
Abstract
Purpose of Review
To provide an overview and highlight recent updates in the field of paraneoplastic neurologic disorders.
Recent Findings
The prevalence of paraneoplastic neurologic disorders is greater than previously reported and the incidence has been rising over time, due to improved recognition in the era of antibody biomarkers. Updated diagnostic criteria that are broadly inclusive and also contain diagnostic risk for clinical presentations (high and intermediate) and diagnostic antibodies (high, intermediate, and low) have replaced the original 2004 criteria. Antibody biomarkers continue to be characterized (e.g., KLHL-11 associated with seminoma in men with brainstem encephalitis). Some paraneoplastic antibodies also provide insight into likely immunotherapy response and prognosis. The rise of immune checkpoint inhibitors as cancer therapeutics has been associated with newly observed immune-mediated adverse effects including paraneoplastic neurological disorders. The therapeutic approach to paraneoplastic neurologic disorders is centered around cancer care and trials of immune therapy.
Summary
The field of paraneoplastic neurologic disorders continues to be advanced by the identification of novel antibody biomarkers which have diagnostic utility, and give insight into likely treatment responses and outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Paraneoplastic neurologic disorders are heterogeneous autoimmune diseases occurring in the context of a non-nervous system cancer (solid organ or hematologic). They can arise as the clinical presentation for a previously undiagnosed cancer (e.g., a woman who presents with ataxia in whom ovarian adenocarcinoma is subsequently detected). Paraneoplastic neurological disorders can also arise during treatment for a recently diagnosed cancer, or can be the clinical presentation alerting the treating providers to a cancer relapse. These disorders can target any part of the neuraxis, rostrocaudally, from cerebral cortex to neuromuscular junction, though certain classical syndromes, now known as “high risk phenotypes,” have the highest risk for accompanying cancer (Table 1) [1••].
The diagnosis of a paraneoplastic neurologic disorder is usually supported by the detection of one or more neural antigen-directed IgG autoantibodies. These biomarkers serve to alert the clinician to the probability of malignancy as well as anatomical site and histologic type. While all these are biomarkers of various autoimmune neurological disease, the significance of each for a paraneoplastic diagnosis varies (Table 2 and Table 3). Some well-established biomarkers, such as antineuronal nuclear antibody type 1 (ANNA-1 [anti-Hu]) which has a > 70% positive predictive value for small cell (lung usually) carcinoma (SCLC) or other neuroendocrine lineage carcinomas, are considered high-risk [2]. More recently described biomarkers are considered either intermediate risk (with 30–70% risk for cancer, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic [AMPA]-receptor [R] antibody) or low risk (with 30% cancer risk, such as glial fibrillary acidic protein [GFAP]-IgG) [3, 4•]. Although novel antibody discoveries have increased diagnostic sensitivity, antibody negativity excludes neither a paraneoplastic cause nor cancer diagnosis [5••]. Antigenic target type (intracellular versus cell surface) may also be informative for predicting treatment response and prognosis [6, 7]. Herein, we provide an update on paraneoplastic neurological disorders focusing on presenting phenotypes, antibody diagnostics, clinical assessment, and management.
Epidemiology
Since early post-mortem studies first postulated a link between limbic encephalitis and carcinoma, appreciation of the prevalence of paraneoplastic neurologic disorders has grown [8]. Initially thought to affect only 0.01% of patients with cancer, it has been demonstrated more recently that paraneoplastic disorders occur in 1 in 300 patients with cancer [5••, 9]. Overall, they are most commonly associated with lung, breast, and ovarian carcinomas but specific syndromes carry their own particular cancer associations [5••]. SCLC, with its inherent diverse neural antigen repertoire, carries the greatest risk for paraneoplastic neurologic disorders, occurring in approximately 10% of patients [10]. The prevalence of paraneoplastic neurologic disorders is 5.4 per 100,000 in the USA but increases to 11 per 100,000 for those > 60 years [11•]. The incidence rate across recent epidemiologic studies ranges from 0.4 to 1 per 100,000 person years [5••, 11•, 12]. Furthermore, the incidence of paraneoplastic neurologic disorders has increased over time, a trend consistent across multiple epidemiological studies [5••, 11•, 12].
In Northeastern Italy, the incidence of paraneoplastic neurologic disorders has almost doubled from 2009–2011 (0.62/100,000 person years) to 2015–2017 (1.22/100,000 person years) [5••]. A similar phenomenon was noted in Olmsted county Minnesota with incidence doubling from 1987–2002 (0.4/100,000 person years) to 2003–2018 (0.8/100,000 person years) [11•]. A similar trend was observed for autoimmune encephalitis which has trebled in incidence over 20 years, and is now more prevalent than infectious encephalitis [13].
These observations may be attributable to improved awareness among clinicians, increased detection through paraneoplastic autoantibody tests and profiles, and the advent of autoimmune complications of checkpoint inhibitor therapies for cancer [14, 15].
Pathophysiology
Paraneoplastic neurologic disorders arise in the context of an immune response generated against antigens expressed on tumor cells which are also expressed natively in the host nervous system [16]. Antigens released following tumor cell apoptosis are presented by antigen-presenting cells to helper T cells in peripheral lymph nodes. CD4 + helper T cells subsequently activate antigen-specific B cells into to antibody-producing plasma cells. Disease mechanisms differ between disorders associated with antibodies to cell-surface antigens and those associated with antibodies directed against intracellular antigens. Antibodies against intracellular antigens and are not directly pathogenic but instead represent biomarkers of cytotoxic T cell–mediated cellular injury. This is supported by neuropathological studies which have revealed CD8 + T cell infiltration of neural tissues of patients with antibodies to intracellular antigens [7]. In contrast, antibodies directed against cell-surface antigens bind in vivo and in many instances pathogenic mechanisms have been characterized. For example, antibodies against n-methyl-d-aspartate (NMDA)-R, AMPA-R, and gamma amino butyric acid (GABA)A-Rs lead to neuronal dysfunction through a process of receptor cross-linking and internalization leading to reduced cell-surface receptor density [17]. GABAB-R antibodies, on the other hand, impair receptor function directly without causing receptor internalization [18]. LGI1 antibodies affect the protein–protein interaction with its receptor ADAM22, whereas aquaporin 4 antibodies mediate antigen internalization and complement-induced cytotoxicity [19–21].
Clinical Presentations
General Principles and Criteria
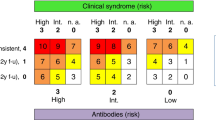
Neurological disorders are subacute in onset (over 6–12 weeks), and rapidly progressive. As per the updated diagnostic criteria, these disorders can be high-risk phenotypes (previously known as classic), and intermediate-risk phenotypes (Table 1). High-risk phenotypic presentations include encephalomyelitis, limbic encephalitis, rapidly progressive cerebellar syndrome, opsoclonus-myoclonus syndrome, sensory neuronopathy, enteric neuropathy, and Lambert-Eaton myasthenic syndrome. Intermediate-risk phenotypes include non-limbic encephalitides (such as anti-NMDA-receptor encephalitis), brainstem encephalitis, Morvan syndrome, isolated myelopathy, stiff-person spectrum disorders, and paraneoplastic polyradiculoneuropathies [1••]. Other neurological disorders (e.g., paraneoplastic chorea, isolated myoclonus) or multifocal disorders (e.g., chorea with polyradiculoneuropathy) may occur in a paraneoplastic context [22, 23]. Thus, it is advisable to have some index of suspicion for a paraneoplastic disorder in patients presenting with a subacute neurological illness where an alternative cause is not immediately obvious.
In cases where an antibody is detected, it is possible to stratify the likelihood of cancer according to the specific antibody. Antibodies are termed “high risk” when the likelihood of cancer is > 70%, “intermediate risk” when there is a 30–70% association with cancer, and “low risk” when the probability of cancer is < 30%, Tables 2 and 3 [1••]. The PNS-Care score allows possible, probable, and definite levels of paraneoplastic diagnostic certainty, based on the level of risk from the neurological phenotype, antibody detected, and cancer found [1••]. For example, a woman with cerebellar ataxia (high risk syndrome, = 3), PCA-1 (high risk antibody, = 3), and ovarian adenocarcinoma (cancer consistent with phenotype and antibody, = 4) would have a score of 10, and thus fulfill definite criteria (score ≥ 8 required).
