
Abstract
Ginsenoside Rg1 is efficient to prevent or treat mental disorders. However, the mechanisms underlying the effects of ginsenoside Rg1 on post-traumatic stress disorder (PTSD) are still not known. In this study, single-prolonged stress (SPS) regime, as well as injection of lipopolysaccharide (LPS), was used to produce PTSD-like behaviors in C57 mice, and the effects of ginsenoside Rg1 (10, 20, 40 mg/kg/d, ip, for 14 days) on PTSD-like behaviors were evaluated. Our results showed that ginsenoside Rg1 promoted fear extinction and prevented depression-like behaviors in both LPS and SPS models. Importantly, ginsenoside Rg1 alleviated LPS- or SPS-stimulated expression of pro-inflammatory cytokines (IL-1β and TNF-α), activation of astrocytes and microglia, and reduction of hippocampal synaptic proteins (PSD95, Arc, and GluA1). Ginsenoside Rg1 also reduced the increase of hippocampal Kir4.1 and GluN2A induced by PTSD regime. Importantly, reducing hippocampal astroglial Kir4.1 expression promoted fear extinction and improved depression-like behaviors in LPS-treated mice. Additionally, intracerebroventricular injection of TNF-α caused an impairment of fear extinction and promoted Kir4.1 expression in the hippocampus. Together, our study reveals novel protective effects of ginsenoside Rg1 against PTSD-like behaviors in mice, likely via promoting synaptic proteins, reducing Kir4.1 and TNF-α in the hippocampus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Post-traumatic stress disorder (PTSD) is a kind of abnormal mental reaction after serious stresses such as war, violent crime, traffic accidents, and natural disasters. The incidence rate of PTSD is increasing year by year. Remarkably, coronavirus disease 2019 (COVID-19) worldwide would leave the patients and medical staff serious mental illness, especially PTSD [1]. The symptoms of PTSD mainly include flashbacks, nightmares, and severe anxiety. Hypersensitivity of fear and depression were also observed in PTSD patients [2]. Cognitive behavioral therapy is the primary selection for the treatment of PTSD, and symptoms-relieving drugs including antidepressant, antianxiety, and anticonvulsant drugs are also effective to relieve the symptoms, but leaving severe adverse effects [2].
Fear extinction refers to the decrease in conditioned fear responses that occur with repeated presentation of the unreinforced conditioned fear stimulus [3]. Fear extinction enables the animals to adapt the traumatic events. However, impairment of fear extinction usually results in severe mental disorders [4]. PTSD is caused by “panic” and “fear” stress and characterized by abnormal fear extinction [5, 6]. Anti-inflammatory therapies that are currently under investigation intended to dampen the cytokine storm of severe COVID-19 infection [7]. Intriguingly, inflammation in fear- and anxiety-based disorders has gained interest as growing literatures indicate that pro-inflammatory markers can directly modulate mental activities [8, 9]. Indeed, high concentrations of inflammatory signals, including cytokines and C-reactive protein, accumulate in the brain of PTSD animals [9].
Inwardly rectifying potassium channel (Kir4.1) is the main potassium channel expressed exclusively in glial cells in the central nervous system (CNS) that regulates the extracellular potassium, transports glutamate, and maintains neuronal homeostasis [10]. Abnormality of Kir4.1 in the brain often participates in the pathophysiological processes of neurological disorders [11, 12]. Astroglial Kir4.1 channel is supposed to be a potential therapeutic target for epilepsy and mental disorders [13, 14]. Moreover, astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression [12], and specific inhibition of Kir4.1 could rapidly eliminate depression-like behaviors [15]. Especially, Kir4.1 performed critical roles in modulating astrocyte-neuron interaction [16]. However, the function of astroglial Kir4.1 in fear extinction in PTSD animals has not been reported.
Ginseng is widely used in the treatment of mental disorders for its functions of “tonifying the five zang organs, calming the mind and soul” [17]. Ginsenoside Rg1 is one of the main active ingredients found in ginseng with various pharmacological activities, including anti-inflammation, anti-oxidation, neuroprotection, etc. [18,19,20]. Ginsenoside Rg1 is well-known for its function in remedying hippocampal synaptic plasticity and memory in Alzheimer’s disease (AD) [21]. In a previous study, we have also reported that chronic administration of ginsenoside Rg1 could promote memory and weak theta-burst stimulation-induced long-term potentiation in middle-aged mice [22]. However, whether ginsenoside Rg1 affects PTSD-like behaviors is still not known. In this study, lipopolysaccharide (LPS)-treated and single-prolonged stress (SPS) mice models were utilized to evaluate the protection of Rg1 against PTSD-like behaviors and to assess the mechanisms.
Materials and Methods
Drugs and Reagents
Ginsenoside Rg1 was obtained from Sigma (HPLC ≥ 98%, Shanghai, China). AAV2/5-GFAP-EGFP-shRNA Kir4.1 and shRNA scramble were purchased from Brain VTA (PT-2054, Wuhan, China). The sequence of shRNA Kir4.1 was 5′-GCCAAGUUCGCACTTCCTACC-3′. Rg1 were dissolved in dimethyl sulfoxide (DMSO) and suspended in saline (DMSO concentration was lower than 0.1%), and mice in control group were injected with the same volume of saline (containing the same amount of DMSO).
Animal Models
Male C57BL/6 mice (22–25 g, 2-month old) were obtained from the Animal Center of the Anhui Medical University (Hefei, China) and housed in plastic cages at room temperature with 12:12-h light-dark cycle and a relative humidity of 45–65%. The animals were fed with rodent pellets and water ad libitum and allowed to acclimatize for at least 7 days before use. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the Ministry of Science and Technology of China (2006), and the experimental procedures were approved by the Ethics Committee of the Anhui University of Chinese Medicine (Hefei, China; Approval No.: SYXK2014-0512). Efforts were made to minimize animal suffering and reduce the number of mice used. The behavioral tests were conducted at 9:00–10:00 am each day.
