
Abstract
COVID-19 infection originated in Wuhan, China in December 2019 and crippled human health globally in no time. The public health emergency required urgent efforts to develop and test the efficacy and safety of vaccines to combat the COVID-19 pandemic. The emergency use approval has been granted to COVID-19 vaccines before the completion of conventional phases of clinical trials. However, there is no comprehensive review of safety data reported from the vaccine trials, which is critical information to inform the policies in order to improve uptake of COVID-19 vaccines and mitigate the risk aversion perceived due to the COVID-vaccine side effects. This study aims to systematically review and synthesize the evidence on the safety data from the published COVID-19 vaccine trials. This study followed PRISMA guidelines. We searched three major electronic databases (PubMed, Embase, and Google Scholar) for published studies between Dec 2019 and 2020. Eligible study designs were randomized trials and pre-and post-intervention evaluations. Descriptive findings of included studies were reported stratified by target population, setting, outcomes, and overall results. From PubMed, Embase, WHO database, and Google Scholar screened titles and abstracts, 11 studies were identified in this review. Most of the reactions reported were mild to moderate whereas a few with severe intensity. All reactions resolved within 3–4 days. The commonly reported local adverse events were pain at the site of injection, swelling, and redness. The systemic reactions included fever, fatigue, myalgia, and headache. Some trials also reported laboratory derangements like decreased hemoglobin, increased bilirubin, altered SGOT and SGPT. None of these alterations were clinically manifested and were self-limiting. Few clinical trials reported serious adverse events, but they were unrelated to vaccination. This systematic review indicates that COVID-19 vaccines can be safe with no serious adverse events. However, long-term post-marketing surveillance data, particularly in high-risk vulnerable populations (elderly and those with co-morbidities, pregnant women, and children) is warranted to ensure the safety of COVID-19 vaccines.
Similar content being viewed by others

Introduction
The year 2020 will be remembered in modern history as the most challenging year in terms to combat SARS CoV-2, a viral infection causing intense respiratory illness. This pandemic burdened the health professionals globally and led to unprecedented paralysis of health care systems and global economic crisis [1]. Healthcare practitioners, researchers, and policymakers around the globe were thrown a challenge to deliver adequate prevention and treatment modalities to combat the pandemic. From the initial stage of this pandemic, scientists were focused on either repurposing the existing drugs or developing vaccines against COVID-19 [2]. The public and private sectors have united together to develop and test the efficacy and safety candidate vaccines. As of January 20th, 2021, one hundred seventy-three vaccines are in preclinical development and 64 in clinical trials [3]. By January 2021, emergency approval was granted to nine vaccines by regulatory authorities in different parts of the globe [4]. The safety data were published for 11 vaccines as interim reports or clinical trial reports [5,6,7,8,9,10,11,12,13,14,15,16,17]. At present, it is very crucial to establish the safety of the COVID-19 vaccines when emergency approval is being granted to these vaccines without completion of all phases of clinical trials. Since vaccines are still being tested in clinical trials, there is no systematic review to our knowledge that reported the profile of COVID-19 vaccines. Therefore, the present study reflects the safety generated from the results of published clinical trials of these vaccines.
Methods
Methods
We followed PRISMA guidelines to conduct this systematic review [18].
Search Strategy
To identify the clinical trials evaluating COVID-19 vaccines, PubMed/Medline, Embase, WHO database, and Google Scholar were systematically screened for medical literature. The articles published up to Dec 22, 2020, were included. The articles were screened by using the search strategy as “(COVID-19 Vaccine)”.
Study Selection
Randomized and nonrandomized Clinical trials assessing the safety of COVID-19 vaccines were included in the study. Trial protocols, observational studies, reviews, meta-analysis, systematic reviews, and commentaries were excluded. Duplicate studies among the clinical trials and trials with different primary objectives or including other interventions other than COVID-19 vaccines were also excluded.
All studies were reviewed for eligibility by two reviewers (RK, SD). Any disagreements and technical uncertainties were discussed and resolved with the third reviewer (JC).
Data extraction
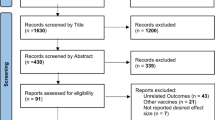
We extracted the vaccine name, type, manufacturer, study phase, number of participants, and safety data from the included clinical trials. The safety information was compared of various COVID-19 vaccines. The data were extracted by two authors (RK, SD) independently from the selected studies. The data were synthesized, disagreements were discussed, and differences were resolved between review authors (RK, SD, JC) (Fig. 1).

PRISMA flow chart of study selection for systematic review
Results
We found 196 records using the search term “COVID-19 vaccine trials”. Out of which 52 were full texts of clinical trials assessing the safety of COVID-19 vaccines. After the removal of 14 duplicate studies and articles reporting different primary outcomes (n = 27), finally, 11 studies were included in this systematic review (Fig. 1). As of 21st January 2021, results /interim reports of eleven clinical trials on COVID-19 vaccines were published [3]. The results could not meta-analyzed as the endpoints of these trials were different. The details of these vaccines and clinical trials reporting the safety of COVID-19 vaccines are summarized in Table 1.
The various vaccine platforms which are being explored to counter SARS CoV-2 are summarized in Table 2. A vaccine that evokes or fabricates the S protein neutralizing antibodies after vaccination is the prime mechanism of action of the majority of the vaccine candidates being developed.
CoronaVac (Sinovac Life Sciences, Beijing, China)
Phase I and phase II randomized, double-blind, placebo-controlled clinical trials conducted in Suining County of Jiangsu Province, China which contained a total of 743 participants with 144 healthy individuals aged 18–59 years were enrolled in the phase 1 trial and 600 in the phase 2 trial. In phase 1 trial, based on the day of the second dose of vaccine, there were two cohorts: days 0 and 14 vaccination cohort (N = 72) and days 0 and 28 cohort (N = 72). In each cohort, there were two blocks with 36 participants in each block. In block 1, participants were randomized to receive either 3 μg of test vaccine (N = 24) or placebo (N = 12) whereas in block two the participants were randomized to 6 μg of test vaccine (N = 24) or placebo (N = 12) [16].