Although paraneoplastic neurologic disorders often precede cancer detection, most cases occur in the context of an existing cancer diagnosis [5••]. The timing of cancer presentation may vary, depending on the antibody specificity. For example, most Purkinje cytoplasmic antibody type 1 (PCA-1, anti-Yo) paraneoplastic cerebellar degeneration cases manifest subsequent to cancer diagnosis, whereas the reverse is true in Kelch-like protein (KLHL)-11 autoimmunity [24–26, 27•]. When the neurological disorder is encountered first, 92% of cancers are detected within 5 years of symptom onset [5••]. However, recent consensus guidelines note that in a majority of cases, cancer is identified within 2 years of the onset of symptoms [1••]. For example, the mean interval to cancer diagnosis is 6 months in KLHL-11 rhombencephalitis, 6.5 months in ANNA-1 encephalomyelitis, and 9 months in ANNA-2-(anti-Ri)-associated disorders [2, 27•, 28]. A list of intracellular and cell-surface antibodies along with their clinical and tumor associations are provided in Tables 2 and 3. Paraneoplastic neurologic disorder phenotypes are considered in rostrocaudal order below.
Central Nervous System
Limbic Encephalitis
Limbic encephalitis is considered a high-risk paraneoplastic phenotype. Patients with limbic encephalitis present with subacute onset and rapid progression over approximately 3 months with working memory deficits, seizures, or psychiatric symptoms. Those with typical limbic encephalitis have bilateral T2 signal abnormalities of limbic structures on MRI, and at least one of CSF pleocytosis or EEG with temporal lobe findings (slow waves, seizures, or both). Patients with a paraneoplastic antibody could meet diagnostic criteria without fulfilling all of those criteria (EEG, CSF white cell count, and MRI findings) [29]. Paraneoplastic limbic encephalitis occurs most commonly in association with ANNA-1 and CRMP-5-IgG. SCLC is the most frequent neoplasm [30]. In the experience of the authors, forme frustes of limbic encephalitis can also occur, mostly isolated limbic seizures without cognitive impairment, though usually this occurs in a non-paraneoplastic context such as LGI1 or GAD65 autoimmunity.[31].
Encephalitis (Extra-limbic)
Extra-limbic encephalitis is considered intermediate risk for a paraneoplastic cause. The term “extra-limbic” refers to clinical, radiological, or EEG findings indicative of neocortical temporal or extra-temporal localization, including multifocal disorders. Examples include anti-NMDA-receptor encephalitis, gamma-aminobutyric acid-A receptor (GABAA-R) encephalitis, dipeptidyl-peptidase-like protein (DPPX) encephalitis, and autoimmune GFAP astrocytopathy. Clinical and radiological presentations are diverse (Table 2). Anti-NMDA-R encephalitis classically manifests with a psychiatric prodrome followed by seizures, movement disorders, autonomic instability, and coma. Normal MRI and CSF pleocytosis are typical and neoplasm (usually ovarian teratoma) occurs in 38% [32, 33]. GABAA-R encephalitis usually presents with a multifocal extra-limbic syndrome with seizures, with multifocal non-enhancing deep or subcortical white matter lesions sometimes in association with thymoma [34, 35]. DPPX encephalitis classically presents with a triad of gastrointestinal disturbance, CNS hyperexcitability (spasms, myoclonus), and encephalopathy, with normal MRI imaging, in association, rarely, with B cell neoplasia [36, 37]. Autoimmune GFAP astrocytopathy presents with various neuropsychiatric symptoms, meningeal symptoms (blurred vision, headache) and sometimes tremor, or myelopathic sensory and autonomic findings, accompanied by CSF pleocytosis. MRI imaging typically includes diverse patterns of post-gadolinium enhancement (classically following the path of deep white matter glial cells running perpendicular to the corpus callosum). Approximately one-third have accompanying neoplasms of diverse histologies [38, 39].
Encephalomyelitis
Patients with encephalomyelitis have a multifocal neurological disorder with symptoms referable to both spinal cord and cerebral cortex, but which may also include symptoms localizing to peripheral nerve, nerve root, or dorsal root ganglia [40]. Terms such as encephalomyeloneuritis or encephalomyeloradiculitis are also employed to more precisely define the clinical phenotype. Paraneoplastic encephalomyelitis is considered a high-risk phenotype and typically has an association with SCLC and one or more of ANNA-1, CRMP-5, and amphiphysin antibodies [2, 41, 42]. Among more recently described syndromes, autoimmune GFAP astrocytopathy (a low risk antibody) may present with encephalomyelitis, though usually with distinctive CSF pleocytosis and inflammatory-appearing T2- or T1-post gadolinium findings within cerebral white matter and central spinal cord. Associated cancers include adenocarcinomas or ovarian teratoma [38]. In myelin oligodendrocyte glycoprotein antibody disease, a report of encephalomyelitis in the context of a MOG protein-expressing teratoma suggests the potential to rarely occur as a paraneoplastic phenomenon [43].
Brainstem Syndromes
Paraneoplastic rhombencephalitis presents with symptoms localizing to the brainstem and cerebellar connections including gait disturbance, postural instability, oscillopsia, vertigo, diplopia, dysarthria, dysphagia, sleep disorders (such as sleep disordered breathing, stridor, dream enactment behavior), and cranial neuropathies [44]. Supranuclear gaze palsies can resemble those of progressive supranuclear palsy [45]. Specific signs suggestive of certain antibody specificities include jaw dystonia and ANNA-2 (in the setting of breast adenocarcinoma) and vestibulocochlear symptoms (vertigo, tinnitus and hearing loss) in men with KLH-11 antibody (seminoma in 70%) [26, 27•, 28]. Listeria rhombencephalitis is an important subacute-onset differential diagnostic consideration [46].
Rapidly Progressive Cerebellar Ataxia
This high-risk phenotype presents with gait, speech, and dysmetric limb findings (a pancerebellar disorder) with significant disability accruing over 12 weeks. More rapid and slowly progressive forms have also been described [47]. For this phenotype, a diverse range of antibodies (paraneoplastic and non-paraneoplastic) could be considered, starting with those more common with higher cancer risk (such as PCA-1 [ovarian and breast adenocarcinoma associated], PCA-Tr and metabotropic glutamate receptor [mGluR]-1, which are both lymphoma associated), while research-based testing could be considered for the less common and infrequently cancer-associated analytes (such as inositol triphosphate receptor [ITPR]-1 and septin-7 antibodies) [25, 48–51]. Distinct clinical features may aid in the diagnosis of certain ataxias such as loss of taste sensation in mGluR1 ataxia and episodic ataxia which can arise in the context of contactin-associated protein-like 2 (CASPR2) autoimmunity (sometimes accompanying thymoma) [52, 53].
Opsoclonus-Myoclonus Syndrome
Opsoclonus-myoclonus syndrome (OMS) is subjectively characterized by generalized tremulousness and oscillopsia, and objectively by arrhythmic, multidirectional conjugate saccadic eye movements (opsoclonus) accompanied by limb and trunk myoclonus. Other clinical features may include ataxia, encephalopathy, and sleep disturbance [54]. In children, opsoclonus-myoclonus is strongly associated with neuroblastoma [55]. Among adults, OMS mostly arises as an idiopathic autoimmune phenomenon. Paraneoplastic antibodies encountered include ANNA-2 antibodies (less commonly ANNA-1). Breast adenocarcinoma and SCLC are the more frequently reported oncological accompaniments [54, 56]. OMS accompanying encephalopathy sometimes occurs in anti-NMDA-R encephalitis.
Myelopathy
Isolated paraneoplastic myelopathy manifests with subacute or insidiously progressive spinal cord signs such as motor weakness, bowel, or bladder disturbance. It can also present with tract-specific signs such as a dorsal column syndrome. Associated antibody specificities include CRMP-5, amphiphysin, ANNA-1, and neuronal intermediate filaments (particularly neurofilament light chain) [57•, 58]. Small cell carcinoma and breast adenocarcinoma are the typical neoplastic associations [58]. Although more commonly an idiopathic autoimmune phenomenon, a paraneoplastic myelitis can occur in the setting of neuromyelitis optica spectrum disorder (NMOSD) mediated by aquaporin-4 (AQP4) antibodies. Neoplastic accompaniments include thymoma, and breast and lung carcinomas [59]. Older age of onset, especially in men, is a risk factor for paraneoplastic NMOSD [60].
Stiff Person Syndrome
Paraneoplastic stiff person syndrome (SPS) accounts for just 1–2% of all SPS cases [61]. Stiff person syndrome arises usually as an idiopathic autoimmune phenomenon associated with glutamic acid decarboxylase (GAD65) or glycine receptor antibodies [61]. The classical SPS clinical presentation includes muscle rigidity and spasms, symmetrically involving the trunk and proximal lower limbs. Symptoms are exacerbated by emotional distress or startle. Partial forms affecting one limb or the trunk in isolation have also been described. In rare cases of SPS, usually among older male patients, GAD65 antibodies may occur in association with lung, breast, or thymic neoplasms [62]. In the setting of glycine receptor autoimmunity, the progressive encephalomyelitis with rigidity and myoclonus (PERM) phenotype is most common [63]. This represents SPS findings in the context of a widespread encephalomyelitis. Though usually idiopathic in etiology, lymphoma and thymoma are among reported neoplastic accompaniments [64].