Experimental Protocol 1
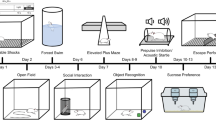
LPS (Sigma, St. Louis, MO, USA) was dissolved in saline. I.p injection of 2 mg/kg LPS was used in our experimental settings [23]. Mice were randomly allocated into five groups: a control group, a LPS group, and three Rg1-treated groups (10, 20, 40 mg/kg) (N = 7 in each group). Mice in control and model groups received the same volume of saline from day 8 to day 21, while the mice in Rg1-treated groups received 10, 20, and 40 mg/kg/d, respectively, from day 8 to day 21 (i.p., one injection per day). Except for the control group, all mice received LPS on day 22 (Fig. 1a).

Schematics illustrating the study design. Experimental procedure for the study of intraperitoneal injection of ginsenoside Rg1 on fear extinction and depression-like behaviors in lipopolysaccharide (LPS)-treated and single-prolonged stress (SPS) mice. (a) The animals were habituated for 7 days and administered with ginsenoside Rg1 for 14 days. LPS was intraperitoneally injected, and the behaviors were tested. Thereafter, hippocampi were collected. (b) The animals were habituated for 7 days, and SPS model was produced on the 8th day. Twenty-four hours later, ginsenoside Rg1 was administered for 14 days, and the behaviors were tested, or their hippocampi were collected
Experimental Protocol 2
The SPS protocol was conducted as previously described [24]. Briefly, the mice were restrained for 2 h and then immediately underwent a 20-min forced swimming in 25 °C water. Following a 15-min recuperation period, the mice were exposed to ether until the loss of consciousness. The mice were then left at home cage. This experiment was divided into four groups: a control group, a SPS group, a SPS + Rg1 group, and a control + Rg1 group (N = 7 in each group). The mice in SPS + Rg1 group and control + Rg1 group received 20 mg/kg/d Rg1 (i.p., one injection per day) from day 8 to day 21, while the mice in control and SPS groups received the same volume of saline (Fig. 1b).
Experimental Protocol 3
Fourteen mice were habituated for 7 days and placed on stereotaxic instrument (RWD, Shenzhen, China) under isoflurane anesthesia. AAV2/5 GFAP-EGFP-shRNA Kir4.1 or AAV2/5 EGFP-shRNA control were bilaterally injected into the hippocampus (− 2.0-mm anterior-posterior, 1.3-mm medial-lateral, and − 1.9-mm dorsal-ventral relative to the bregma) with 10-μL Hamilton syringe at a rate of 0.2 μL/min (2 μL on each side) (N = 7 in each group) [25]. After injection, the needles were retained for 5 min to allow the drug to be fully absorbed. 2 mg/kg LPS was injected 25 days after the viral injection. Behavioral tests were conducted, and hippocampal tissues were collected 24 h after LPS injection following decapitation (Fig. 7a).
Experimental Protocol 4
An intracerebral injection of TNF-α was performed as described previously [26]. Briefly, lateral ventricle cannulas were placed under isoflurane anesthesia, using a stereotactic alignment instrument at the following coordinates relative to bregma: − 1.0 mm, − 0.5 mm, and − 2.25 mm. In each mouse, 500 ng mouse tumor necrosis factor (TNF-α, Abcam, USA) were given in 4 μL total volume. The control mice received similar volume of vehicle. In each group, there were 7 mice. The rate of injection was set at a speed of 500 nL/min with an infusion pump. Behavioral tests were conducted, and hippocampal tissues were collected 24 h later (Fig. 8a).
Behavioral Tests
Fear Extinction Protocol
Fear memory and extinction were conducted using the typical conditioned stimulus (CS)/unconditioned stimulus (US)-CS protocol as previously described [27]. The fear conditioning box (UgoBasile, Gemonio, Italy) with a camera was connected with ANY-Maze software (Stoelting Co., Wood Dale, IL, USA). After drug administrations, the mice underwent a fear memory and extinction protocol. The mice were placed in the fear conditioning box for 5 min and exposed to three electric foot shocks (1.0 mA, 2 s). Twenty-four hours later, the mice were put back into the fear conditioning box and exposed to the similar context for 15 min without electric shock. The freezing time in the initial 5 min was statistically analyzed as the memory consolidation. The mice were put into the fear conditioning box for 5 min at the same time every day for 3 extinction days. The conditioning chamber was cleaned with 70% ethanol to remove the background odor (Fig. 1).
Open Field Test
The mice were tested in an open field test (OFT) (Shanghai Ruanlong Information Technology Co. Ltd., Shanghai, China, BW-OF302). In a quiet environment, the mice were placed in the center of the hood (40 cm × 40 cm × 65 cm). The behaviors were captured by a video camera for 5 min. After that, the box was cleaned with 70% ethanol. The border and central distances were analyzed by SuperMaze software.
Tail Suspension Test
The tail suspension test (TST) protocol was performed as previously described [27]. Each mouse was suspended on the edge of a shelf ~ 50 cm above the bottom of the sound insulation box using adhesive tape placed ~ 1 cm from the tip of the tail (Shanghai Ruanlong Information Technology Co. Ltd., Shanghai, China, YLS-18A). The animals were allowed to move for 6 min, and behavior was recorded with SuperMaze software. The time it took until it remained immobile was measured. The duration of immobility was recorded in the last 4 min.
Forced Swimming Test
After TST, the mice were allowed to rest for 24 h. A single testing session was applied in the forced swimming test (FST) according to the previous publication [28]. The mice were then individually placed in a 2 L glass container with water temperature at 22 ± 3 °C (Shanghai Ruanlong Information Technology Co. Ltd., Shanghai, China, BW-DFS201.) as previously described [27]. SuperMaze software was used to record the movement. The mouse was judged to be immobile when it ceased struggling and remained floating motionless in the water, making only those movements necessary to keep its head above water. The duration of immobility was recorded during the last 4 min of the test.
Measurement of Cytokines
Hippocampal levels of IL-1β and TNF-α were examined by ELISA using IL-1β (JL18442) and TNF-α (JL10484) ELISA assay kits (Shanghai Jianglai, Shanghai, China). The hippocampal tissue was weighed. An appropriate volume of PBS (1 mL/10 mg) was added to a portion of the hippocampal tissue, which was then rapidly homogenized, and the cells were disrupted by an ultrasonic cell disrupter (Scietz Biotechnology Co. Ltd., Ningbo, China). After centrifugation (5870×g for 5 min), the supernatant was collected, and protein quantification was performed. Absorbance was quickly read on the microplate reader at a detection wavelength of 450 nm. The concentrations of IL-1β and TNF-α were calculated according to the standard curve.
