS J 275:4252–4262
Castellani RJ, Smith MA (2011) Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is “too big to fail.”. J Pathol 224:147–152
Mangiarini L, Sathasivam K, Seller M, et al. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506
Myers RH (2004) Huntington’s disease genetics. NeuroRx 1:255–262
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium (2015) Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 162:516–526
Moss DJH, Pardiñas AF, Langbehn D, et al. (2017) Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol 16:701–711
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium, Lee J-M, Correia K, et al. (2019) Huntington’s disease onset is determined by length of uninterrupted CAG, not encoded polyglutamine, and is modified by DNA maintenance mechanisms. bioRxiv 529768
Bettencourt C, Hensman-Moss D, Flower M, et al. (2016) DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol 79:983–990
Lee J-M, Chao MJ, Harold D, et al. (2017) A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet 26:3859–3867
Morales F, Vásquez M, Santamaría C, Cuenca P, Corrales E, Monckton DG (2016) A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair 40:57–66
Wright GEB, Collins JA, Kay C, et al. (2019) Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased Somatic Instability, Hastening Onset of Huntington Disease. Am J Hum Genet 104:1116–1126
Jiang B, Glover JNM, Weinfeld M (2017) Neurological disorders associated with DNA strand-break processing enzymes. Mech Ageing Dev 161:130–140
Takashima H, Boerkoel CF, John J, et al. (2002) Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32:267–272
Moreira MC, Barbot C, Tachi N, et al. (2001) The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 29:189–193
Shen J, Gilmore EC, Marshall CA, et al. (2010) Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 42:245–249
Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T, Sequeiros J, Hardy J, Coutinho P, Guerreiro R (2015) Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet 96:474–479
Gao R, Liu Y, Silva-Fernandes A, et al. (2015) Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet 11:e1004834
Hoch NC, Hanzlikova H, Rulten SL, et al. (2017) XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541:87–91
Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, Aghamohammadi A (2019) Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatr Allergy Immunol. https://doi.org/10.1111/pai.13020
Stagni V, Cirotti C, Barilà D (2018) Ataxia-Telangiectasia Mutated Kinase in the Control of Oxidative Stress, Mitochondria, and Autophagy in Cancer: A Maestro With a Large Orchestra. Front Oncol 8:73
Paules RS, Levedakou EN, Wilson SJ, Innes CL, Rhodes N, Tlsty TD, Galloway DA, Donehower LA, Tainsky MA, Kaufmann WK (1995) Defective G2 checkpoint function in cells from individuals with familial cancer syndromes. Cancer Res 55:1763–1773
Cheng Q, Chen L, Li Z, Lane WS, Chen J (2009) ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J 28:3857–3867
Lu X-H, Mattis VB, Wang N, et al. (2014) Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med 6:268ra178
Giuliano P, De Cristofaro T, Affaitati A, Pizzulo GM, Feliciello A, Criscuolo C, De Michele G, Filla A, Avvedimento EV, Varrone S (2003) DNA damage induced by polyglutamine-expanded proteins. Hum Mol Genet 12:2301–2309
Illuzzi J, Yerkes S, Parekh-Olmedo H, Kmiec EB (2009) DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res 87:733–747
Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WMC, Truant R (2017) Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet 26:395–406
Askeland G, Dosoudilova Z, Rodinova M, et al. (2018) Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci Rep 8:9817
Castaldo I, De Rosa M, Romano A, et al. (2019) DNA damage signatures in peripheral blood cells as biomarkers in prodromal huntington disease. Ann Neurol 85:296–301
Elfawy HA, Das B (2019) Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci 218:165–184
DiGiovanni LF, Mocle AJ, Xia J, Truant R (2016) Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum Mol Genet 25:3937–3945
Bowie LE, Maiuri T, Alpaugh M, et al. (2018) N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc Natl Acad Sci U S A 115:E7081–E7090
Hung CL-K, Maiuri T, Bowie LE, et al. (2018) A Patient-Derived Cellular Model for Huntington’s Disease Reveals Phenotypes at Clinically Relevant CAG Lengths. Mol Biol Cell 29(23):2809-2820. mbc E18090590
Gao R, Chakraborty A, Geater C, et al. (2019) Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife. https://doi.org/10.7554/eLife.42988
Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, Adam S, Greenberg C, Ives EJ, Clarke LA (1994) Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet 6:409–414
Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, Wheeler VC (2009) Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 18:3039–3047
Shelbourne PF, Keller-McGandy C, Bi WL, et al. (2007) Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet 16:1133–1142
Pinto RM, Dragileva E, Kirby A, et al. (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet 9:e1003930
Manley K, Shirley TL, Flaherty L, Messer A (1999) Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet 23:471–473
Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG (2004) Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet 13:1815–1825
Tomé S, Manley K, Simard JP, et al. (2013) MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet 9:e1003280
Schmidt MHM, Pearson CE (2016) Disease-associated repeat instability and mismatch repair. DNA Repair 38:117–126
Hashida H, Goto J, Kurisaki H, Mizusawa H, Kanazawa I (1997) Brain regional differences in the expansion of a CAG repeat in the spinocerebellar ataxias: dentatorubral-pallidoluysian atrophy, Machado-Joseph disease, and spinocerebellar ataxia type 1. Ann Neurol 41:505–511
Peters AC, Young LC, Maeda T, Tron VA, Andrew SE (2003) Mammalian DNA mismatch repair protects cells from UVB-induced DNA damage by facilitating apoptosis and p53 activation. DNA Repair 2:427–435
Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14:927–939
Luo Y, Lin F-T, Lin W-C (2004) ATM-mediated stabilization of hMutL DNA mismatch repair proteins augments p53 activation during DNA damage. Mol Cell Biol 24:6430–6444
Meyers M, Hwang A, Wagner MW, Bruening AJ, Veigl ML, Sedwick WD, Boothman DA (2003) A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene 22:7376–7388
Arora S, Huwe PJ, Sikder R, et al. (2017) Functional analysis of rare variants in mismatch repair proteins augments results from computation-based predictive methods. Cancer Biol Ther 18:519–533
Siehler SY, Schrauder M, Gerischer U, Cantor S, Marra G, Wiesmüller L (2009) Human MutL-complexes monitor homologous recombination independently of mismatch repair. DNA Repair 8:242–252
Kato N, Kawasoe Y, Williams H, et al. (2017) Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway. Cell Rep 21:1375–1385
Limpose KL et al. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. - PubMed - NCBI. https://www.ncbi.nlm.nih.gov/pubmed/28629773. Accessed 24 Jun 2019
Fishel ML et al. DNA repair in neurons: so if they don’t divide what’s to repair? - PubMed - NCBI. https://www.ncbi.nlm.nih.gov/pubmed/16879837. Accessed 24 Jun 2019
Madabhushi R, Gao F, Pfenning AR, et al. (2015) Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161:1592–1605
Jin H, Cho Y (2017) Structural and functional relationships of FAN1. DNA Repair 56:135–143
Derheimer FA, Kevin Hicks J, Paulsen MT, Canman CE, Ljungman M (2009) Psoralen-Induced DNA Interstrand Cross-Links Block Transcription and Induce p53 in an Ataxia-Telangiectasia and Rad3-Related-Dependent Manner. Mol Pharmacol 75:599
Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS (2014) Interstrand DNA-DNA cross-link formation between adenine residues and abasic sites in duplex DNA. J Am Chem Soc 136:3483–3490
Zhao X-N, Usdin K (2018) FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair 69:1–5
Goold R, Flower M, Moss DH, et al. (2019) FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet 28:650–661
Maiuri T, Bowie LE, Truant R (2019) DNA Repair Signaling of Huntingtin: The Next Link Between Late-Onset Neurodegenerative Disease and Oxidative DNA Damage. DNA Cell Biol 38:1–6
Aiken CT, Steffan JS, Guerrero CM, et al. (2009) Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J Biol Chem 284:29427–29436
Di Pardo A, Maglione V, Alpaugh M, et al. (2012) Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc Natl Acad Sci U S A 109:3528–3533
Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64:828–840
Barciszewski J, Mielcarek M, Stobiecki M, Siboska G, Clark BF (2000) Identification of 6-furfuryladenine (kinetin) in human urine. Biochem Biophys Res Commun 279:69–73
Barciszewski J, Siboska GE, Pedersen BO, Clark BF, Rattan SI (1997) A mechanism for the in vivo formation of N6-furfuryladenine, kinetin, as a secondary oxidative damage product of DNA. FEBS Lett 414:457–460