Peripheral Nervous System, Neuromuscular Junction, and Muscle
Polyradiculopathy
Paraneoplastic polyradiculopathy or polyradiculoneuropathy occurs in isolation or as part of a multifocal encephalomyeloneuropathic disorder. CRMP-5 polyradiculoneuropathy usually presents with a painful, asymmetric axonal neuropathy. Amphiphysin autoimmunity is associated with a symmetric axonal neuropathy [66–68]. Microtubule-associated protein 1B (MAP1B) autoimmunity more commonly presents with a painless polyradiculoneuropathy [69•]. The most common oncological association for MAP1B and CRMP-5 IgGs is small cell carcinoma. Adenocarcinoma of breast or SCLC are the usual oncologic accompaniments of amphiphysin-IgG [67, 68, 69•].
Subacute Sensory Neuronopathy
Subacute sensory neuronopathy is a high-risk paraneoplastic phenotype in which the disorder localizes to the dorsal root ganglion. Patients develop sensory loss subacutely over weeks or months, initially affecting vibration and proprioception, followed by involvement of the other sensory modalities. The clinical picture is one of profound sensory ataxia often with accompanying pseudoathethosis on clinical examination [70]. The presence of ANNA-1 and accompanying SCLC is prototypic, but subacute sensory neuronopathy can also arise in the setting of CRMP-5, amphiphysin, and MAP1B antibodies [67, 68, 69•, 71].
Autonomic Neuropathy
Chronic gastric pseudo-obstruction represents a focal form of autoimmune autonomic neuropathy. This disorder presents with abdominal distension, cramping, nausea, vomiting, and weight loss not explained by mechanical obstruction. Other or additional gastroenterologic localizations may be encountered, namely small-bowel pseudo-obstruction or large-bowel obstipation. One or more other features of dysautonomia may accompany the gastroenterologic complaints (orthostatic hypotension, mydriasis, heat intolerance due to anhidrosis, erectile dysfunction, and urinary retention) [72, 73]. Serological findings include ANNA-1, in the setting of small cell carcinoma, or (occasionally in the authors’ experience) thymoma. Autoimmune autonomic ganglionopathy with ganglionic nicotinic acetylcholine receptor (α3-AChR) antibody detected is occasionally paraneoplastic in etiology, associated with diverse cancer types, including small cell carcinoma and thymoma [73, 74]. When autonomic neuropathy and sensorimotor neuropathy co-exist, the typical oncological accompaniment is SCLC [74].
Neuromuscular Junction Disorders
Lambert Eaton myasthenic syndrome (LEMS) is a high-risk paraneoplastic phenotype accompanied by small cell carcinoma in 50% generally, with an even higher risk in smokers older than 50 with weight loss [75]. Though P/Q-type calcium channel antibody is a general biomarker for LEMS, coexisting antiglial/neuronal nuclear antibody (AGNA, or Sry-like high-mobility group box protein 1 [SOX-1]-IgG) or ANNA-1 positivity is more predictive of small cell carcinoma in those cases [76]. In a study assessing for risk for SCLC in LEMS patients, age at onset, smoking behavior, weight loss, Karnofsky performance status, bulbar involvement, male sexual impotence, and the presence of SOX-1 antibody were independent predictors [77]. A DELTA-P score was derived allocating 1 point for the presence of each of the following items at or within 3 months from onset: age at onset ≥ 50 years, smoking at diagnosis, weight loss ≥ 5%, bulbar involvement, erectile dysfunction, and Karnofsky performance status lower than 70. A DELTA-P score of 0 or 1 corresponded to a < 3% chance of SCLC, whereas a score of ≥ 4 or more had a positive predictive value for SCLC of > 90% [77]. Myasthenia gravis is associated with thymoma in 10–15% of cases [78].
Neuromyotonia
Peripheral nerve excitability, or neuromyotonia, is characterized by stiffness, muscle cramps, fasciculations, and/or myokymia. Dysautonomia including hypohidrosis, orthostatic hypotension, and gastric dysmotility may co-occur in patients with LGI1 or CASPR2 antibodies [79]. Morvan’s syndrome is the co-occurrence of neuromyotonia with central nervous system dysfunction such as encephalopathy, seizures, and insomnia (termed “agrypnia excitata”). Morvan’s syndrome carries a 50% risk of thymoma, usually accompanying CASPR2 antibodies [80, 81].
Myopathy
Dermatomyositis is a form of immune-mediated myopathy which carries a 15% risk of malignancy [82]. It presents with proximal muscle weakness and cutaneous findings such as a periorbital heliotrope rash or violaceous papules over the dorsal aspect of the metacarpophalangeal joints (“Gottron papules”). Dermatomyositis is associated with a wide range of malignancies and the histologic type varies according to ethnicity and underlying population risk profile [83]. Certain antibodies such as nuclear matrix protein 2, transcription intermediary factor 1 gamma, and small ubiquitin-like modifier 1 activating enzyme subunit occur more frequently in paraneoplastic dermatomyositis [84]. Necrotizing autoimmune myopathy is characterized by painful, proximal muscle weakness and can occur as a paraneoplastic phenomenon in 10% of cases, some of whom are signal recognition particle (SRP) antibody positive. The most frequent cancer association is gastrointestinal adenocarcinoma [85].
Diagnostic Evaluation
Clinical Assessment
The neurological clinical assessment should pay particular attention to the trajectory of symptom onset and progression. In almost all circumstances, onset is subacute. Neurological exam should assist in localizing the disorder to one or more anatomic regions of the nervous system. Risk factors to consider when evaluating paraneoplastic neurologic disorders include a current or remote history of malignancy, smoking history, coexisting non-neurologic autoimmune disease history, and exposure to immune checkpoint inhibitors.
Imaging
MRI
In paraneoplastic limbic encephalitis, T2 hyperintensity of the mesial temporal lobes is typical, occasionally with contrast enhancement (Fig. 1) [86]. In extra-limbic paraneoplastic encephalitides, MRI may demonstrate extra-temporal lobar abnormalities that can resemble infectious or neoplastic processes [87]. Linear, radial enhancement extending from the lateral ventricles is a characteristic MRI imaging finding in GFAP astrocytopathy [38]. Distinctive radiological features also arise in GABAA-R encephalitis characterized by multi-lobar deep white matter and juxtacortical lesions on FLAIR imaging (without enhancement), Fig. 1 [88]. In many instances, MRI of the brain can be entirely normal, including most patients with anti-NMDA receptor encephalitis [32].

MRI and antibody test findings in paraneoplastic disorders. Top row, axial MRI images demonstrate (left, T2 FLAIR) bilateral mesial temporal hyperintensities in a patient with ANNA-1-associated paraneoplastic limbic encephalitis (middle, T2 FLAIR) multifocal extra-limbic hyperintensities in a patient with GABA-A receptor (R) encephalitis, and (right, T1 post-gadolinium) spinal cord tractopathies in a patient with seronegative renal cell carcinoma-associated paraneoplastic myelopathy. Bottom row, indirect immunofluorescence assay using mouse brain tissue as substrate reveals a typical synaptic pattern of IgG staining of hippocampus (left) and cerebellum (right) produced by CSF from a patient with GABA-A-R encephalitis. GABA-A-R specificity was confirmed by GABA-A-R alpha 1 subunit specific cell-based assay (not shown). Top right image from reference 58 reproduced with permission from American Academy of Neurology
The hallmark radiographic feature of paraneoplastic myelitis is longitudinally extensive, gadolinium-enhancing T2 signal change selective for individual spinal tracts, sometimes with enhancement post-gadolinium (Fig. 1). This finding most commonly affects the dorsal and lateral columns. However, the MRI may be normal in half of all cases [66, 89].
Nerve root enhancement is a radiological feature present in one-third of patients with paraneoplastic myeloneuropathy [66]. In paraneoplastic myopathy, MRI can support the diagnosis, localize the affected muscles for a targeted biopsy, and in certain cases distinguish between myositis subtypes [90].
Neurophysiology
Neurophysiological testing can aid in localizing a paraneoplastic neurologic disorder. The extreme delta brush pattern on EEG occurring in one-third of patients with anti-NMDA receptor encephalitis is associated with more severe illness [91]. Slow wave or epileptiform activity localized to the temporal lobe may aid reaching a diagnosis of limbic encephalitis [29]. Continuous motor-unit activity on EMG, agonist–antagonist co-contraction, exaggerated acoustic startle, and exteroceptive responses are characteristic of stiff person syndrome [92]. In neuromyotonia, nerve conduction studies reveal stimulus-induced after discharges and EMG characteristically demonstrates spontaneous firing of doublet or multiplet motor unit discharges indicative of peripheral nerve hyperexcitability [93]. Pre- and post-synaptic neuromuscular junction disorders can be distinguished by compound muscle action potential amplitude responses to repetitive nerve stimulation (increment in LEMS, decrement in myasthenia gravis) [94].