Immunofluorescence
The brain was washed 3 times with PBS, and antigen retrieval was performed using citrate buffer; samples were then permeabilized and blocked in PBS containing 0.5% Triton X-100 for 1 h and 5% goat serum (Hyclone, Thermo Fisher Scientific) at room temperature for 1 h. Sections were incubated with primary antibodies in blocking buffer overnight at 4 °C. After washing, secondary antibodies were added to the blocking buffer and incubated with the sections for 1 h at room temperature. Samples were then washed and counterstained with DAPI. Images were taken with an FV1000 Olympus Confocal Laser Scanning Microscope (Olympus, Tokyo, Japan). Primary antibodies used for immunostaining included glial fibrillary acidic protein (GFAP) (mouse, 1:100; Beyotime, AG259), Kir4.1 (rabbit, 1:100; Abcam, ab240876), and ionized calcium-binding adapter molecule 1 (Iba-1) (mouse, 1:100; Santa Cruz, SC-32725). Anti-mouse/rabbit secondary antibodies (1:1000) were purchased from ORIGENE. In each mouse, the images of dentate gyrus (DG) region were conducted in 8 sections at bregma − 1.22 mm, − 1.46 mm, − 1.70 mm, − 1.94 mm, − 2.18 mm, − 2.42 mm, − 2.66 mm, and − 2.90 mm. The intensities of the 8 sections were averaged to reduce the bias of position.
Western Blotting
The hippocampi were homogenized in lysis buffer (RIPA) on ice for 30 min and subsequently centrifuged at 12000 rpm for 5 min at 4 °C. Protein concentrations were determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, USA). Equivalent amounts of proteins were processed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot as previously described [29]. The primary antibodies used in this experiment were Kir4.1 (1:1000, Abcam), AKT (1:1000, CST), Arc (1:1000, CST), PSD95 (1:1000, CST), GluN2A (1:1000, Abcam), GluA1 (1:1000, Abcam), and β-actin (1:1000, CST). Band intensity was densitometrically quantified using Image J software.
Statistical Analyses
Data were presented as means ± SEM. Statistical analyses were performed using GraphPad Prism 4.02. The differences between two groups were analyzed by Student’s t test and one-way or two-way ANOVA followed by Bonferroni test. P values less than 0.05 were considered to indicate statistical significance.
Results
Rg1 Ameliorated the Impairment of Fear Extinction and Depression-Like Behaviors in LPS-Treated and SPS Mice
In Fig. 2a, there was no significant difference in freezing time of the first day among different groups (P > 0.05), suggesting that LPS did not influence the consolidation of fear memory. With the increasing time, apparent fear extinction was observed in control mice, but not in LPS-treated mice. By contrast, pre-treatment with Rg1 (20, 40 mg/kg) ameliorated the impairment of fear extinction compared with model group (the fourth day: P < 0.05, F (4, 30) = 14.38; Fig. 2b). We further assessed the depression-like behaviors in those mice using TST and FST. The motor function was detected by OFT. As shown in Fig. 2c, the total distance in each group was comparable, which indicated that the motor function was not affected by either LPS or Rg1 treatment (P > 0.05, F (4, 30) = 0.4057). Compared with control group, the immobility times of TST (P < 0.05, F (4, 30) = 26.72 Fig. 2d) and FST (P < 0.05, F (4, 30) = 62.46, Fig. 2e) in LPS mice increased significantly, which were prevented by Rg1 treatment (20, 40 mg/kg).

Ginsenoside Rg1 facilitates fear extinction and reduces depression-like behaviors in LPS-treated mice. (a) Extinction of fear memory, (b) the freezing value of the fourth day, (c) total distance traveled in each group was comparable. Behavior analysis of tail suspension test (d) and forced swim test (e). #P < 0.05 vs. control group. *P < 0.05 vs. LPS. Values are presented as means ± SEM from 7 animals in each group
We also determined whether Rg1 prevented fear extinction and depression-like behaviors in SPS mice. As shown in Fig. 3a, fear extinction in SPS mice were impaired compared with control mice. By contrast, treatment with Rg1 (20 mg/kg) could apparently prevent the impairment of fear extinction compared with SPS mice (the fourth day: P < 0.05, F (3, 24) = 25.23; Fig. 3b). The motor function in each group was comparable as the total distance in the OFT was not different among groups (P > 0.05, F (3, 24) = 0.4104; Fig. 3c). Moreover, Rg1 treatment significantly decreased the immobility times of TST (P < 0.05, F (3, 24) = 22.61; Fig. 3d) and FST (P < 0.05, F (3, 24) = 11.26; Fig. 3e) compared with SPS group. Together, these results suggest that LPS treatment, as well as SPS regime, could impair fear extinction and cause depression-like behaviors, while Rg1 could improve those abnormal behaviors.

Ginsenoside Rg1 facilitates fear extinction and reduces depression-like behaviors in SPS mice. (a) Extinction of fear memory, (b) the freezing value of the fourth day, (c) total distance traveled in each group was comparable. Behavior analysis of tail suspension test (d) and forced swimming test (e). #P < 0.05 vs. control group. *P < 0.05 vs. SPS. Values are presented as means ± SEM from 7 animals in each group
Rg1 Reduced Hippocampal Pro-inflammatory Cytokines and Glia Activation in LPS-Treated and SPS Mice
We assessed the effect of Rg1 on the levels of TNF-α and IL-1β in the hippocampus using ELISA. The results showed that the levels of TNF-α (P < 0.05, F (4, 20) = 19.25) and IL-1β (P < 0.05, F (4, 20) = 24.25) were dramatically enhanced in LPS group compared with control group. By contrast, Rg1 treatment attenuated the increase of pro-inflammatory cytokines (vs. model, both P < 0.05) (Fig. 4a, b).