In phase 2, 600 participants were enrolled with 300 participants each in two cohorts, days 0 and 14 vaccination cohort and days 0 and 28 vaccination cohort. In each cohort, participants were randomized to receive either 3 μg (N = 120) of test vaccine or 6 μg (N = 120) of test vaccine or placebo (N = 60). In phase 1, out of 144 participants enrolled, one patient withdrew from the trial, and thus 143 participants who were administered at least one dose of the investigational product were included in the safety analysis. The safety endpoint for both the phases was the occurrence of adverse reactions within 28 days of vaccination in participants receiving at least one dose of the test vaccine [16].
The incidence of adverse reactions on day 0 and 14 cohorts were 7 of 24 (29%), 9 of 24 (38%), and 2 of 24 (8%) in 3 μg, 6 μg, and placebo group respectively. In the days 0 and 28 cohorts, the incidence of adverse reactions was 3 of 24 (13%),4 of 24 (17%), and 3 of 23 (13%) in the 3 μg, 6 μg, and placebo groups respectively. Among the local adverse reactions, injection-site pain was the commonest. In the day 0 and 14-day vaccination cohort, it was reported in the four (17%), five (21%) one (4%) patient in the 3 μg, 6 μg, and placebo group respectively. Within the cohort of 0 and 28-day vaccination, injection site pain was reported in the three (13%) each in 3 μg, 6 μg, and placebo groups. The majority of the adverse reactions reported were grade 1 in terms of severity and almost all patients recovered within 48 h of occurrence, except single case of severe adverse reaction was reported in the 6 μg group of 0 and 14-day cohort where the participant had acute hypersensitivity reaction manifested as urticaria. The patient recovered within 3 days on the administration of chlorphenamine and dexamethasone. It was not followed by a similar reaction after the second dose. No serious adverse events were reported in phase 1. There were clinically significant alternations in the laboratory indicators three days post-vaccination in 10 out of 143 (7%) in phase 1 participants, these derangements were considered to be unrelated to the vaccination [16].
In phase 2, the overall incidence of adverse reaction in the 0 and 14-day cohort were 40 of 120 (33%), 42 of 120 (35%), and 13 of 60 (22%) in the 3 μg, 6 μg and placebo group respectively and in the days 0 and 28 cohort, 23 of 120 (19%), 23 of 120 (19%) and 11 of 60 (18%) in 3 μg group, 6 μg group and placebo respectively. The commonest local adverse reaction was injection site pain which was observed in 25 of 120 (21%), 31 of 120 (26%), 6 of 60 (10%) in the 3 μg, 6 μg and placebo group respectively in the 0 and 14 days vaccination cohort and 12 of 120 (10%), 13 of 120 (11%), 6 of 60 (10%) in 3 μg, 6 μg and placebo group respectively in the days 0 and 28 days cohort. As in the case of phase 1, most of the adverse reactions were of grade 1 severity, and the affected participants recovered within 48 h. No serious adverse event was reported in phase 2 [16].
Sinopharma whole virus vaccine
The interim analysis results were published for phase 1 and phase 2, double-blind, randomized, placebo-controlled clinical trial conducted in Henan Province, China. There were in total 320 healthy participants aged 18–59 years with 96 participants and 224 participants in phase 1 and phase 2 trials respectively. In phase 1, the assignment of the participants was done randomly to one of the three-dose groups (low dose 2.5 μg, medium-dose 5 μg and high dose 10 μg group) or the control group, with 24 participants in each group. The intervention was administered intramuscularly at days 0, 28, and 56 days. In phase 2, there were 2 cohorts: 0 and 14-day program (N = 112) and 0 and 28-day program (N-112). In each cohort, the assignment of the participants was done randomly to either the medium-dose 5 μg group (N = 84) or to the control group (N = 28 in each cohort). The primary safety outcome was taken as the occurrence of combined adverse reactions after seven days of each dose of vaccination [14].
The overall combined adverse events reported after 7 days of vaccination for phases 1 and 2 were 48 of 320 (15%). Distinctly in phase 1, the adverse reaction was observed in the 5 of 24 (20.8%) in low dose, 4 of 24 (16.7%) in medium dose, 6 of 24(25.0%) in high dose, and 3 of 24 (12.5%) in control group. 7 In phase 2, in the cohort days 0–14 program, the adverse reactions were reported in 5 of 84 (6.0%) for medium dose 5 μg and 4 of 28 (14.3%) for the control group; the adverse reactions reported for the second cohort were 6 of 84 (19.0%) in medium dose 5 μg dose group and 5 of 28 (17.9%) in the control group [14].
The most common adverse reaction reported was the injection site pain and fever. Injection site pain was suffered by 14 of 96 (14.5%) and 21 of 224 (9.37) in phase 1 and phase 2 respectively. Fever was observed in 2 of 96 (2.0%) in phase 1 and 8 of 224 (3.57) in phase 2 [14].
In phase 1, laboratory safety testing was performed before and 4 days after vaccination. A transient derangement in the laboratory parameters such as total lymphocyte count: 2 of 24 (8%) 2.5 μg dose group; 1 of 24 (4.2%) in 5 μg dose group, 1 of 24 (4.2%) in 10 μg dose group, and 2 of 24 (8.4%) in the control group was observed. Total bilirubin was raised in 1 of 24 (4.2%) in low and medium dose, 3 of 24 (12.5%) in high dose, and none in the control group. Urinary red blood cells were abnormal in 2 of 24 (8.3%) in the low dose, 1 of 24 (4.2%) in medium dose, 4 of 24 (16.7%), and 2 of 24 (8.3%) in the control group. All these altered laboratory parameters self-resolved without any treatment [14].
It is mentioned in the published article that there were four severe adverse events incidents during the follow-up and they were concluded to be not related to the vaccination. However, the details of these reactions could not be found in the published article as well as in the supplementary documents provided [14].
BBIBP-CorV Vaccine
This was a randomized, double-blind, placebo-controlled, phase 1 and phase 2 trial conducted in Henan Province, China. In the phase 1 trial, 192 healthy volunteers of age 18–80 were divided into two groups: 18–59 years (N = 96) and above age 60 years (N = 96). The participants of both groups were randomly assigned (1:1:1:1) to a 2 μg cohort, 4 μg cohort, or 8 μg cohort and placebo. In each cohort, 24 participants were administered test vaccine and 8 were given placebo in two doses on days 0 and 28. Two participants withdrew from the trial before completion. In phase 2, 448 healthy participants aged 18–59 year were randomly assigned to administer vaccine or placebo in a single dose of 8 μg on day 0 or two dosing schedules of 4 μg on days 0 and 14, 0 and 21 or 0 and 28 with 112 participants in each cohort, 84 receiving vaccine dose and 24 in the placebo group. Four participants from the dosing schedule 4 μg on days 0 and 21 withdrew from the trial and 1 from the dosing schedule 4 μg days 0 and 28 [15].