Immunological testing
Antibody testing
Neural antibodies, which are of IgG class, belong to 2 broad groups, those mostly “high risk” and reactive with linear epitopes of nuclear (e.g., Hu), nucleolar (e.g., Ma2), or cytoplasmic (e.g., Yo) antigens, and those of “intermediate” or “low” risk mostly reactive with cell surface protein conformation-dependent extracellular epitopes, such as GluN1 subunit of NMDA receptor (Tables 2 and 3). Antibody testing should be performed in both serum and CSF in a laboratory with expertise diagnosing autoimmune neurologic disorders. Certain IgGs are more readily detected in serum (e.g., LGI1 antibody) and others are more readily and specifically detected in CSF (e.g., NMDA-R and GFAP antibodies) [38, 79, 95]. Tissue-based immunohistochemistry or immunofluorescence serves as a screening test for most antibodies; a high degree of interpretative experience is necessary to precisely identify these (Fig. 1). IgG specificities identified should be confirmed by protein-specific methods, such as western blot, ELISA or radioimmunoprecipitation assays for intracellular antigens, and transfected cell-based assays (observer-based or flow cytometry) for cell-surface antibodies. It is not recommended to rely on commercial line blots alone and positive results, where possible, should be correlated with tissue immunohistochemistry [96•]. In the authors’ experience, certain antibodies are not sensitively detected by tissue-based assays (e.g., LGI1, CASPR2, AQP4-IgG, and MOG-IgG), but are detectable sensitively and specifically by optimized cell-based assays, either by microscopy or by flow cytometry [97]. The absence of an antibody does not preclude the diagnosis of a paraneoplastic neurologic disorder. Where a high index of suspicion persists in seronegative cases, serum and CSF samples could also be referred to a laboratory with research expertise for novel antibody testing [1••].
CSF
Routine findings on CSF that are supportive of a paraneoplastic neurologic disorder include pleocytosis, the presence of CSF-exclusive oligoclonal bands or a raised CSF IgG index or synthesis rate. Cytology should be performed to exclude direct rather than remote effect of systemic cancer. CSF glucose levels are not abnormal in paraneoplastic neurologic disorders but can be reduced in metastatic cancer or infection [98]. Normal CSF parameters do not exclude a paraneoplastic diagnosis [99].
Cancer Screening
Cancer testing may be quite selective, when guided by the antibody detected, or could be a broad search in seronegative patients or in those with an antibody with less specific cancer associations. CT imaging of chest, abdomen, and pelvis is usually pursued. This could be supplemented, as appropriate, with CT of the neck, ultrasound, or MRI of the gynecologic tract, ultrasound of testes, pelvic MRI, mammography, and gastroenterologic endoscopies. Fluorodeoxyglucose-PET-CT (orbits to thighs) is useful for detecting carcinoma or lymphoma in primary screening, or after normal CT imaging [100]. High-resolution CT or MRI is preferred for thymoma detection. Paraneoplastic antibodies may direct the search for cancer and related management decisions. For example, in a man with KLHL-11 or Ma2 antibodies, testicular seminoma or other germinoma would be suspected [27•, 44]. For patients with Ma2 antibodies, a compatible neurologic phenotype and testicular microcalcifications on ultrasound, orchiectomy would be considered for patients under 50 years [1••]. Similarly, in post-menopausal women with PCA-1, exploratory surgery or prophylactic hysterectomy and bilateral salpingo-oophorectomy might be considered even if imaging tests were negative [1••]. While improvements may be encountered in KLHL-11 patients (50%), they are almost always absent in PCA-1 patients (85%) [25, 26]. For patients with a high-risk antibody accompanied by a compatible neurologic syndrome without underlying cancer, it is recommended that tumor screening should be repeated every 4–6 months for 2 years. The same applies for intermediate-risk antibodies but only if accompanied by a high-risk phenotype (Table 1). For low-risk antibodies, cancer screening at the time of diagnosis is deemed sufficient; however, decisions should be guided by individual patient assessment, presence of risk factors, and clinical judgment [1••].
Pathology
In the context of suspected autoimmune encephalitis, occasional patients meet the criteria for seronegative probable autoimmune encephalitis on the basis of inflammatory brain biopsy findings, and one other supportive finding (inflammatory CSF or MRI) [29]. In that setting, a search for cancer might be undertaken, though the overall risk for cancer in a seronegative non-limbic encephalitis is low [101]. In myositis, specific features on muscle biopsy aid in arriving at a diagnosis of dermatomyositis or immune-mediated necrotising myopathy [102]. Though not done routinely, a paraneoplastic diagnosis may be strengthened by detecting the cognate antigen for a low-risk antibody in the patient neoplasm by immunohistochemistry [1••].
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors (ICIs) are a class of monoclonal antibodies which inhibit the immune-checkpoints that exert a negative regulatory effect on the immune system [103]. Targets include cytotoxic T-lymphocyte antigen 4 (CTLA-4: ipilimumab), programmed cell death protein 1 (rembrolizumab, nivolumab), and programmed cell death ligand 1 (durvalumab) [104]. These therapies restore host antitumour immunity, promote tumor cell death, and have been associated with improved outcomes even in patients with advanced malignancy [105]. Novel toxicities termed immune-related adverse events (irAEs) have emerged in association with ICIs. Neurologic irAEs (nirAE) are thought to affect 1% of patients and include myositis, myasthenia gravis, and inflammatory neuropathies including Guillain-Barré syndrome [106]. This risk increases when anti-CTLA-4 is used in combination with anti-PD-1/anti-PD ICIs [107, 108]. Neuromuscular and peripheral nirAEs are more common than disorders with central nervous system involvement, and myasthenia gravis has the highest fatality rate among all nirAEs [109].
In 2019, Graus and colleagues defined three clinical scenarios where nirAEs can be considered to meet criteria for paraneoplastic neurologic disorders: (i) when the symptoms of the nirAE are identical to that of a classical paraneoplastic neurologic disorders, irrespective of the presence of paraneoplastic antibodies; (ii) any nirAE in association with the detection of paraneoplastic antibodies provided other causes are excluded; (iii) nirAEs in the presence of cell-surface protein or synaptic antibodies in the presence of a typical tumor [110••].
Paraneoplastic neurologic syndromes often respond poorly to systemic immunotherapy due to irreversible cytotoxic T cell–driven neuronal injury [16]. However, in cases associated with ICI use, withdrawal of the ICI in addition to treatment with one or more of corticosteroids, IVIg and plasma exchange can be beneficial [14]. Administration of corticosteroids in particular has been associated with favorable clinical outcomes, even in disorders that do not typically demonstrate a favorable response to steroids (e.g., Guillain-Barré syndrome) [14, 109]. Seronegative cases have been reported in the context of ICI use and therefore the absence of high or intermediate-risk antibodies does not exclude the diagnosis [111].
Treatment
Treatment includes nervous system–directed immunotherapy, cancer-specific treatment, and symptomatic therapy.
Immunotherapy
In general, disorders associated with cell-surface antibodies respond to immunotherapy better than other paraneoplastic disorders, whereas those associated with intracellular anti-neuronal antibodies respond less favorably [16]. Class 1 evidence for immunosuppressant therapy is lacking and treatment strategies are guided by antibody type (cell surface-directed versus intracellular), expert opinion, and small case series. Improvements may be modest in the case of disorders associated with antibodies to intracellular antigens and arresting further neurological decline is often the therapy goal. It is recommended that baseline neurological assessments be obtained and recorded prior to initiation of therapy to evaluate the degree of treatment response. A trial of immunotherapy typically consists of IV methylprednisolone 1 g and/or intravenous immunoglobulin (IVIg) 0.4 g/kg daily for 5 days and can occur during or after cancer treatment. This may be followed by weekly doses for 6–12 weeks, or a slow oral taper of prednisone. Plasmapheresis every second day for 5–7 treatments can also be trialed upfront or used in treatment-resistant cases. In syndromes associated with intracellular antigen-directed antibodies, neuronal injury is evidentially cytotoxic T cell–mediated and therefore cyclophosphamide can be used [112]. Rituximab has generally been considered a second-line agent in patients with disorders associated with cell-surface antibodies, though in anti-NMDA-R encephalitis it is frequently used early in the disease course of patients with severe neurological disease without waiting for outcomes of first-line therapy (steroids and IVIg or plasma exchange) [113].
Relapsing disease is uncommon, though it can occur in 20–30% of patients with certain intermediate or low-risk antibodies such as anti-NMDA-R encephalitis, autoimmune GFAP astrocytopathy, PERM, and LgI1 encephalitis [32, 38, 64, 79]. For those relapsing patients, maintenance immunotherapy considerations include intravenous rituximab or oral agents such as azathioprine, mycophenolate mofetil, or methotrexate. For cases requiring chronic therapy, the weaning of intravenous methylprednisolone or IVIg should occur in a cautious manner, extending the dosing interval gradually over 3–6 months, to mitigate against the risk of relapse. For paraneoplastic disorders in the setting of ICI use, the treatment involves withdrawal of the ICI and administration of corticosteroids (IV regimen as detailed above or oral prednisolone 60–80 mg for 1–2 weeks) [14]. There is no established consensus regarding the duration of chronic immunotherapy in paraneoplastic neurologic disorders.