Ginsenoside Rg1 reduces pro-inflammatory cytokines and glia activation in the hippocampus of PTSD mice. TNF-α and IL-1β levels following LPS treatment (a, b) and SPS regime (c, d) were measured by ELISA. Ginsenoside Rg1 administration reduced hippocampal GFAP and Iba-1 expression in DG region of LPS-injected mice (e). Scale bar: 50 μm. Values are presented as means ± SEM (n = 4) in each group. #P < 0.05 vs. control group. *P < 0.05 vs. LPS or SPS
We further detected TNF-α and IL-1β after Rg1 administration in SPS model. As shown in Fig. 4c, d, TNF-α (P < 0.05, F (3, 16) = 54.98) and IL-1β (P < 0.05, F (3, 16) = 71.2) were significantly enhanced in SPS group compared with control group. However, Rg1 (20 mg/kg) treatment reversed the increase of TNF-α and IL-1β caused by SPS (vs. model, both P < 0.05).
Astrocytes and microglia are involved in the production of pro-inflammatory cytokines [30]. To evaluate the activation of astrocyte and microglia in the hippocampus, we performed a quantitative analysis of astrocytic and microglial response using immunohistochemistry with GFAP and Iba-1 antibodies. Immunostaining revealed that LPS triggered the expression of cellular markers of astrocytic and microglia in the DG region, which was reduced by Rg1 treatment (both P < 0.05; GFAP: F (2, 6) = 29.95; Iba-1: F (2, 6) = 27.08; Fig. 4e).
Rg1 Promoted Synaptic Proteins in the Hippocampus of Mice Treated by LPS or SPS Regime
Glia provide neurons with morphogenic and metabolic support and are fundamental to the functional regulation of dendritic spines [31]. The results showed that the expression of PSD95 (P < 0.05, F (4, 15) = 13.77), Arc (P < 0.05, F (4, 15) = 48.57), and GluA1 (P < 0.05, F (4, 15) = 12.4) in LPS group were lower than those in control group (Fig. 5a, b). However, GluN2A (P < 0.05, F (4, 15) = 15.99) was significantly higher in LPS mice than that in control group. Treatment with Rg1 restored the expression of synaptic proteins of PSD95, Arc, GluN2A, and GluA1 (Fig. 5a, b). We further verified whether Rg1 was able to regulate synaptic protein expression in SPS mice. The results showed that the expression of hippocampal PSD95 (P < 0.05, F (3, 12) = 48.44), Arc (P < 0.05, F (3, 12) = 19.57), and GluA1 (P < 0.05, F (3, 12) = 15.68) were reduced in SPS mice compared with control mice (Fig. 5c, d). By contrast, the expression of GluN2A increased in SPS mice compared with control mice (P < 0.05, F (3, 12) = 12.24). Rg1 treatment reduced the changes of PSD95, Arc, GluA1, and GluN2A caused by SPS regime (Fig. 5c, d).

Ginsenoside Rg1 promotes synaptic protein expression in the hippocampus of PTSD mice. The levels of PSD95, Arc, GluN2A, and GluA1 in LPS (a, b) and SPS (c, d) models were analyzed using western blotting. Values are presented as means ± SEM (n = 4) in each group. #P < 0.05 vs. control group. *P < 0.05 vs. LPS or SPS
Rg1 Reduced LPS-Induced Increase of Kir4.1 in the Hippocampus
We also detected hippocampal Kir4.1 expression in LPS-treated mice using Kir4.1/GFAP double staining. Our data showed that LPS-induced activation of astrocytes was co-localized with Kir4.1 expression (Fig. 6a). We further confirmed the expression of Kir4.1 and GFAP protein levels in the hippocampus by western blotting. As shown in Fig. 6b, c, the expression of Kir4.1 (P < 0.05, F (4,15) = 7.109) and GFAP (P < 0.05, F (4,15) = 46.53) in the hippocampus of LPS group was significantly higher than those in control group, but those effects were reduced by treatment with 20 mg/kg Rg1 (P < 0.05 vs. model).

Ginsenoside Rg1 reduces the increase of hippocampal Kir4.1 and GFAP in the LPS-treated mice. (a) Representative image of double-immunofluorescence staining for GFAP and Kir4.1. Green, Kir4.1; red, GFAP; scale bar, 50 μm; (b–c) ginsenoside Rg1 downregulated the expression level of Kir4.1 and GFAP in the hippocampus. Values are presented as means ± SEM (n = 4) in each group. #P < 0.05 vs. control; *P < 0.05 vs. LPS group
Reducing Hippocampal Kir4.1 Ameliorated the Impairment of Fear Extinction and Depression-like Behaviors in LPS-Treated Mice
As LPS promoted hippocampal Kir4.1 expression and induced PTSD-like behaviors, we assessed the effect of reducing hippocampal Kir4.1 on PTSD-like behaviors. Viral-encoding Kir4.1 was designed and bilaterally injected into the hippocampus (Fig. 7a). As shown in Fig. 7b, the viruses specifically targeted GFAP+ cells (Fig. 7b). On the fourth day of extinction, the freezing time in shRNA Kir4.1-injected mice was significantly lower than shRNA scramble mice (P < 0.05, t = 6.15, df = 6), Fig. 7c). Compared with mice receiving scrambled control, the immobility times of the mice injected with shRNA Kir4.1 in the TST (t = 4.292, df = 6, Fig. 7d) and FST (t = 2.866, df = 6, Fig. 7e) were significantly lower. Interestingly, shRNA Kir4.1 could significantly reduce hippocampal Kir4.1 expression compared with scrambled control (t = 7.505, df = 3, Fig. 7f).

Reducing Kir4.1 prevents PTSD-like behaviors in LPS-treated mice. (a) A schematic illustrating the injection of viral encoding Kir4.1 shRNA or scramble. Fourteen C57 mice were habituated for 7 days and received intrahippocampal injection of viral expression of shRNA Kir4.1 or scramble. Four weeks later, LPS was intraperitoneally injected. Twenty-four hours later, behavioral tests were conducted, or hippocampi were collected. (b) The viruses encoding shRNA Kir4.1 carrying GFAP promoter. (c) Viral-encoding shRNA Kir4.1 facilitates fear extinction in LPS mice. (d) Immobility time in TST. (e) Immobility time in FST. (f) Expression of Kir4.1 was reduced by viral encoding shRNA Kir4.1. Values are presented as means ± SEM (n = 4–7) in each group. *P < 0.05 vs. LPS + scramble group
Intracerebral Injection of TNF-α Promoted Kir4.1 Expression and Caused PTSD-Like Behaviors
To distinguish the exact cytokine associated with PTSD-like behaviors, we carried out intracerebral injection of TNF-α (Fig. 8a). As shown in Fig. 8b, the expression of Kir4.1 was promoted after TNF-α injection compared with control (t = 7.503, df = 3). Further, intracerebral injection of TNF-α could also impair fear extinction (the fourth day: P < 0.05, t = 3.66, df = 6; Fig. 8c). Moreover, the immobility times of the mice receiving TNF-α in the TST (t = 3.333, df = 6, Fig. 8d) and FST (t = 4.561, df = 6, Fig. 8e) were significantly increased compared with control. Together, these results suggested that TNF-α could promote astroglial Kir4.1 expression and cause PTSD-like behaviors.