Wyszko E, Barciszewska MZ, Markiewicz M, Szymański M, Markiewicz WT, Clark BFC, Barciszewski J (2003) “Action-at-a distance” of a new DNA oxidative damage product 6-furfuryl-adenine (kinetin) on template properties of modified DNA. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression 1625:239–245
Carrette LLG, Madder A (2014) A synthetic oligonucleotide model for evaluating the oxidation and crosslinking propensities of natural furan-modified DNA. Chembiochem 15:103–107
Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM (2013) A Neo-Substrate that Amplifies Catalytic Activity of Parkinson’s-Disease-Related Kinase PINK1. Cell 154:737–747
Brace LE, Vose SC, Stanya K, et al. (2016) Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech Dis 2:16022
Montenarh M (2016) Protein kinase CK2 in DNA damage and repair. Transl Cancer Res 5:49–63
Carrette LLG, Gyssels E, De Laet N, Madder A (2016) Furan oxidation based cross-linking: a new approach for the study and targeting of nucleic acid and protein interactions. Chem Commun 52:1539–1554
Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010) NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 30:2967–2978
Andrabi SA, Umanah GKE, Chang C, Stevens DA, Karuppagounder SS, Gagné J-P, Poirier GG, Dawson VL, Dawson TM (2014) Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A 111:10209–10214
Sasaki Y, Hozumi M, Fujimori H, Murakami Y, Koizumi F, Inoue K, Masutani M (2016) PARG Inhibitors and Functional PARG Inhibition Models. Curr Protein Pept Sci 17:641–653
Fasullo M, Endres L (2015) Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int J Mol Sci 16:9431–9449
Andrabi SA, Dawson TM, Dawson VL (2008) Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci 1147:233–241
Kam T-I, Mao X, Park H, et al. (2018) Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science. https://doi.org/10.1126/science.aat8407
Dahl J-U, Gray MJ, Jakob U (2015) Protein quality control under oxidative stress conditions. J Mol Biol 427:1549–1563
Weids AJ, Ibstedt S, Tamás MJ, Grant CM (2016) Distinct stress conditions result in aggregation of proteins with similar properties. Sci Rep 6:24554
Berger NA, Besson VC, Boulares AH, et al. (2018) Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br J Pharmacol 175:192–222
Levin AA (2019) Treating Disease at the RNA Level with Oligonucleotides. N Engl J Med 380:57–70
Gorman GS, Pitceathly RDS, Turnbull DM, Taylor RW (2013) RRM2B-Related Mitochondrial Disease. Mitochondrial Disorders Caused by Nuclear Genes 171–182
Cho EC, Yen Y (2016) Novel regulators and molecular mechanisms of p53R2 and its disease relevance. Biochimie 123:81–84
Chang L, Zhou B, Hu S, Guo R, Liu X, Jones SN, Yen Y (2008) ATM-mediated serine 72 phosphorylation stabilizes ribonucleotide reductase small subunit p53R2 protein against MDM2 to DNA damage. Proc Natl Acad Sci U S A 105:18519–18524
Pfeiffer A, Luijsterburg MS, Acs K, et al. (2017) Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J 36:1066–1083
Thompson TC, Li L (2017) Connecting androgen receptor signaling and the DNA damage response: Development of new therapies for advanced prostate cancer. Mol Cell Oncol 4:e1321167
Cortes CJ, La Spada AR (2018) X-Linked Spinal and Bulbar Muscular Atrophy: From Clinical Genetic Features and Molecular Pathology to Mechanisms Underlying Disease Toxicity. Adv Exp Med Biol 1049:103–133
Vichi P, Coin F, Renaud JP, Vermeulen W, Hoeijmakers JH, Moras D, Egly JM (1997) Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. EMBO J 16:7444–7456
Taniguchi JB, Kondo K, Fujita K, et al. (2016) RpA1 ameliorates symptoms of mutant ataxin-1 knock-in mice and enhances DNA damage repair. Hum Mol Genet 25:4432–4447
Ito H, Fujita K, Tagawa K, et al. (2015) HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol Med 7:78–101
Gabbita SP, Lovell MA, Markesbery WR (1998) Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem 71:2034–2040
Love S, Barber R, Wilcock GK (1999) Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 122 ( Pt 2):247–253
Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME (2010) Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci U S A 107:17757–17762
Murphy MP, Hartley RC (2018) Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. https://doi.org/10.1038/nrd.2018.174
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 499 kb)
Rights and permissions
About this article

Cite this article
Maiuri, T., Suart, C.E., Hung, C.L.K. et al. DNA Damage Repair in Huntington’s Disease and Other Neurodegenerative Diseases. Neurotherapeutics 16, 948–956 (2019). https://doi.org/10.1007/s13311-019-00768-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00768-7