Cancer Treatment
The treatment of cancer, as guided by oncology and surgery, may coincide with stabilization or improvement of neurological symptoms. Some patients with anti-NMDA-R encephalitis have marked neurological improvements after ovarian teratoma resection [32]. However, because this response is variable, immune therapy is universally recommended in addition to teratoma removal [113]. In many cases, cancer treatment may at best result in stabilization of the neurological disorder such as in PCA-1 cerebellar ataxia [25].
Symptomatic Therapy
Anti-seizure medications should be trialed for symptomatic seizures but immunotherapy is characteristically more effective for seizure control [114]. Benzodiazepines (usually diazepam) are used for SPS [61]. Other movement disorders may respond to symptomatic therapy such as parkinsonism (levodopa), myoclonus (benzodiazepines), or dystonia (trihexyphenidyl or botulinum toxin). Carbamazepine or phenytoin are employed to manage muscle cramps and stiffness in paraneoplastic neuromyotonia [115]. Pyridostigmine is used in gastrointestinal pseudo-obstruction, myasthenia gravis, and LEMS (with 3,4 diaminopyridine for the latter) [115,116,117]. Neuropathic pain medications such as gabapentin, pregabalin, and tricyclic anti-depressants are employed in paraneoplastic neuropathy, but cancer treatment remains the optimal means of stabilizing symptoms [117].
Conclusions
The field of paraneoplastic neurologic disorders has been advanced through updated diagnostic criteria aiding classification. The prevalence of these disorders is greater than previously reported. With increased use of immune-checkpoint inhibitors, it is possible that the incidence of paraneoplastic neurologic disorders will continue to rise. IgG biomarkers have diagnostic, therapeutic, and prognostic utility and can guide management decisions. Many paraneoplastic neurologic disorders remain seronegative.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Graus F, Vogrig A, Muñiz-Castrillo S, Antoine JCG, Desestret V, Dubey D, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm 2021;8:e1014. https://doi.org/10.1212/NXI.0000000000001014. An important consensus document on the updated diagnostic classification and terminology of paraneoplastic neurologic disorders.
Graus F, Keime-Guibert F, Reñe R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138–48. https://doi.org/10.1093/brain/124.6.1138.
Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424–34. https://doi.org/10.1002/ana.21589.
• Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 2016;73:1297–307. https://doi.org/10.1001/jamaneurol.2016.2549. Although more often an idiopathic autoimmune disorder, GFAP astrocytopathy can occur as a paraneoplastic disorder in approximately one third of cases.
•• Vogrig A, Gigli GL, Segatti S, Corazza E, Marini A, Bernardini A, et al. Epidemiology of paraneoplastic neurological syndromes: a population-based study. J Neurol. 2020;267:26–35. https://doi.org/10.1007/s00415-019-09544-1. The first epidemiological study of paraneoplastic neurologic disorders which highlighted a much higher prevalence of these disorders than previously reported.
Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. 2017;97:839–87. https://doi.org/10.1152/physrev.00010.2016.
Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain. 2012;135:1622–38. https://doi.org/10.1093/brain/aws082.
Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain. 1968;91:481–96. https://doi.org/10.1093/brain/91.3.481.
Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543–54. https://doi.org/10.1056/NEJMra023009.
Gozzard P, Woodhall M, Chapman C, Nibber A, Waters P, Vincent A, et al. Paraneoplastic neurologic disorders in small cell lung carcinoma. Neurol. 2015;85:235–9. https://doi.org/10.1212/WNL.0000000000001721.
• Shah S, Flanagan EP, Paul P, Smith CY, Bryant SC, Devine MF, et al. Population-based epidemiology study of paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm 2022;9:e1124. https://doi.org/10.1212/NXI.0000000000001124. Prevalence and incidence of paraneoplastic neurologic disorders in Olmsted county, Minnesota.
Hébert J, Riche B, Vogrig A, Muñiz-Castrillo S, Joubert B, Picard G, et al. Epidemiology of paraneoplastic neurologic syndromes and autoimmune encephalitides in France. Neurol Neuroimmunol Neuroinflamm 2020;7:e883. https://doi.org/10.1212/NXI.0000000000000883.
Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. 2018;83:166–77. https://doi.org/10.1002/ana.25131.
Dubey D, David WS, Reynolds KL, Chute DF, Clement NF, Cohen JV, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing spectrum. Ann Neurol. 2020;87:659–69. https://doi.org/10.1002/ana.25708.
Sechi E, Markovic SN, McKeon A, Dubey D, Liewluck T, Lennon VA, et al. Neurologic autoimmunity and immune checkpoint inhibitors: autoantibody profiles and outcomes. Neurol. 2020;95:e2442–52. https://doi.org/10.1212/WNL.0000000000010632.
McKeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol. 2011;122:381–400. https://doi.org/10.1007/s00401-011-0876-1.
Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;76:108–19. https://doi.org/10.1002/ana.24195.
Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9:67–76. https://doi.org/10.1016/S1474-4422(09)70324-2.
Kornau HC, Kreye J, Stumpf A, Fukata Y, Parthier D, Sammons RP, et al. Human Cerebrospinal fluid monoclonal LGI1 autoantibodies increase neuronal excitability. Ann Neurol. 2020;87:405–18. https://doi.org/10.1002/ana.25666.
Debanne D, el Far O. Pre- and postsynaptic effects of LGI1 autoantibodies in a murine model of limbic encephalitis. Brain. 2018;141:3084–97. https://doi.org/10.1093/brain/awy271.
Hinson SR, Romero MF, Popescu BFGh, Lucchinetti CF, Fryer JP, Wolburg H, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci. 2012;109:1245–50. https://doi.org/10.1073/pnas.1109980108.
McKeon A, Pittock SJ, Glass GA, Josephs KA, Bower JH, Lennon VA, et al. Whole-body tremulousness. Arch Neurol. 2007;64:1318. https://doi.org/10.1001/archneur.64.9.1318.
O’Toole O, Lennon VA, Ahlskog JE, Matsumoto JY, Pittock SJ, Bower J, et al. Autoimmune chorea in adults. Neurol. 2013;80:1133–44. https://doi.org/10.1212/WNL.0b013e3182886991.
Shams’Ili S, Grefkens J, de Leeuw B, van den Bent M, Hooijkaas H, van der Holt B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126:1409–18. https://doi.org/10.1093/brain/awg133.
McKeon A, Tracy JA, Pittock SJ, Parisi JE, Klein CJ, Lennon VA. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: the cerebellum and beyond. Arch Neurol. 2011;68:1280–7. https://doi.org/10.1001/archneurol.2011.128.
Dubey D, Wilson MR, Clarkson B, Giannini C, Gandhi M, Cheville J, et al. Expanded clinical phenotype, oncological associations, and immunopathologic insights of paraneoplastic Kelch-like protein-11 encephalitis. JAMA Neurol. 2020;77:1420–9. https://doi.org/10.1001/jamaneurol.2020.2231.
• Mandel-Brehm C, Dubey D, Kryzer TJ, O’Donovan BD, Tran B, Vazquez SE, et al. Kelch-like protein 11 antibodies in seminoma-associated paraneoplastic encephalitis. N Engl J Med. 2019;381:47–54. https://doi.org/10.1056/nejmoa1816721. (A novel paraneoplastic rhombencephalitis which predominantly affects men with testicular cancer.)
Pittock SJ, Lucchinetti CF, Lennon VA. Anti-neuronal nuclear autoantibody type 2: paraneoplastic accompaniments. Ann Neurol. 2003;53:580–7. https://doi.org/10.1002/ana.10518.
Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15:391–404. https://doi.org/10.1016/S1474-4422(15)00401-9.
Lawn ND, Westmoreland BF, Kiely MJ, Lennon VA, Vernino S. Clinical, magnetic resonance imaging, and electroencephalographic findings in paraneoplastic limbic encephalitis. Mayo Clin Proc. 2003;78:1363–8. https://doi.org/10.4065/78.11.1363.
Quek AML, Britton JW, McKeon A, So E, Lennon VA, Shin C, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69:582–93. https://doi.org/10.1001/archneurol.2011.2985.
Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–65. https://doi.org/10.1016/S1474-4422(12)70310-1.
Dalmau J, Armangué T, Planagumà J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019;18:1045–57. https://doi.org/10.1016/S1474-4422(19)30244-3.