Intracerebral injection of TNF-α promotes Kir4.1 expression and causes PTSD-like behaviors. (a) A schematic for the study design of intraventricular injection of TNF-α. (b) Intraventricular injection of TNF-α promoted Kir4.1 protein expression. (c) Intracerebroventricular injection of TNF-α impaired fear extinction. (d) Immobility time in TST. (e) Immobility time in FST. Values are presented as means ± SEM (n = 4–7) in each group. *P < 0.05 vs. control
Discussion
In this study, we demonstrated that Rg1 administration promoted fear extinction and relieved depression-like behaviors in both SPS mice and LPS-treated mice. More importantly, Rg1 promoted synaptic protein expression and reduced Kir4.1 and TNF-α level. Specific reduction of astroglial Kir4.1 in the hippocampus could ameliorate the PTSD-like behaviors. On the contrary, injection of TNF-α mimicked the effect of SPS and LPS injection (Fig. 9). Our study would provide novel evidence for the preventive effects of Rg1 on PTSD-like behaviors.

A schematic diagram of ginsenoside Rg1 in preventing PTSD-like behaviors in mice. The mechanism may be related to the neuroinflammation caused by TNF-α and IL-1β, the expression of Kir4.1 protein, and NMDAR (GluN2A), AMPAR (GluA1), Arc, and PSD95-related synaptic plasticity. PTSD, post-traumatic stress disorder; TNF-α, tumor necrosis factor-α; Kir4.1, inwardly rectifying potassium channel; IL-1β, interleukin 1β; NMDAR, N-methyl-D-aspartic acid receptor; AMPAR, α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor
Rg1 Ameliorated the PTSD-Like Behaviors in Mice
Rg1 is notable for its neuroprotective effects on memory impairment, such as in Alzheimer’s disease [32], D-galactose-induced senescence [33], and LPS model [34]. Our previous study also demonstrated that Rg1 treatment improved memory in middle-aged mice by regulating the AKT pathway, altering apical spines, and facilitating hippocampal long-term potentiation [22]. In addition, researchers have discovered that Rg1 prevented PTSD-like behaviors by decreasing serum corticosterone (CORT) and hypothalamus corticotrophin-releasing hormone (CRH) levels [35]. In this study, we utilized the typical CS/US-CS model to test the fear extinction. In fact, the memory consolidations after LPS or SPS were not changed. However, fear extinction was different among groups. Especially, fear extinction was impaired in LPS and SPS groups but ameliorated by ginsenoside Rg1.
As different doses of LPS could elicit different features of behaviors, we selected a dose of LPS, which did not affect fear memory consolidation. LPS (2 mg/kg) could impair fear extinction, which might mimic the impairment of fear extinction in PTSD [5, 36]. Moreover, this dose of LPS did not affect the motor function, which might explain that the impairment of fear extinction was not attributed to impairment of motor function. SPS is a pre-clinical model that displays behavioral, molecular, and physiological alterations that recapitulate many of the same alterations observed in PTSD [37]. Most importantly, Rg1 could remedy the impairment of fear extinction induced by LPS, as well as by SPS regime. In our preliminary experiments, we found an increase of inflammatory reaction in SPS mice model. Interestingly, fear extinction was impaired in LPS-treated mice. Therefore, we proposed that SPS model and LPS model might encompass some similarities. We thus selected LPS-treated mice model to further investigate the potential mechanisms underlying the impairment of fear extinction. However, whether LPS could be used to model PTSD is still questionable. In future study, LPS-induced impairment of fear extinction should be more deeply investigated.
Rg1 Ameliorated Pro-inflammatory Cytokines and Glia Activation
Inflammatory storms are important factors leading to cognitive impairment in LPS and SPS animal models [38, 39]. Neuroinflammation has been reported as one of the causes of the cognitive impairment [38, 39]. It plays a critical role in the development of Parkinson’s disease, Huntington’s disease, and other neurodegenerative diseases [40]. Injection of LPS activates glia to synthesize and secrete the pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) [41]. Microglia and astrocytes mediate the production of pro-inflammatory cytokines, which drive the pathogenesis of depressive-like phenotypes [42]. Our study demonstrated that Rg1 reduced the overproduction of hippocampal TNF-α and IL-1β induced by LPS and SPS. Astrocytes regulate the micro-environment of the adult neurogenic niche and the synaptic integration of the newly formed neurons into existing hippocampal circuits and promote the formation of new memories [43]. Further, immunofluorescence demonstrated that GFAP and Iba-1 were increased in LPS-injected mice. By contrast, treatment with Rg1 could reduce the numbers of GFAP and Iba-1-positive cells.
Although our study mainly concentrated on the PTSD mice model, we are still not sure of the function of pro-inflammatory cytokines TNF-α and IL-1β in fear extinction. Therefore, we directly perfused TNF-α into ventricular, and the results indicated that the immobility time of TST and FST was longer than control. Especially, fear extinction is impaired by injection of TNF-α, which indicated that TNF-α might be a key factor causing abnormality of fear extinction.
Ginsenoside Rg1 Reduced Hippocampal Kir4.1 Expression
In the CNS, astrocytes play a crucial role in maintaining the structural and functional integrity of the brain, which includes formation of maintenance of water and ion homeostasis, metabolism of neurotransmitters, and secretion of various neuroactive molecules. Among these functions, spatial potassium (K+) buffering by astrocytes is an essential system for controlling extracellular K+ concentration and neuronal excitability [13], whereas astrocyte hyperactivity leads to the production of pro-inflammatory cytokines such as IL-1β, which is a key driver in the clinical manifestation of depressive symptoms. In our present study, we demonstrated that LPS treatment promoted hippocampal Kir4.1, while Rg1 treatment could reduce the increase of Kir4.1 expression. Further, the impairment of fear extinction caused by LPS was improved by shRNA Kir4.1. After extinction, immobility times of TST and FST in shRNAKir4.1 were also lower than those in scrambled control, which indicated that reducing Kir4.1 might promote extinction of fear memory and reduce depression-like behaviors in LPS-treated mice.