In the phase 1 trial, the adverse reactions were reported within 7 days after vaccination in 42 of 144 (29%) out of 144 participants who received the vaccine in comparison to 8 of 48 (17%) volunteers who received a placebo. In the group aged 18–59, in 2 μg cohort adverse reactions were reported in 11 of 25 (46%) with 2 μg dose compared to 3of 8 (38%) in placebo; 8 of 24 (33%) with 4 μg compared to 2 of 8 (25%) in the 4 μg cohort, and 11 of 24 (46%) with 8 μg compared to 1 of 8 (13%) with placebo. In the age group above 60 years of age, the adverse reactions were in 2 μg cohort:1 of 24 (4%) compared with 1 of 8 (13%) with placebo; in 4 μg cohort: 6 of 24 (25%) with test vaccine compared to zero in placebo; in 8 μg cohort: 5 out of 24 (21%) with the vaccine in comparison to 1 of 8 (13%) with placebo. The most common local adverse reaction reported in both age groups was pain at the injection site with an overall occurrence of 34 of 144 (24%) participants in comparison to placebo were 3 of 48 (6%) participants reported adverse reactions. In the participants who received test vaccine in the age group 18–59, the pain was reported in 9 of 24 (38%) with 2 μg, 7 of 24 (29%) with 4 μg, and 9 of 24 (38%). Swelling at the site of injection was reported by the 1 of 72 (3%) and itch in 1 of 72 (1%) was reported [15].
In the vaccine participants in age 60 years and above, pain at the site of injection was reported by 2 of 24 (4%) in 2 μg group, 4 of 24 (17%) in 4 μg group, and 4 of 24 (17%) in 8 μg group. Another adverse reaction reported at the site of injection by this age group was in duration in 2 (3%) out of 72 participants [15].
In case of systematic adverse reactions, fever was most commonly reported in 5 of 144 (4%) vaccine recipients in comparison to 3 (6%) out of 48 placebo recipients. In the 18–59 age group, fever was observed in 1 of 24 (4%) in 2 μg cohort, 1 of 24 (4%) in 4 μg cohort and 2 of 24 (8%) in 8 μg cohort. The other systemic adverse events reported in this age group were fatigue: 2 in 72 (3%), nausea 1 of 72 (1%) inappetence in 1 of 72 (1%), vomiting 1 of 72 (1%), constipation 1 of 72 (1%), headache 1 of 72 (1%) and itch 1 of 72 (1%). In the age group 60 years (N = 72): fever in one (1%) and fatigue in one (1%) was reported in the 8 μg cohort; joint pain in 1 (1%) and headache in one (1%) was reported in 4 μg cohort. Muscle pain was reported by one (1%) in the placebo group. All adverse reactions encountered in phase 1 were mild or moderate in severity and no serious adverse drug reaction was reported within 28 days after vaccination. Laboratory derangements of mild to moderate severity such as abnormal hemoglobin 1 (1%), blood urea nitrogen 1 (1%), blood glucose 7 (10%), serum total bilirubin 14 (19%), urinary glucose 1 (1%), urinary protein 1 (1%) were observed in the group 18–59 receiving the vaccine (N = 72). In the group aged 60 and above abnormal hemoglobin 3 (4%), white blood cells 1 (1%), ALT 2 (3%), AST 4 (6%), blood urea nitrogen 4 (6%), blood glucose 10 (14%), serum total bilirubin 9(12%), and urinary protein 2 (4%) of mild or moderate severity were reported [15].
In the phase 2 trial, the overall adverse reaction was observed in 76 of 336 (23%) vaccine recipients in comparison to 19 of 112 (17%) with placebo. The overall adverse reactions were 33 of 112 (33%) in vaccine recipients in comparison to 3of 28 (11%) in placebo recipients in the 8 μg day 0 group; 18 of 84 (21%) in the vaccine in comparison to 5 of 28 (18%) in placebo in the 4 μg days 0 and 14; 15 of 84 (18%) in the vaccine in comparison to 5 of 28 (18%) in placebo recipients in the 4 μg days 0 and 21 group; 10 of 84 (12%) in vaccine and 6 of 28 (21%) in the placebo recipients in the 4 μg 0- and 28-day cohort. As in phase 1, the most common adverse reaction was pain at the injection site in 53 of 336 (16%) vaccine recipients in comparison to 4 of 112 (4%) in the placebo group. The most common systematic adverse reaction in the group receiving the vaccine was fever in 7 of 336 (2%). All reactions in phase 2 were also of mild to moderate severity [15].
ChAdOx1 n CoV-19 (AZD1222) Vaccine
It consisted of 4 multicentric trials: COV001 phase 1 trial in the UK (n = 1077), COV002 in the UK (n = 10,673), COV003 in Brazil (10,002), and COV005 in South Africa (n = 1049). These were randomized controlled trials, three of them were single-blinded and COV005 was double-blinded. In these trials, the test vaccine ChAdOx1 CoV-19 was compared with the control MenACWY vaccine. The interim analysis of the safety and efficacy of the pooled data of these trials was published [6, 13].
In COV001 is a continuing single-blinded phase 1/2 study with 1077 healthy participants were randomized (1:1) to receive either test vaccine ChAdOx1 n CoV-19 (N = 544) in a dose of 5 × 1010 viral particles or to the control MenACWY (N = 533). In the COV002 trial, there were 5489 participants in the test vaccine group and 5184 in the control group. In the vaccine group, there were two dose cohorts: one dose cohort received 2.2 × 1010 (low dose) as the first dose followed by a booster dose of 5 × 1010 (standard dose) and another cohort received two standard doses. Each of these dose groups was compared with the control vaccine groups. In the COV003 group, two doses of 3.5 − 65 × 1010 of test vaccine (N = 5000) were administered 12 weeks apart and compared with control (N = 5002). In COV005, two doses of test vaccine (N = 1008) were administered in 3.5 − 6.5 × 1010 viral particles injected 4 weeks apart and compared with the control vaccine (N = 1005) [6, 13].