Spatola M, Petit-Pedrol M, Simabukuro MM, Armangue T, Castro FJ, Barcelo Artigues MI, et al. Investigations in GABA A receptor antibody-associated encephalitis. Neurol. 2017;88:1012–20. https://doi.org/10.1212/WNL.0000000000003713.
Pettingill P, Kramer HB, Coebergh JA, Pettingill R, Maxwell S, Nibber A, et al. Antibodies to GABAA receptor 1 and 2 subunits: clinical and serologic characterization. Neurol. 2015;84:1233–41. https://doi.org/10.1212/WNL.0000000000001326.
Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M, Roberts K, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73:120–8. https://doi.org/10.1002/ana.23756.
Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurol. 2014;83:1797–803. https://doi.org/10.1212/WNL.0000000000000991.
Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. 2017;81:298–309. https://doi.org/10.1002/ana.24881.
Gravier-Dumonceau A, Ameli R, Rogemond V, Ruiz A, Joubert B, Muñiz-Castrillo S, et al. Glial fibrillary acidic protein autoimmunity. Neurology. 2022;98:e653–68. https://doi.org/10.1212/wnl.0000000000013087.
Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135–40. https://doi.org/10.1136/jnnp.2003.034447.
Honnorat J, Cartalat-Carel S, Ricard D, Camdessanche JP, Carpentier AF, Rogemond V, et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry. 2008;80:412–6. https://doi.org/10.1136/jnnp.2007.138016.
Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol. 2005;58:96–107. https://doi.org/10.1002/ana.20529.
Wildemann B, Jarius S, Franz J, Ruprecht K, Reindl M, Stadelmann C. MOG-expressing teratoma followed by MOG-IgG-positive optic neuritis. Acta Neuropathol. 2021;141:127–31. https://doi.org/10.1007/s00401-020-02236-5.
Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127:1831–44. https://doi.org/10.1093/brain/awh203.
Adams C, McKeon A, Silber MH, Kumar R. Narcolepsy, REM sleep behavior disorder, and supranuclear gaze palsy associated with Ma1 and Ma2 antibodies and tonsillar carcinoma. Arch Neurol. 2011;68:521. https://doi.org/10.1001/archneurol.2011.56.
Weinstein AJ. Listeria rhombencephalitis. Arch Neurol. 1982;39:514. https://doi.org/10.1001/archneur.1982.00510200056012.
Simard C, Vogrig A, Joubert B, Muñiz-Castrillo S, Picard G, Rogemond V, et al. Clinical spectrum and diagnostic pitfalls of neurologic syndromes with Ri antibodies. Neurol Neuroimmunol Neuroinflamm 2020;7:e699. https://doi.org/10.1212/NXI.0000000000000699.
Alfugham N, Gadoth A, Lennon VA, Komorowski L, Scharf M, Hinson S, et al. ITPR1 autoimmunity: frequency, neurologic phenotype, and cancer association. Neurol - Neuroimmunol Neuroinflammation. 2018;5:e418. https://doi.org/10.1212/NXI.0000000000000418.
Bernal F, Shams’ili S, Rojas I, Sanchez-Valle R, Saiz A, Dalmau J, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurol. 2003;60:230–4. https://doi.org/10.1212/01.WNL.0000041495.87539.98.
Spatola M, Petit Pedrol M, Maudes E, Simabukuro M, Muñiz-Castrillo S, Pinto A-L, et al. Clinical features, prognostic factors, and antibody effects in anti-mGluR1 encephalitis. Neurol. 2020;95:e3012–25. https://doi.org/10.1212/WNL.0000000000010854.
Hinson SR, Honorat JA, Grund EM, Clarkson BD, Miske R, Scharf M, et al. Septin-5 and - 7-IgGs : neurologic, serologic, and pathophysiologic characteristics. Ann Neurol. 2022. https://doi.org/10.1002/ana.26482.
Lopez-Chiriboga AS, Komorowski L, Kümpfel T, Probst C, Hinson SR, Pittock SJ, et al. Metabotropic glutamate receptor type 1 autoimmunity. Neurol. 2016;86:1009–13. https://doi.org/10.1212/WNL.0000000000002476.
Joubert B, Gobert F, Thomas L, Saint-Martin M, Desestret V, Convers P, et al. Autoimmune episodic ataxia in patients with anti-CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm 2017;4:e371. https://doi.org/10.1212/NXI.0000000000000371.
Klaas JP, Ahlskog JE, Pittock SJ, Matsumoto JY, Aksamit AJ, Bartleson JD, et al. Adult-onset opsoclonus-myoclonus syndrome. Arch Neurol. 2012;69:1598–607. https://doi.org/10.1001/archneurol.2012.1173.
Antunes NL, Khakoo Y, Matthay KK, Seeger RC, Stram DO, Gerstner E, et al. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol. 2000;22:315–20. https://doi.org/10.1097/00043426-200007000-00007.
Oh SY, Kim JS, Dieterich M. Update on opsoclonus–myoclonus syndrome in adults. J Neurol. 2019;266:1541–8. https://doi.org/10.1007/s00415-018-9138-7.
• Basal E, Zalewski N, Kryzer TJ, Hinson SR, Guo Y, Dubey D, et al. Paraneoplastic neuronal intermediate filament autoimmunity. Neurol. 2018;91:E1677–89. https://doi.org/10.1212/WNL.0000000000006435. A novel paraneoplastic disorder which is characterised most often by ataxia or encephalopathy, and has an association with neuroendocrine tumours.
Flanagan EP, McKeon A, Lennon VA, Kearns J, Weinshenker BG, Krecke KN, et al. Paraneoplastic isolated myelopathy: clinical course and neuroimaging clues. Neurol. 2011;76:2089–95. https://doi.org/10.1212/WNL.0b013e31821f468f.
Pittock SJ, Lennon VA. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch Neurol 2008;65:629–632. https://doi.org/10.1001/archneur.65.5.629.
Sepúlveda M, Sola-Valls N, Escudero D, Rojc B, Barón M, Hernández-Echebarría L, et al. Clinical profile of patients with paraneoplastic neuromyelitis optica spectrum disorder and aquaporin-4 antibodies. Mult Scler J. 2018;24:1753–9. https://doi.org/10.1177/1352458517731914.
McKeon A, Robinson MT, McEvoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69:230–8. https://doi.org/10.1001/archneurol.2011.991.
Ariño H, Höftberger R, Gresa-Arribas N, Martínez-Hernández E, Armangue T, Kruer MC, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72:874. https://doi.org/10.1001/jamaneurol.2015.0749.
Carvajal-González A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain. 2014;137:2178–92. https://doi.org/10.1093/brain/awu142.
Hinson SR, Lopez-Chiriboga AS, Bower JH, Matsumoto JY, Hassan A, Basal E, et al. Glycine receptor modulating antibody predicting treatable stiff-person spectrum disorders. Neurol - Neuroimmunol Neuroinflammation. 2018;5:e438. https://doi.org/10.1212/NXI.0000000000000438.
de Camilli P, Thomas A, Cofiell R, Folli F, Lichte B, Piccolo G, et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of Stiff-Man syndrome with breast cancer. J Exp Med. 1993;178:2219–23. https://doi.org/10.1084/jem.178.6.2219.
Shah S, Vazquez Do Campo R, Kumar N, McKeon A, Flanagan EP, Klein C, et al. Paraneoplastic myeloneuropathies: clinical, oncologic, and serologic accompaniments. Neurol. 2021;96:e632-9. https://doi.org/10.1212/WNL.0000000000011218.
Dubey D, Lennon VA, Gadoth A, Pittock SJ, Flanagan EP, Schmeling JE, et al. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurol. 2018;90:e103–10. https://doi.org/10.1212/WNL.0000000000004803.
Dubey D, Jitprapaikulsan J, Bi H, do Campo RV, McKeon A, Pittock SJ, et al. Amphiphysin-IgG autoimmune neuropathy. Neurol. 2019;93:e1873-80. https://doi.org/10.1212/WNL.0000000000008472.
• Jitprapaikulsan J, Klein CJ, Pittock SJ, Gadoth A, Mckeon A, Mills JR, et al. Phenotypic presentations of paraneoplastic neuropathies associated with MAP1B-IgG. J Neurol Neurosurg Psychiatry. 2019. https://doi.org/10.1136/jnnp-2019-322175. A novel paraneoplastic neurologic disorder, first described in 2017, most often manifesting with neuropathy and cerebellar ataxia.
Chalk CH, Windebank AJ, Kimmel DW, McManis PG. The distinctive clinical features of paraneoplastic sensory neuronopathy. Can J Neurol Sci / J Can Sci Neurol. 1992;19:346–51. https://doi.org/10.1017/S0317167100041974.
Camdessanché J, Antoine J, Honnorat J, Vial C, Petiot P, Convers P, et al. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. Brain. 2002;125:166–75. https://doi.org/10.1093/brain/awf006.
Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurol. 1998;50:652–7. https://doi.org/10.1212/WNL.50.3.652.