Potential Interaction Among Inflammation, Kir4.1, and Synaptic Plasticity in PTSD Models
Studies have shown that abnormalities of synaptic are associated with the increase of IL-1β, IL-6, and TNF-α and NF-κB activation [44]. At different stages of neurogenesis, the activation of microglia is beneficial or harmful to neurons depending on the secretion regulation of pro-inflammatory and anti-inflammatory factors [45]. TNF-ɑ may lead to hearing loss and synaptic imbalance [46]. Glutamate is the main excitatory neurotransmitter in the mammalian brain, playing a key role in learning and memory and neuroplasticity [47]. However, the molecular mechanism contributing to the increase of astrocyte Kir4.1 protein following LPS treatment requires further verification. Elevated extracellular potassium concentration ([K+]out) causes neuronal depolarization that can lead to enhanced glutamate release from the presynaptic and enhanced N-methyl-D-aspartic acid (NMDA) receptor activation in the postsynaptic [48]. In addition, glutamate uptake was inhibited by knocking down of Kir4.1 [49]. Using the Kir4.1 knock-out mouse, Brasko et al. demonstrated that Kir4.1 subunit was expressed in the glial plasma lemma and supported a role of PSD95 in their membrane localization [50]. These in vitro findings were corroborated in the hippocampal CA1 and DG of Arc knockout mice, where a reduction of spine density and decreased abundance of thin spines were found [51]. Aberrant Arc expression in the hippocampus in response to chronic NMDA receptors hypo-function decreased spine density [52]. GluA1 and GluN2A are important subunits of α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors and NMDA receptors, which possess different functions in the excitability of neurons. Although both receptors are cation dependent, AMPA receptors are more important for the basal synaptic transmission, while NMDA receptors are critical for synaptic plasticity [53]. Moreover, NMDA receptors might also contribute to inflammation [54]. In this study, we found that GluA1 decreased, while GluN2A increased followed by LPS or SPS treatment. Especially, ginsenoside Rg1 reversed the changes caused by LPS or SPS. Nevertheless, the specific roles of AMPA receptors and NMDA receptors in PTSD deserved future investigation. These results suggested that Rg1 increased synaptic-associated proteins by preventing the activation of TNF-α and IL-1β [55]. However, the precise mechanisms driving those effects and the relationship between Kir4.1 and synaptic plasticity are still not completely clear.
In summary, we reported a distinct role of Rg1 in remedying PTSD-like behaviors in mice. The mechanism may be related to the recovery of hippocampal synaptic plasticity, reduction of Kir4.1 level, and pro-inflammatory cytokines. Our study would provide theoretical evidence for the selection of ginseng in the treatment of mental disorders.
Data Availability
The datasets used during the present study are available from the corresponding author upon reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor
- COVID-19:
-
Coronavirus disease 2019
- IL-1β:
-
Interleukin 1β
- Kir4.1:
-
Inwardly rectifying potassium channel
- NMDAR:
-
N-methyl-D-aspartic acid receptor
- PTSD:
-
Post-traumatic stress disorder
- SPS:
-
Single-prolonged stress
- TNF-α:
-
tumor necrosis factor-α
References
Brooks SK, Webster RK, Smith LE, Woodland L, Wessely S, Greenberg N, Rubin GJ (2020) The psychological impact of quarantine and how to reduce it: rapid review of the evidence. Lancet 395(10227):912–920. https://doi.org/10.1016/S0140-6736(20)30460-8
Yang Z, Gu S, Honnorat N, Linn KA, Shinohara RT, Aselcioglu I, Bruce S, Oathes DJ et al (2018) Network changes associated with transdiagnostic depressive symptom improvement following cognitive behavioral therapy in MDD and PTSD. Mol Psychiatry 23(12):2314–2323. https://doi.org/10.1038/s41380-018-0201-7
Ji G, Yakhnitsa V, Kiritoshi T, Presto P, Neugebauer V (2018) Fear extinction learning ability predicts neuropathic pain behaviors and amygdala activity in male rats. Mol Pain 14:1744806918804441. https://doi.org/10.1177/1744806918804441
Yabuki Y, Fukunaga K (2019) Clinical therapeutic strategy and neuronal mechanism underlying post-traumatic stress disorder (PTSD). Int J Mol Sci 20(15). https://doi.org/10.3390/ijms20153614
Baek J, Lee S, Cho T, Kim SW, Kim M, Yoon Y, Kim KK, Byun J et al (2019) Neural circuits underlying a psychotherapeutic regimen for fear disorders. Nature 566(7744):339–343. https://doi.org/10.1038/s41586-019-0931-y
Mary A, Dayan J, Leone G, Postel C, Fraisse F, Malle C, Vallee T, Klein-Peschanski C et al (2020) Resilience after trauma: the role of memory suppression. Science 367(6479):eaay8477. https://doi.org/10.1126/science.aay8477
Kempuraj D, Selvakumar GP, Ahmed ME, Raikwar SP, Thangavel R, Khan A, Zaheer SA, Iyer SS et al (2020) COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist 26:402–414. https://doi.org/10.1177/1073858420941476
Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T (2017) Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology 42(1):254–270. https://doi.org/10.1038/npp.2016.146
Tursich M, Neufeld RW, Frewen PA, Harricharan S, Kibler JL, Rhind SG, Lanius RA (2014) Association of trauma exposure with proinflammatory activity: a transdiagnostic meta-analysis. Transl Psychiatry 4:e413. https://doi.org/10.1038/tp.2014.56
Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML (2016) The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol 132(1):1–21. https://doi.org/10.1007/s00401-016-1553-1