For the interim safety analysis, data of 12,021 participants were included from the test vaccine group (534 from COV001, 5479 from COV002, 5000 from COV003, and 1008 from COV005) and 11,724 from the control group (533 in COV001, 5184 in COV002, 5002 in COV003 and 1005 in COV005) [6, 13].
There were 168 serious adverse events (SAE) of 23,745 (0.7%) in the trial participants, 79 of 12,021 (0.7%) in the ChAdOx1 nCoV group, and 89 of 11,723 (0.8%) in the participants in the control group. Some serious adverse events as per MedDRA system organ class belonged to cardiac disorders (5 in test vaccine, 6 in control), eye disorders (2 in test vaccine, zero in control), gastrointestinal disorders ( 8 in test and 11 in control), infections and infestations (18 in test and 27 in control), neoplasm benign, malignant and unspecified (4 in test and 5 in control), nervous system disorders (7 in test and 4 in control), musculoskeletal and connective tissue disorders ( 5 in test and 2 in control), renal and urinary disorders (4 in test and 6 in control), reproductive system and breast disorders (7 in test and 2 in control) [6, 13].
The number of total adverse events was 175 (84 in the test vaccine group and 91 in control), out of these only three were considered to be related to the test or control vaccine. This included hemolytic anemia after 10 days of administration of control vaccine, a case of transverse myelitis 14 days after a booster dose of the experimental vaccine, fever higher than 40 °C reported 14 days after vaccination. There were four non-COVID-19 deaths, three in the control group and 1 in the experimental vaccine group but these were considered unrelated to the vaccines. The cause of death determined for these deaths were blunt force trauma, fungal pneumonia, homicide, and road traffic accident. Ten participants were hospitalized due to COVID-19 infection 21 days after the first dose, two had severe infections including one death. All these 10 cases were from the control group [6, 13].
Adenovirus Type-5 (Ad5)-Vectored COVID-19 Vaccine
It was a phase 1 open-label, non-randomized, single centered, dose-escalation study with 108 participants which were recruited for three dose groups: low dose 5 × 1010 (n = 36), middle dose 1 × 1011 (n = 36) and high dose 1.5 × 10 11 (n = 36). The primary safety outcome was any adverse events within 7 days of vaccination [17].
The phase 2 randomized, double-blind placebo-controlled single centered trial of AD5-vectored vaccine for COVID-19 was conducted in China. Total 508 healthy adults, more than 18 years were enrolled with 382 in the two doses of test vaccine group (1 × 1011 viral particles, n = 253; 5 × 1010 viral particles, n = 129) and 126 in the placebo control group. The primary objective of this study was to evaluate the immunogenicity and safety of the test vaccine. The primary outcome for the safety evaluation was the incidence of adverse reactions within 14 days of immunization [17].
In phase 1, there was at least one adverse reaction in each group 87 of 108 (81%) with 30 out of 36 (83%) in the low dose group, 30 of 36 (83%) in the middle dose group, and 27 of 36 (75%) in the high dose group. The most common local adverse reaction was pain at the site of injection in 58 of 108 (54%) with 17 of 36 (47%) in the low dose group, 20 of 36 (56%) in the middle dose group, and 21 of 36 (58%) in the high dose group. The most common overall systemic adverse reactions were fever 50 of 108 (46%), fatigue 47 of 108 (44%), headache 42 of 108 (39%), and muscle pain 18 of 108 (17%). Fever occurred in 15 of 36 (42%) in the low dose group, 15 of 36 (42%) of the middle dose group, and 20 of 36 (56%) in the high dose group. Muscle pain was reported 7 of 36 (10%), 3 of 36 (8%), and 8 of 36 (22%) in low dose group, middle dose group, and high dose group respectively. Headache was reported in 14 of 36 (39%) in the low dose group, 11 of 36 (31%) in the middle dose group, and 17 of 36 (47%) in the high dose group. The majority of the adverse reactions were of mild to moderate intensity. Severe adverse reaction of high fever along with fatigue, dyspnoea, and pain in muscles was reported in nine participants with 2 of 36 (6%) in low dose group, 2 of 30 (6%) in the middle dose, and 5 of 36 (14%) in high dose group. Joint pain and severe fatigue were reported in high-dose participants. All these adverse reactions were self-limiting. On day seven after vaccination, the laboratory derangements were of mild to moderate including increase in bilirubin in 9 of 108 (8%), rise in ALT in 10 of 108 (9%), and fasting hyperglycemia in 4 of 108 (4%) [17].
In the phase 2 trial, in the test vaccine group, the solicited adverse reactions were reported 14 days post-vaccination in 183 of 253 (72%) in 1 × 1011 and 96 of 129 (74%) in 5 × 1010 in comparison to the 46 of 126 (37%) in the control group. In the 1 × 1011, the systemic solicited adverse reactions were reported in 34% with fever in 16% and headache in 28% and 5 × 1010 doses, the systemic solicited adverse reactions were reported in 42% with fever in 32%, headache in 29%. The local adverse reaction included injection site pain 57% in 1 × 1011 and 56% in 5 × 1010. Most of the adverse reactions were mild or moderate in severity. The severe adverse reactions of grade 3 were reported in 24 of 253 (9%) of 1 × 1011 and 1 of 129 (1%) of the 5 × 1010 vaccine group. The most common severe adverse reaction was fever in 20 of 253 ( 8%) in the 1 × 1011 dose group and 1 of 129 (1%) in the 5 × 1010. Increasing age, male gender, and preexisting Ad5 immunity were associated with a lesser incidence of fever after vaccination. These grade 3 reactions were self-limiting and resolved within 72–96 h without any intervention. The unsolicited adverse reactions reported within 14 days after vaccination were 19 of 129 (8%) in the 1 × 1011 dose group and 7 of 253 (6%) in the 5 × 1010 dose group and 7 of 126 (6%) in the placebo group. After 28 days of vaccination, 196 of 253 (77%), 98 of 129, and 61 of 126 (48%) reported at least one adverse reaction in the 1 × 1011 doses, 5 × 1010 doses, and placebo respectively. There were no serious adverse reactions reported in this study [17].