Cutsforth-Gregory JK, McKeon A, Coon EA, Sletten DM, Suarez M, Sandroni P, et al. Ganglionic antibody level as a predictor of severity of autonomic failure. Mayo Clin Proc. 2018;93:1440–7. https://doi.org/10.1016/j.mayocp.2018.05.033.
Lee H-R, Lennon VA, Camilleri M, Prather CM. Paraneoplastic gastrointestinal motor dysfunction: clinical and laboratory characteristics. Am J Gastroenterol. 2001;96:373–9. https://doi.org/10.1111/j.1572-0241.2001.03454.x.
Maddison P, Lipka AF, Gozzard P, Sadalage G, Ambrose PA, Lang B, et al. Lung cancer prediction in Lambert-Eaton myasthenic syndrome in a prospective cohort. Sci Rep. 2020;10:10546. https://doi.org/10.1038/s41598-020-67571-9.
Zalewski NL, Lennon VA, Lachance DH, Klein CJ, Pittock SJ, Mckeon A. P/Q- and N-type calcium-channel antibodies: oncological, neurological, and serological accompaniments. Muscle Nerve. 2016;54:220–7. https://doi.org/10.1002/mus.25027.
Titulaer MJ, Maddison P, Sont JK, Wirtz PW, Hilton-Jones D, Klooster R, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) tumor association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;29:902–8. https://doi.org/10.1200/JCO.2010.32.0440.
Gilhus NE, Nacu A, Andersen JB, Owe JF. Myasthenia gravis and risks for comorbidity. Eur J Neurol. 2015;22:17–23. https://doi.org/10.1111/ene.12599.
Gadoth A, Pittock SJ, Dubey D, McKeon A, Britton JW, Schmeling JE, et al. Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG-positive patients. Ann Neurol. 2017;82:79–92. https://doi.org/10.1002/ana.24979.
Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72:241–55. https://doi.org/10.1002/ana.23577.
Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133:2734–48. https://doi.org/10.1093/brain/awq213.
Sigurgeirsson B, Lindelöf B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med. 1992;326:363–7. https://doi.org/10.1056/NEJM199202063260602.
Hsu JL, Liao MF, Chu CC, Kuo HC, Lyu RK, Chang HS, et al. Reappraisal of the incidence, various types and risk factors of malignancies in patients with dermatomyositis and polymyositis in Taiwan. Sci Rep 2021;11:4545. https://doi.org/10.1038/s41598-021-83729-5.
Yang H, Peng Q, Yin L, Li S, Shi J, Zhang Y, et al. Identification of multiple cancer-associated myositis-specific autoantibodies in idiopathic inflammatory myopathies: a large longitudinal cohort study. Arthritis Res Ther 2017;19:259. https://doi.org/10.1186/s13075-017-1469-8.
Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol. 2015;72:996–1003. https://doi.org/10.1001/jamaneurol.2015.1207.
Heine J, Prüss H, Bartsch T, Ploner CJ, Paul F, Finke C. Imaging of autoimmune encephalitis - relevance for clinical practice and hippocampal function. Neurosci. 2015;309:68–83. https://doi.org/10.1016/j.neuroscience.2015.05.037.
McKeon A, Ahlskog JE, Britton JA, Lennon VA, Pittock SJ. Reversible extralimbic paraneoplastic encephalopathies with large abnormalities on magnetic resonance images. Arch Neurol 2009;66:268–271. https://doi.org/10.1001/archneurol.2008.556.
Deng B, Cai M, Qiu Y, Liu X, Yu H, Zhang X, et al. MRI Characteristics of autoimmune encephalitis with autoantibodies to GABAA receptor. Neurol - Neuroimmunol Neuroinflammation. 2022;9:e1158. https://doi.org/10.1212/NXI.0000000000001158.
Flanagan EP, Keegan BM. Paraneoplastic myelopathy. Neurol Clin. 2013;31:307–18. https://doi.org/10.1016/j.ncl.2012.09.001.
Pinal-Fernandez I, Casal-Dominguez M, Carrino JA, Lahouti AH, Basharat P, Albayda J, et al. Thigh muscle MRI in immune-mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann Rheum Dis. 2017;76:681–7. https://doi.org/10.1136/annrheumdis-2016-210198.
Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurol. 2012;79:1094–100. https://doi.org/10.1212/WNL.0b013e3182698cd8.
Armon C, McEvoy KM, Westmoreland BF, McManis PG. Clinical neurophysiologic studies in stiff-man syndrome: use of simultaneous video-electroencephalographic-surface electromyographic recording. Mayo Clin Proc. 1990;65:960–7. https://doi.org/10.1016/S0025-6196(12)65157-X.
Bashford J, Chan WK, Coutinho E, Norwood F, Mills K, Shaw CE. Demystifying the spontaneous phenomena of motor hyperexcitability. Clin Neurophysiol. 2021;132:1830–44. https://doi.org/10.1016/j.clinph.2021.03.053.
Howard JF, Sanders DB, Massey JM. The electrodiagnosis of myasthenia gravis and the Lambert-Eaton myasthenic syndrome. Neurol Clin. 1994;12:305–30. https://doi.org/10.1016/S0733-8619(18)30099-9.
Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–77. https://doi.org/10.1016/S1474-4422(13)70282-5.
• Ruiz-García R, Martínez-Hernández E, Saiz A, Dalmau J, Graus F. The diagnostic value of onconeural antibodies depends on how they are tested. Front Immunol 2020;11:1482. https://doi.org/10.3389/fimmu.2020.01482. This paper demonstrates the diagnostic inaccuracy that can arise when solely using commercial line blots for antibody identification and advocates a dual approach with tissue immunohistochemistry.
Fryer JP, Lennon VA, Pittock SJ, Jenkins SM, Fallier-Becker P, Clardy SL, et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol - Neuroimmunol Neuroinflammation. 2014;1:e11. https://doi.org/10.1212/NXI.0000000000000011.
Chow E, Troy SB. The differential diagnosis of hypoglycorrhachia in adult patients. Am J Med Sci. 2014;348:186–90. https://doi.org/10.1097/MAJ.0000000000000217.
Escudero D, Guasp M, Ariño H, Gaig C, Martínez-Hernández E, Dalmau J, et al. Antibody-associated CNS syndromes without signs of inflammation in the elderly. Neurol. 2017;89:1471–5. https://doi.org/10.1212/WNL.0000000000004541.
McKeon A, Apiwattanakul M, Lachance DH, Lennon VA, Mandrekar JN, Boeve BF, et al. Positron emission tomography-computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67:322–9. https://doi.org/10.1001/archneurol.2009.336.
Lee W-J, Lee H-S, Kim D-Y, Lee H-S, Moon J, Park K-I, et al. Seronegative autoimmune encephalitis: clinical characteristics and factors associated with outcomes. Brain. 2022;145:3509–21. https://doi.org/10.1093/brain/awac166.
Vattemi G, Mirabella M, Guglielmi V, Lucchini M, Tomelleri G, Ghirardello A, et al. Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Autoimmun Highlights. 2014;5:77–85. https://doi.org/10.1007/s13317-014-0062-2.
Astaras C, de Micheli R, Moura B, Hundsberger T, Hottinger AF. Neurological adverse events associated with immune checkpoint inhibitors: diagnosis and management. Curr Neurol Neurosci Rep 2018;18:3. https://doi.org/10.1007/s11910-018-0810-1.
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94. https://doi.org/10.1200/JCO.2014.56.2736.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. https://doi.org/10.1038/nrc3239.
Cuzzubbo S, Javeri F, Tissier M, Roumi A, Barlog C, Doridam J, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. 2017;73:1–8. https://doi.org/10.1016/j.ejca.2016.12.001.
Farooq MZ, Aqeel SB, Lingamaneni P, Pichardo RC, Jawed A, Khalid S, et al. Association of immune checkpoint inhibitors with neurologic adverse events. JAMA Netw Open. 2022;5:e227722. https://doi.org/10.1001/jamanetworkopen.2022.7722.
Vogrig A, Muñiz-Castrillo S, Desestret V, Joubert B, Honnorat J. Pathophysiology of paraneoplastic and autoimmune encephalitis: genes, infections, and checkpoint inhibitors. Ther Adv Neurol Disord 2020;13:1–15. https://doi.org/10.1177/1756286420932797.
Marini A, Bernardini A, Gigli GL, Valente M, Muñiz-Castrillo S, Honnorat J, et al. Neurologic adverse events of immune checkpoint inhibitors: a systematic review. Neurol. 2021;96:754–66. https://doi.org/10.1212/WNL.0000000000011795.
•• Graus F, Dalmau J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2019;16:535–48. https://doi.org/10.1038/s41571-019-0194-4. A review of paraneoplastic neurologic disorders triggered by ICI use which provides a number of case definitions.