Wu W, Yao H, Zhao HW, Wang J, Haddad GG (2018) Down-regulation of inwardly rectifying K(+) currents in astrocytes derived from patients with Monge’s disease. Neuroscience 374:70–79. https://doi.org/10.1016/j.neuroscience.2018.01.016
Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, Ma S, Sang K et al (2018) Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554(7692):323–327. https://doi.org/10.1038/nature25752
Ohno Y (2018) Astrocytic Kir4.1 potassium channels as a novel therapeutic target for epilepsy and mood disorders. Neural Regen Res 13(4):651–652. https://doi.org/10.4103/1673-5374.230355
Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I et al (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17(5):694–703. https://doi.org/10.1038/nn.3691
Cui Y, Hu S, Hu H (2019) Lateral Habenular burst firing as a target of the rapid antidepressant effects of ketamine. Trends Neurosci 42(3):179–191. https://doi.org/10.1016/j.tins.2018.12.002
Kelley KW, Ben Haim L, Schirmer L, Tyzack GE, Tolman M, Miller JG, Tsai HH, Chang SM et al (2018) Kir4.1-dependent astrocyte-fast motor neuron interactions are required for peak strength. Neuron 98(2):306–319 e307. https://doi.org/10.1016/j.neuron.2018.03.010
Oliynyk S, Oh S (2013) Actoprotective effect of ginseng: Improving mental and physical performance. J Ginseng Res 37(2):144–166. https://doi.org/10.5142/jgr.2013.37.144
Cheng W, Jing J, Wang Z, Wu D, Huang Y (2017) Chondroprotective effects of ginsenoside Rg1 in human osteoarthritis chondrocytes and a rat model of anterior cruciate ligament transection. Nutrients 9(3). https://doi.org/10.3390/nu9030263
Chen J, Zhang X, Liu X, Zhang C, Shang W, Xue J, Chen R, Xing Y et al (2019) Ginsenoside Rg1 promotes cerebral angiogenesis via the PI3K/Akt/mTOR signaling pathway in ischemic mice. Eur J Pharmacol 856:172418. https://doi.org/10.1016/j.ejphar.2019.172418
Fan C, Song Q, Wang P, Li Y, Yang M, Yu SY (2018) Neuroprotective effects of ginsenoside-Rg1 against depression-like behaviors via suppressing glial activation, synaptic deficits, and neuronal apoptosis in rats. Front Immunol 9:2889. https://doi.org/10.3389/fimmu.2018.02889
Fang F, Chen X, Huang T, Lue LF, Luddy JS, Yan SS (2012) Multi-faced neuroprotective effects of ginsenoside Rg1 in an Alzheimer mouse model. Biochim Biophys Acta 1822(2):286–292. https://doi.org/10.1016/j.bbadis.2011.10.004
Zhu G, Wang Y, Li J, Wang J (2015) Chronic treatment with ginsenoside Rg1 promotes memory and hippocampal long-term potentiation in middle-aged mice. Neuroscience 292:81–89. https://doi.org/10.1016/j.neuroscience.2015.02.031
Francois A, Terro F, Quellard N, Fernandez B, Chassaing D, Janet T, Rioux Bilan A, Paccalin M et al (2014) Impairment of autophagy in the central nervous system during lipopolysaccharide-induced inflammatory stress in mice. Mol Brain 7:56. https://doi.org/10.1186/s13041-014-0056-z
Tanaka KI, Yagi T, Nanba T, Asanuma M (2018) Application of single prolonged stress induces post-traumatic stress disorder-like characteristics in mice. Acta Med Okayama 72(5):479–485. https://doi.org/10.18926/AMO/56245
Jeon SG, Kang M, Kim YS, Kim DH, Nam DW, Song EJ, Mook-Jung I, Moon M (2018) Intrahippocampal injection of a lentiviral vector expressing neurogranin enhances cognitive function in 5XFAD mice. Exp Mol Med 50(3):e461. https://doi.org/10.1038/emm.2017.302
Braun TP, Grossberg AJ, Veleva-Rotse BO, Maxson JE, Szumowski M, Barnes AP, Marks DL (2012) Expression of myeloid differentiation factor 88 in neurons is not requisite for the induction of sickness behavior by interleukin-1beta. J Neuroinflammation 9:229. https://doi.org/10.1186/1742-2094-9-229
Yang SJ, Song ZJ, Wang XC, Zhang ZR, Wu SB, Zhu GQ (2019) Curculigoside facilitates fear extinction and prevents depression-like behaviors in a mouse learned helplessness model through increasing hippocampal BDNF. Acta Pharmacol Sin 40(10):1269–1278. https://doi.org/10.1038/s41401-019-0238-4
Castagne V, Moser P, Roux S, Porsolt RD (2011) Rodent models of depression: forced swim and tail suspension behavioral despair tests in rats and mice. Curr Protoc Neurosci Chapter 8:Unit 8 10A. https://doi.org/10.1002/0471142301.ns0810as55
Song ZJ, Yang SJ, Han L, Wang B, Zhu G (2019) Postnatal calpeptin treatment causes hippocampal neurodevelopmental defects in neonatal rats. Neural Regen Res 14(5):834–840. https://doi.org/10.4103/1673-5374.249231
Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 109(4):E197–E205. https://doi.org/10.1073/pnas.1111098109
Allen NJ (2014) Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol 30:439–463. https://doi.org/10.1146/annurev-cellbio-100913-013053
Nie L, Xia J, Li H, Zhang Z, Yang Y, Huang X, He Z, Liu J et al (2017) Ginsenoside Rg1 ameliorates behavioral abnormalities and modulates the hippocampal proteomic change in triple transgenic mice of Alzheimer’s disease. Oxidative Med Cell Longev 2017:6473506–6473517. https://doi.org/10.1155/2017/6473506
Chen L, Yao H, Chen X, Wang Z, Xiang Y, Xia J, Liu Y, Wang Y (2018) Ginsenoside Rg1 decreases oxidative stress and down-regulates Akt/mTOR signalling to attenuate cognitive impairment in mice and senescence of neural stem cells induced by D-galactose. Neurochem Res 43(2):430–440. https://doi.org/10.1007/s11064-017-2438-y
Jin Y, Peng J, Wang X, Zhang D, Wang T (2017) Ameliorative effect of ginsenoside Rg1 on lipopolysaccharide-induced cognitive impairment: role of cholinergic system. Neurochem Res 42(5):1299–1307. https://doi.org/10.1007/s11064-016-2171-y
Wang Z, Zhu K, Chen L, Ou Yang L, Huang Y, Zhao Y (2015) Preventive effects of ginsenoside Rg1 on post-traumatic stress disorder (PTSD)-like behavior in male C57/B6 mice. Neurosci Lett 605:24–28. https://doi.org/10.1016/j.neulet.2015.08.017