Sputnik Vaccine
These were two phase 1/2 non-randomized studies of two formulations (frozen and lyophilized) conducted in Russia on a heterologous COVID-19 vaccine compromising of a recombinant adenovirus type 26 (rAd26-S) vector a recombinant adenovirus type 5 (rAd5-S) vector. It included 76 healthy participants of age 18–60 with 38 participants in each study. In each study, in phase 1, nine participants were administered rAd26-S or rAd5-S and in phase 2, 20 participants were administered rAd26-S on day 0 and rAd5-S on day 21 in phase 2. The primary safety outcome was measured by the number of participants developing adverse reactions [9].
In both the studies, the most common adverse reaction was pain at the site of injection 44 of 76 (58%), headache 32 of 75 (42%), hyperthermia 38 of 76 (50%), asthenia 21 of 76 (28%) and muscle and joint pain 18 of 76 (24%) [9].
In phase 1 for Gam-COVID-Vac formulation, 8 of 9 (89%) and 3 of 9 (22%) had hyperthermia in rAd26-S and rAd5-S respectively; headache 6 of 9 (67%) in rAd26-S and 3 of 9 (33%) in rAd5-S. For Gam-COVID-Vac-Lyo, 1 of 9 (11%) each of rAd26-S and rAd5-S; headache in 3 of 9 (33%) and 4 of 9 (44%) for rAd26-S and rAd5-S respectively [9].
In phase 2, for 20 of 20 (100%) and 7 of 20 (35%) hyperthermia, 11 of 20 (55%) and 5 of 20 (25%) headache, 11 of 20 (55%), and 4 of 20 (20%) asthenia, 5 of 20 (25%) and 6 of 20 (30%) muscle and joint pain for Gam-COVID-Vac and Gam-COVID-Vac-Lyo respectively. Additional adverse reactions diarrhea 3 of 20 (15%), rhinorrhoea 4 of 20 (20%), loss of appetite 1 of 20 (5%), malaise 2 of 20 (10%) were observed for Gam-COVID-Vac formulation.
Transient alternation in laboratory variables was noted for both formulations. For Gam-COVID-Vac, 9 of 9 (100%) for both rAd26-S and rAd5-S in phase 1 and 20 of 20 (100) in phase 2. For Gam-COVID-Vac-Lyo, 7 of 9 (78%) for rAd26-S and 6 of 9 (67%) for rAd5-S in phase 1 and 18 of 20 (90%) in phase 2 [9]. The majority of adverse events were mild and there were no serious adverse events reported in both the trials. None of the adverse reactions led to the withdrawal of study participants. Most of the adverse reactions occurred after the second vaccination [9].
Janssen Ad26.COV2.S COVID-19 Vaccine
It is a multi-centric phase 1/2 a randomized double-blind placebo-control trial. The interim results of three cohorts 1a and 1b of participants aged 18–55 (n = 402) and cohort 3 of aged 65 and above (n = 403) were published to report the safety of Ad26 COV2-S. The test vaccine was administered in two doses 5 × 1010 (low dose) or 1 × 1011 (high dose) viral particles per vaccination, as a single dose or in two doses spaced at 56 days. The participants of both cohorts were randomly assigned to any of the vaccination groups: low dose followed by low dose, low dose followed by placebo, high dose followed by high dose, high dose followed by placebo, and placebo followed by placebo. The endpoint for safety was the occurrence of adverse reactions 28 days after vaccination, local and systemic reactogenicity for 7 days post-vaccination, and serious adverse reactions during the study [12].
The local adverse reactions in cohort 1 were 288 of 402 (72%) and cohort 3 were 183 of 394 (46%) participants. Out of these, 235 (58%) in cohort 1a and 1b and 108 (27%) in cohort 3 were of mild to moderate intensity. In cohort 1, solicited local adverse events were observed in 103 of 162 (64%) in low dose, 123 of 158 (78%) in high dose, and 7 of 82 (9%) in the placebo group. In cohort 3, the solicited adverse reaction was reported in 66 of 161 (41%), 68 of 161 (42%), 11 of 81 (14%) in the low dose, medium dose, and high dose respectively [12].
In the three participants, the tenderness at the site of infection was of grade 3 severity. The commonest local adverse reaction reported was injection site pain. The solicited systemic adverse reactions were recorded in 64% (258) were mild to moderate of grade 1 and grade 2 but in 11% (46) participants observed grade 3 adverse reactions [12].
In both the cohorts, the systemic adverse reaction was of mild to moderate intensity and the commonly reported were headache, myalgia, and fatigue. In cohort 1, 105 of 162 (65%) in low dose group, 133 of 158 (84%) in high dose, and 21 of 82 (26%) in the placebo group. In cohort 3 the solicited systemic adverse events were reported in 74 of 162 (46%), 88 of 158 (55%), and 19 of 82 (23%) in the low dose, high dose, and placebo group respectively. In cohort 1, 15 of 162 (9%), 32 of 158 (20%), and zero in low dose, high dose, and placebo respectively, and in cohort 3, solicited systemic adverse reactions were reported in 1 of 162 (1%) in low dose, 4 of 158 (4%) in high dose and zero in the placebo group [12].
The overall reporting of fever was 19% (76) and the grade 3 fever was recorded in 5% (22) patients. In cohort 1, fever was reported in 25 of 162 (15%) in low dose and 62 of 158 (39%) in high dose with grade 3 fever was reported in 5% in low dose and 9% in high dose. In cohort 3, fever was reported in 7 of 162 (4%) in low dose and 14 of 158 (9%) in high dose. No incident of fever was reported in the placebo group. Two days post-vaccination, fever was reported which got resolved within 1–2 days [12].
In cohort 3, 183 of 394 (46%) participants reported solicited adverse events. The local solicited adverse reaction was reported in 27% (108 of 394) mostly of mild or moderate intensity with one participant reporting swelling and erythema of grade 3. In cohort 3 also the commonest local adverse reaction was injection site pain. The solicited systemic adverse reactions were reported in 36% (140 of 394), mostly of grade 1 and grade 2 severity [12].
The unsolicited adverse events with low dose were reported in 34 of 162 (21%), 56 of 158 (35%) in high dose, and in 14 of 82 (17%) in the placebo group in cohort 1 and cohort 3, 27 of 162 (17%) in low dose, 38 of 158 (24%) and 13 of 82 (16%) in the placebo group [12].
Five serious adverse events were reported: one case of hypotension, one of bilateral nephrolithiasis in a patient with a history of kidney stones, one of legionella pneumonia, one case of worsening of multiple sclerosis, and one case of fever leading to hospitalization. All these SAE were deemed unrelated to the vaccine except fever which resolved within 12 h [12].
Novarax NVX-CoV2373 Vaccine
It was a randomized placebo-controlled phase 1/2 trial of NVX-CoV2373 vaccine with doses of 5 μg and 25 μg, with or without Matrix-M1 adjuvant. There were 131 healthy participants of 18 to 59 years were enrolled in the study, out of which 23 received placebo (Group A), 25 received 25 µg of test vaccine (Group B), 29 received 5 µg of test vaccine along with MatrixMI (Group C), 28 received 25 µg of test vaccine plus Matrix-M1 (Group D) and 26 received a single dose of test vaccine plus Matrix-MI followed by a single dose of placebo (Group E). Thus, out of 131, 83 participants received the vaccine with adjuvant, 25 without adjuvant, and 23 were given a placebo. All 131 participants were given two doses at the gap of 21 days. In the study published the results of the phase 1 trial conducted in Australia are published. The primary safety endpoints were the number and percentage of participants with solicited and systemic reactogenicity after 7 days of vaccination and their intensity and duration between days 0 to 7 and days 21 to 28 after vaccination [8].
The adverse reactions after the first vaccination with either absent or of mild intensity. The local adverse events were 100%, 96%, 89%, 84%, and 88% and the systemic was observed in 91%, 92%, 96%, 68%, and 89% in the group A, B, C, D, and E respectively. Two participants, one each in group D and E had severe adverse events- headache, fatigue, and malaise. After the second vaccination, similar to the first dose the adverse reaction was absent or of mild intensity with local adverse reactions as 100%, 100%, 65%, 67%, and 100%; systemic adverse reactions as 86%, 84%, 73%, 58%, and 96% respectively. In one participant of group D, there was a severe local event of tenderness and in 8 participants, one or two from each group had a severe systemic reaction and the most common systematic reactions were fatigue and joint pain. Fever of 38.1 °C was reported in one participant of group D. All the adverse reactions resolved within 2 days of occurrence [8].
The laboratory derangements of grade 2 or higher were observed in 13 of 131 (10%) in total with 9 after the first dose and 4 after the second dose of vaccine. These were not associated with any clinical manifestation. Hemoglobin was found to be lowered (Grade 2) in six participants which resolved with 7 to 21 days. In four participants with one from the placebo group, there were increased liver enzymes after the first dose and vaccine and resolved within 7 to 14 days. There was no serious adverse reaction reported for this study [8].
Moderna (mRNA-1273) Vaccine
This was a phase 1, dose-escalation, open-label clinical trial to assess the safety and efficacy of mRNA-1273 (Moderna) vaccine in 45 healthy adults of 18 to 55 age. The test vaccine was administered in two doses at the gap of 28 days in the dose of 25 μg, 100 μg, and 250 μg. There were 15 participants in each dose group. The safety endpoint was the occurrence of any adverse event after seven days of each dose [7].
After both vaccinations, the common solicited systemic adverse reactions were of mild to moderate intensity (included headache, chills, fatigue, myalgia, and pain at the site of injection). Local adverse events were of mild to moderate intensity and the most commonly reported local reaction was pain at the site of injection. The systemic adverse reaction was in 5 of 15 (33%) in 25 μg, 10 out of 15 (67%) in 100 μg, and 8 of 15 (53%) in the 250 μg dose group. All the systemic reactions were mild and were common after the second vaccination with 7 of 13 (54%) in the 25 μg group, 15 of 15 (100%) in the 100 μg group, and 14 of 14 (100%) in 250 μg dose group. There was no incidence of fever in any participant after the first vaccination but after the second vaccination 6 out of 15 (40%) in 100 μg and 8 of 14 (57%) in the 100 μg dose group. One participant had a fever of 39.6 °C which was graded as a severe adverse reaction. One participant was withdrawn from the study due to the occurrence of transient urticarial after the first dose in the 25 μg dose group. There was no serious adverse reaction reported during this clinical trial [7].
BNT162b1 vaccine
In the phase 1 placebo control observer-blinded dose-escalation study there were 195 participants were administered either of the two formulations of the test vaccine- BNT162b1 or BNT162b2 in the dose10 μg, 20 μg and 30 μg, and 100 μg in two age groups (18 to 55 and 65 to 85 years). Out of 195 participants, cohort 1 (N = 105) was assigned to receive BNT162b1 or placebo and cohort 2 was assigned to receive BNT162b2 or placebo. Each cohort was further divided into two groups based on age: group 1 had patients aged 18–55 years (N = 60 in cohort 1 and N = 45 in cohort 2) and group 2 had patients aged 65 to 85 (N = 45 in cohort 1 and N = 45 cohort 2).
In group 1 (n = 60) of cohort 1, 12 participants each received 2 doses of 10 μg, 20 μg, and 100 μg and 12 received a single dose of 100 μg and 12 received one dose of placebo and 9 received a second dose of placebo. In group 2, 12 participants each received 2 doses of 10 μg, 20 μg, and 100 μg, and 9 received two doses of placebo. In cohort 2, the group 1 and 2: 12 each received two doses of BNT162b2 in 10 μg, 20 μg, 30 μg each and 9 received two doses of placebo.
There were in total 13 groups with 15 participants in each group. In each group, 12 participants received a test vaccine and 3 were administered a placebo. In phase 1, the primary safety endpoint was solicited local and systemic reaction and the use of the antipyretic drug within 7 days of administration of test vaccine or placebo [10].
Phase 2 was a placebo-controlled, observer-blinded, pivotal efficacy trial in which the participants received two doses of the vaccine at the gap of 21 days. In this study 43,548 patients were randomized, 21,720 received the test vaccine BNT162b1 and 21,728 received placebo. At the time of analysis, the safety data for 37,706 participants were available [11].