Desmedt A, Marighetto A, Piazza PV (2015) Abnormal fear memory as a model for posttraumatic stress disorder. Biol Psychiatry 78(5):290–297. https://doi.org/10.1016/j.biopsych.2015.06.017
Lisieski MJ, Eagle AL, Conti AC, Liberzon I, Perrine SA (2018) Single-prolonged stress: a review of two decades of progress in a rodent model of post-traumatic stress disorder. Front Psych 9:196. https://doi.org/10.3389/fpsyt.2018.00196
Lopes PC (2016) LPS and neuroinflammation: a matter of timing. Inflammopharmacology 24(5):291–293. https://doi.org/10.1007/s10787-016-0283-2
Lee B, Shim I, Lee H, Hahm DH (2018) Effects of epigallocatechin gallate on behavioral and cognitive impairments, hypothalamic-pituitary-adrenal axis dysfunction, and alternations in hippocampal BDNF expression under single prolonged stress. J Med Food 21(10):979–989. https://doi.org/10.1089/jmf.2017.4161
Pizza V, Agresta A, D'Acunto CW, Festa M, Capasso A (2011) Neuroinflamm-aging and neurodegenerative diseases: an overview. CNS Neurol Disord Drug Targets 10(5):621–634. https://doi.org/10.2174/187152711796235014
Song Z, Shen F, Zhang Z, Wu S, Zhu G (2020) Calpain inhibition ameliorates depression-like behaviors by reducing inflammation and promoting synaptic protein expression in the hippocampus. Neuropharmacology 174:108175. https://doi.org/10.1016/j.neuropharm.2020.108175
Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP (2014) Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 76(7):575–584. https://doi.org/10.1016/j.biopsych.2013.10.014
Anacker C, Hen R (2017) Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat Rev Neurosci 18(6):335–346. https://doi.org/10.1038/nrn.2017.45
Liu Y, Zhang Y, Zheng X, Fang T, Yang X, Luo X, Guo A, Newell KA et al (2018) Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J Neuroinflammation 15(1):112. https://doi.org/10.1186/s12974-018-1141-5
Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158(3):1021–1029. https://doi.org/10.1016/j.neuroscience.2008.06.052
Wang W, Zhang LS, Zinsmaier AK, Patterson G, Leptich EJ, Shoemaker SL, Yatskievych TA, Gibboni R et al (2019) Neuroinflammation mediates noise-induced synaptic imbalance and tinnitus in rodent models. PLoS Biol 17(6):e3000307. https://doi.org/10.1371/journal.pbio.3000307
Deutschenbaur L, Beck J, Kiyhankhadiv A, Muhlhauser M, Borgwardt S, Walter M, Hasler G, Sollberger D et al (2016) Role of calcium, glutamate and NMDA in major depression and therapeutic application. Prog Neuro-Psychopharmacol Biol Psychiatry 64:325–333. https://doi.org/10.1016/j.pnpbp.2015.02.015
Poolos NP, Kocsis JD (1990) Elevated extracellular potassium concentration enhances synaptic activation of N-methyl-D-aspartate receptors in hippocampus. Brain Res 508(1):7–12. https://doi.org/10.1016/0006-8993(90)91110-3
Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55(3):274–281. https://doi.org/10.1002/glia.20455
Brasko C, Hawkins V, De La Rocha IC, Butt AM (2017) Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct Funct 222(1):41–59. https://doi.org/10.1007/s00429-016-1199-8
Peebles CL, Yoo J, Thwin MT, Palop JJ, Noebels JL, Finkbeiner S (2010) Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci U S A 107(42):18173–18178. https://doi.org/10.1073/pnas.1006546107
Balu DT, Coyle JT (2014) Chronic D-serine reverses arc expression and partially rescues dendritic abnormalities in a mouse model of NMDA receptor hypofunction. Neurochem Int 75:76–78. https://doi.org/10.1016/j.neuint.2014.05.015
Zhu G, Yang S, Xie Z, Wan X (2018) Synaptic modification by L-theanine, a natural constituent in green tea, rescues the impairment of hippocampal long-term potentiation and memory in AD mice. Neuropharmacology 138:331–340. https://doi.org/10.1016/j.neuropharm.2018.06.030
Sil S, Ghosh T, Ghosh R (2016) NMDA receptor is involved in neuroinflammation in intracerebroventricular colchicine-injected rats. J Immunotoxicol 13(4):474–489. https://doi.org/10.3109/1547691X.2015.1130760
Li Y, Pehrson AL, Waller JA, Dale E, Sanchez C, Gulinello M (2015) A critical evaluation of the activity-regulated cytoskeleton-associated protein (Arc/Arg3.1)'s putative role in regulating dendritic plasticity, cognitive processes, and mood in animal models of depression. Front Neurosci 9:279. https://doi.org/10.3389/fnins.2015.00279
Funding
This study was supported by the National Natural Science Foundation of China (81673716, 81874417), Anhui Natural Science Foundation (1808085J15), the National Key R&D Program of China (No. 2019YFC1708701), and Opening Project of Zhejiang Provincial Preponderant and Characteristic Subject of Key University Zhejiang Chinese Medical University (No. ZYXZD2019005).
Author information
Authors and Affiliations
Contributions
GQZ and ZRZ designed the research; ZRZ, ZJS, FMS, PX, and JW performed the research and analyzed the data; ASZ provided suggestions for the experimental design and manuscript; and GQZ and ZRZ wrote the manuscript.
Corresponding authors
Ethics declarations
The experimental procedures were approved by the Ethics Committee of Anhui University of Chinese Medicine (Hefei, China: Approval No.: SYXK2014-0512).
Conflict of Interest
The authors declare that they have no conflict of interest.
Consent to Participate
Not applicable.
Consent for Publication
All authors consent to the publication of this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, Z., Song, Z., Shen, F. et al. Ginsenoside Rg1 Prevents PTSD-Like Behaviors in Mice Through Promoting Synaptic Proteins, Reducing Kir4.1 and TNF-α in the Hippocampus. Mol Neurobiol 58, 1550–1563 (2021). https://doi.org/10.1007/s12035-020-02213-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02213-9