In phase 1, participants reported mild to moderate local adverse reactions at the injection site as most common in the BNT162b1. The local adverse reactions were found to be more common with the second dose. A similar pattern of adverse reaction was observed with BNT162b2. The systemic effect in the participants 18–55 of age who received 10 μg, 20 μg, or 30 μg of BNT162b1 reported fever and chills of mild-to-moderate intensity. In the patients 65 to 85 years of age, the systemic events were of milder intensity in comparison to the younger age group. Fatigue and headache were the most common systemic adverse reaction reported by older adults. Both the local and systemic reactions were dose depended. With the BNT162b2 the systemic events were milder. Severe adverse reactions like headache, fatigue, joint, and muscle pain were reported by the age group 18–55 but not by the older adults. The use of antipyretic or pain medications with both the formulation in both the age groups increased with an increase in dose. The use of these medications was lower with the BNT162b1. 50% and 8% of the reported adverse events were found to be associated with the vaccine or placebo respectively in the participants of 18–55 age group receiving 30 μg of BNT162b2. In the case of BNT162b1 dose 30 μg, the adverse events were found to be associated with the vaccine in 17% and 25% in 65–85 years and 18–55 years respectively. No serious adverse event was reported with both the formulations in any dose or age group [10].
In phase 2, the local reactogenicity subset included 8183 participants. The most commonly reported adverse reaction was mild to moderate intensity pain at the site of injection. The reporting of pain at the site of injection was less among participants above 55 years with 77% after the first dose and 66% after the second dose. The pain was reported to a lesser extent by the patients above age 55 (83% after the first dose in comparison to 14% with placebo and 78% after the second dose in comparison to 12% in placebo in age 16 to 55 and 71% after the first dose in comparison to 9% in placebo and 66% after the second dose in comparison to 8% with placebo in the age above 55. Other local adverse reactions included redness (11% in the age group 16–55 and 12% in the age above 55). After the second dose, the proportion of local adverse reactions did not change. The local reactions were mostly mild to moderate in intensity [11].
The systemic adverse events were more common after the second dose and were reported more by the younger participants of 16–55 age than the older vaccine recipients of age above 55. The systemic adverse events followed by the second dose were fatigue and headache. Headache was reported in 52% in vaccine recipients and 24% in placebo in the age group 18–55 and 59% and 14% in vaccine recipients respectively in the age group above 55. Fatigue was reported by 59% in vaccine recipients and 23% in the placebo group in a group of age 16–55 and 39% and 17% in vaccine and placebo group respectively in the participants above age 65. The severe systemic adverse reaction was reported in less than 2% of vaccine recipients after either dose except for fatigue and headache which was reported in 3.8% and 2.0% respectively after the second dose. Fever was reported by 16% and 11% of younger and older vaccine recipients respectively after the second dose [11].
The adverse event analysis after the first dose was done for 43,252 participants with 21,621 participants in the BNT162b2 group and 21,631 in the placebo group. The adverse events were reported more in the BTN162b2 recipients 5770 of 21,621 (27%) than the placebo recipients 2638 of 21,631. Lymphadenopathy was reported by 64 of 21,621 (0.3%) vaccine recipients and in 6 of 21,621 (0.15%) placebo group. Serious adverse events reported in the vaccine group were 126 of 21,621(0.6%) and 11 of 21,631 (0.5%) in the placebo group. Out of these only four (shoulder injury, right axillary lymphadenopathy, paroxysmal ventricular arrhythmia, and right leg paresthesia) were considered to be related to the vaccine. There were six deaths during the trial including two from the vaccine recipient group due to arteriosclerosis and cardiac arrest and four in the placebo group (2 due to unknown causes, one by myocardial infarction, and one due to hemorrhagic stroke). None of these deaths were considered to be related to trial interventions [11].
Covaxin (BBV152) Vaccine
It was a double-blinded randomized controlled phase 1 clinical trial conducted in India. The test vaccine BBV152 is formulated with two adjuvant-Algel and Algel-IMDG. Total 375 participants aged between 18 and 55 were randomized into four groups, three groups (n = 100) to be administered one of the three test vaccine formulations: 3 μg with Algel-IMDG, 6 μg with Algel-IMDG, 6 μg with Algel and an Algel only control arm (n = 75). The primary safety outcome the number of participants with local and systemic adverse reactions within 2 h, 7 days, 14 days and 28 days after vaccination [5].
The local and systemic adverse reactions reported after both doses of vaccine were mild or moderate in intensity. The local and systemic adverse events in the test vaccine and placebo group were equally distributed [5].
After the first dose, the solicited local adverse reactions were reported in five each of 100 (5%) in the 3 μg Algel-IMDG and 6 μg Algel-IMDG group and 3 of 75 (4%) in the control group. The solicited systemic adverse reactions were reported in the 5 of 100 (5%) in the 3 μg Algel-IMDG group and 14 of 100 (14%) in the 6 μg Algel-IMGD group and 7 of 75 (9%) in the algel only control group. The common adverse events across all groups included 17 of 375 (5%); headache 13 of 375 (3%); fatigue 11 of 375 (3%); fever 9 of 375 (2%) and nausea and vomiting 7 of 375 (2%). After both doses of vaccine, solicited local and systemic adverse reactions were reported by 17 of 100 (17%) in the 3 μg with algel-IMDG group, 21 of 100 (21%) in the 6 μg with algel-IMDG, and 14 of 100 (14%) in the 6 μg with algel group and 10 (13%) with algel only group. One patient each with fever and chills of severe intensity was reported in the 3 μg with algel-IMGD group. Both these events were considered unrelated to the test vaccine [5].
The important abnormal laboratory parameters after vaccination included derangements in bilirubin, SGOT, SGPT, Cholesterol C-reactive protein levels. These findings had no corroborating clinical manifestations [5].
There was one serious adverse event reported in the 6 μg with algel group due to which the patient was hospitalized. Five days post-vaccination the participant tested positive for SARS-CoV-2 and the symptoms included fever and headache. This event was not causally associated with the vaccine [5].
Discussion
In this review, we synthesized the safety data of eleven published clinical trials of COVID-19 vaccines. Due to the heterogeneity in the safety endpoints across trials, we could not perform meta-analysis. In this review, we found the adverse reactions reported in the 11 trials were mild to moderate with few severe reactions which were unrelated to the test vaccine. Common adverse events were pain at the site of injection, fever, myalgia, fatigue, and headache. Serious adverse events were reported in four trials: COVID-19 Vaccine AstraZeneca (AZD1222)—168 SAE with only three related to the vaccine; Ad26.COV2.S- four with none related to the testing vaccine; five with Comirnaty (BNT162b1) and one with Covaxin (BBV152) vaccine.
As per the United States Food and Drug Administration guidelines for “Emergency Use Authorization for Vaccines to Prevent COVID-19 Guidance for Industry October 2020, it is recommended that pharmaceutical company must inform the regulatory authority within 24 h of completion of any interim analysis which is intended to be used for attaining Emergency Use Approval (EUA) [19]. The EUA may be granted based on the interim analysis of clinical endpoint from phase 3 efficacy study and the safety data with a minimum follow-up of two months. This data should be supplemented with the safety data of all safety data of phase 1 and phase 2 including the details of serious adverse events, adverse events of special events and cases of severe COVID-19 infection, and longer safety follow-up [19].
Recognizing the urgent need for the COVID-19 vaccine, the analysis of the safety data of phase 1 and 2 is of prime importance for COVID-19 vaccines and in this pandemic scenario when the emergency use approval is being granted to COVID-19 vaccines. Thus, this systematic review of the safety data reported in clinical trial reports of COVID-19 was conducted to assess the probability of adverse events with the COVID-19 vaccines.
To our knowledge, this is the first study to systematically review the safety of COVID-19 vaccines and we have individually reviewed the published safety data of each vaccine and complied it in this review article. The limitation of this study is that the data is taken from the published articles/ interim reports and the supplementary data provided with them. These documents had limited information in the summarized format and the individual patient data was not available, thus there is a possibility to miss some important aspects.
Based on the systematic analysis of the published safety data of the eleven COVID-19, we conclude that these vaccines are safe. It is relevant to document that the Emergency Use Approval is different from marketing approval. At present all the COVID-19 vaccines are granted EUA and not marketing authorization. In the EUA, the vaccine is available in a controlled way and the government makes it available to the population on a priority basis [20]. Further safety data from a larger sample size and of longer duration are warranted to establish safety and attain licensing so that vaccine is available to the general population.
References
World Health Organisation: COVID-19 vaccines: safety surveillance manual. https://www.who.int/vaccine_safety/committee/Module_Regulatory_reliance.pdf?ua=1 (2020). Accessed 28 Jan 2021.
World Health Organisation: WHO coronavirus disease (COVID-19) dashboard. https://covid19.who.int/ (2021). Accessed 28 Jan 2021.
World Health Organisation: Draft landscape and tracker of COVID-19 candidate vaccines. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (2021). Accessed 22 Jan 2021.
Craven J. RAPS: COVID-19 vaccine tracker. https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-vaccine-tracker (2021). Accessed 23 Jan 2021.
Ella R, Vadrevu KM, Jogdand H, Prasad S, Reddy S, Sarangi V, et al. A phase 1: safety and immunogenicity trial of an inactivated SARS-CoV-2 vaccine-BBV152. J MedRxiv. 2020. https://doi.org/10.1101/2020.12.11.20210419.
Folegatti PM, Ewer KJ, Aley PK, Angus B, Becker S, Belij-Rammerstorfer S, et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020;396(10249):467–78. https://doi.org/10.1016/s0140-6736(20)31604-4.
Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, et al. An mRNA vaccine against SARS-CoV-2—preliminary report. New Engl J Med. 2020;383(20):1920–31. https://doi.org/10.1056/NEJMoa2022483.
Keech C, Albert G, Cho I, Robertson A, Reed P, Neal S, et al. Phase 1–2 trial of a SARS-CoV-2 recombinant spike protein nanoparticle vaccine. New Engl J Med. 2020;383(24):2320–32. https://doi.org/10.1056/NEJMoa2026920.
Logunov DY, Dolzhikova IV, Zubkova OV, Tukhvatulin AI, Shcheblyakov DV, Dzharullaeva AS, et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: two open, non-randomised phase 1/2 studies from Russia. Lancet. 2020;396(10255):887–97. https://doi.org/10.1016/s0140-6736(20)31866-3.
Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature. 2020;586(7830):589–93. https://doi.org/10.1038/s41586-020-2639-4.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. New Engl J Med. 2020;383(27):2603–15. https://doi.org/10.1056/NEJMoa2034577.
Sadoff J, Le Gars M, Shukarev G, Heerwegh D, Truyers C, de Groot AM, et al. Safety and immunogenicity of the Ad26.COV2.S COVID-19 vaccine candidate: interim results of a phase 1/2a, double-blind, randomized, placebo-controlled trial. J MedRxiv. 2020. https://doi.org/10.1101/2020.09.23.20199604.
Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397(10269):99–111. https://doi.org/10.1016/s0140-6736(20)32661-1.
Xia S, Duan K, Zhang Y, Zhao D, Zhang H, Xie Z, et al. Effect of an inactivated vaccine against SARS-CoV-2 on safety and immunogenicity outcomes: interim analysis of 2 randomized clinical trials. JAMA. 2020;324(10):951–60. https://doi.org/10.1001/jama.2020.15543.
Xia S, Zhang Y, Wang Y, Wang H, Yang Y, Gao GF, et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: a randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet Infect Dis. 2021;21(1):39–51. https://doi.org/10.1016/s1473-3099(20)30831-8.
Zhang Y, Zeng G, Pan H, Li C, Hu Y, Chu K, et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect Dis. 2020. https://doi.org/10.1016/s1473-3099(20)30843-4.
Zhu FC, Guan XH, Li YH, Huang JY, Jiang T, Hou LH, et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2020;396(10249):479–88. https://doi.org/10.1016/s0140-6736(20)31605-6.
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700. https://doi.org/10.1136/bmj.b2700%JBMJ.
Administration FaD: Emergency use authorization for vaccines to prevent COVID-19: guidance for industry. https://www.fda.gov/media/142749/download (2020). Accessed 31 Jan 2021
Food and Drug Administration: Emergency use authorization for vaccines explained. https://www.fda.gov/vaccines-blood-biologics/vaccines/emergency-use-authorization-vaccines-explained (2020). Accessed 31 Jan 2021.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaur, R.J., Dutta, S., Bhardwaj, P. et al. Adverse Events Reported From COVID-19 Vaccine Trials: A Systematic Review. Ind J Clin Biochem 36, 427–439 (2021). https://doi.org/10.1007/s12291-021-00968-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-021-00968-z