Wilson R, Menassa DA, Davies AJ, Michael S, Hester J, Kuker W, et al. Seronegative antibody-mediated neurology after immune checkpoint inhibitors. Ann Clin Transl Neurol. 2018;5:640–5. https://doi.org/10.1002/acn3.547.
Vernino S, O’Neill BP, Marks RS, O’Fallon JR, Kimmel DW. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro Oncol. 2004;6:55–62. https://doi.org/10.1215/S1152851703000395.
Nosadini M, Eyre M, Molteni E, Thomas T, Irani SR, Dalmau J, et al. Use and safety of immunotherapeutic management of N -methyl- d -aspartate receptor antibody encephalitis: a meta-analysis. JAMA Neurol. 2021;78:1333–44. https://doi.org/10.1001/jamaneurol.2021.3188.
Husari KS, Dubey D. Autoimmune epilepsy. Neurother. 2019;16:685–702. https://doi.org/10.1007/s13311-019-00750-3.
van Sonderen A, Wirtz PW, Verschuuren JJGM, Titulaer MJ. Paraneoplastic syndromes of the neuromuscular junction: therapeutic options in myasthenia gravis, Lambert-Eaton myasthenic syndrome, and neuromyotonia. Curr Treat Options Neurol. 2013;15:224–39. https://doi.org/10.1007/s11940-012-0213-6.
Taverna JA, Babiker HM, Yun S, Bishop MC, Lau-Braunhut S, Meyer PN, et al. The great masquerader of malignancy: chronic intestinal pseudo-obstruction. Biomark Res 2014;2:23. https://doi.org/10.1186/s40364-014-0023-y.
Antoine JC, Camdessanché JP. Treatment options in paraneoplastic disorders of the peripheral nervous system. Curr Treat Options Neurol. 2013;15:210–23. https://doi.org/10.1007/s11940-012-0210-9.
Simard C, Vogrig A, Joubert B, Muñiz-Castrillo S, Picard G, Rogemond V, et al. Clinical spectrum and diagnostic pitfalls of neurologic syndromes with Ri antibodies. Neurol - Neuroimmunol Neuroinflammation. 2020;7:e699. https://doi.org/10.1212/NXI.0000000000000699.
Chan KH, Vernino S, Lennon VA. ANNA-3 anti-neuronal nuclear antibody: marker of lung cancer-related autoimmunity. Ann Neurol. 2001;50:301–11. https://doi.org/10.1002/ana.1127.
Ortega Suero G, Sola-Valls N, Escudero D, Saiz A, Graus F. Anti-Ma and anti-Ma2-associated paraneoplastic neurological syndromes. Neurol (English Edition). 2018;33:18–27. https://doi.org/10.1016/j.nrleng.2016.05.004.
Graus F, Vincent A, Pozo-Rosich P, Sabater L, Saiz A, Lang B, et al. Anti-glial nuclear antibody: marker of lung cancer-related paraneoplastic neurological syndromes. J Neuroimmunol. 2005;165:166–71. https://doi.org/10.1016/j.jneuroim.2005.03.020.
Yu Z, Kryzer TJ, Griesmann GE, Kim K-K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49:146–54. https://doi.org/10.1002/1531-8249(20010201)49:2%3c146::AID-ANA34%3e3.0.CO;2-E.
Dalmau J, Tüzün E, Wu H, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. https://doi.org/10.1002/ana.21050.
Ruiz-García R, Martínez-Hernández E, Joubert B, Petit-Pedrol M, Pajarón-Boix E, Fernández V, et al. Paraneoplastic cerebellar ataxia and antibodies to metabotropic glutamate receptor 2. Neurol Neuroimmunol Neuroinflamm 2020;7:e658. https://doi.org/10.1212/NXI.0000000000000658.
Loehrer PA, Timmermann L, Pehl A, Bien CI, Pfestroff A, Pedrosa DJ. Rhombencephalitis associated with isolated Zic4-antibodies in Paraneoplastic cerebellar degeneration: a case report. BMC Neurol. 2020;20:208. https://doi.org/10.1186/s12883-020-01788-z.
Bataller L, Wade DF, Graus F, Stacey HD, Rosenfeld MR, Dalmau J. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurol. 2004;62:778–82. https://doi.org/10.1212/01.WNL.0000113749.77217.01.
Zekeridou A, Kryzer T, Guo Y, Hassan A, Lennon V, Lucchinetti CF, et al. Phosphodiesterase 10A IgG: a novel biomarker of paraneoplastic neurologic autoimmunity. Neurol. 2019;93:e815–22. https://doi.org/10.1212/WNL.0000000000007971.
Sabater L, Bataller L, Carpentier AF, Aguirre-Cruz ML, Saiz A, Benyahia B, et al. Protein kinase C autoimmunity in paraneoplastic cerebellar degeneration and non-small-cell lung cancer. J Neurol Neurosurg Psychiatry. 2006;77:1359–62. https://doi.org/10.1136/jnnp.2006.097188.
Valencia-Sanchez C, Knight AM, Hammami MB, Guo Y, Mills JR, Kryzer TJ, et al. Characterisation of TRIM46 autoantibody-associated paraneoplastic neurological syndrome. J Neurol Neurosurg Psychiatry. 2022;93:196–200. https://doi.org/10.1136/jnnp-2021-326656.
van Coevorden-Hameete MH, van Beuningen SFB, Perrenoud M, Will LM, Hulsenboom E, Demonet JF, et al. Antibodies to TRIM46 are associated with paraneoplastic neurological syndromes. Ann Clin Transl Neurol. 2017;4:680–6. https://doi.org/10.1002/acn3.396.
Do LD, Gupton SL, Tanji K, Bastien J, Brugière S, Couté Y, et al. TRIM9 and TRIM67 are new targets in paraneoplastic cerebellar degeneration. Cerebellum. 2019;18:245–54. https://doi.org/10.1007/s12311-018-0987-5.
Prevezianou A, Tzartos JS, Dagklis IE, Bentenidi E, Angelopoulos P, Bostantjopoulou S. Paraneoplastic cerebellar degeneration in a patient with breast cancer associated with carbonic anhydrase–related protein VIII autoantibodies. J Neuroimmunol. 2020;344:577242. https://doi.org/10.1016/j.jneuroim.2020.577242.
Bataller L, Sabater L, Saiz A, Serra C, Claramonte B, Graus F. Carbonic anhydrase-related protein VIII: autoantigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2004;56:575–9. https://doi.org/10.1002/ana.20238.
Sabater L, Gómez-Choco M, Saiz A, Graus F. BR serine/threonine kinase 2: a new autoantigen in paraneoplastic limbic encephalitis. J Neuroimmunol. 2005;170:186–90. https://doi.org/10.1016/j.jneuroim.2005.08.011.
Spatola M, Sabater L, Planagumà J, Martínez-Hernandez E, Armangué T, Prüss H, et al. Encephalitis with mGluR5 antibodies: symptoms and antibody effects. Neurol. 2018;90:e1964–72. https://doi.org/10.1212/WNL.0000000000005614.
Wallwitz U, Brock S, Schunck A, Wildemann B, Jarius S, Hoffmann F. From dizziness to severe ataxia and dysarthria: new cases of anti-Ca/ARHGAP26 autoantibody-associated cerebellar ataxia suggest a broad clinical spectrum. J Neuroimmunol. 2017;309:77–81. https://doi.org/10.1016/j.jneuroim.2017.05.011.
Dubey D, Honorat JA, Shelly S, Klein CJ, Komorowski L, Mills JR, et al. Contactin-1 autoimmunity: serologic, neurologic, and pathologic correlates. Neurol Neuroimmunol Neuroinflamm 2020;7:e771. https://doi.org/10.1212/NXI.0000000000000771.
Querol L, Nogales-Gadea G, Rojas-Garcia R, Martinez-Hernandez E, Diaz-Manera J, Suárez-Calvet X, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. 2013;73:370–80. https://doi.org/10.1002/ana.23794.
Shelly S, Kryzer TJ, Komorowski L, Miske R, Anderson MD, Flanagan EP, et al. Neurochondrin neurological autoimmunity. Neurol Neuroimmunol Neuroinflamm 2019;6:e612. https://doi.org/10.1212/NXI.0000000000000612.
Honorat JA, Sebastian Lopez-Chiriboga A, Kryzer TJ, Komorowski L, Scharf M, Hinson SR, et al. Autoimmune gait disturbance accompanying adaptor protein-3B2-IgG. Neurol. 2019;93:E954–63. https://doi.org/10.1212/WNL.0000000000008061.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Gilligan is funded by the Irish Clinical Academic Training (ICAT) Programme, supported by the Wellcome Trust and the Health Research Board (Grant Number 203930/B/16/Z), and the Health Service Executive National Doctors Training and Planning and the Health and Social Care, Research and Development Division, Northern Ireland. Dr. McGuigan reports no disclosures. Dr. McKeon is funded by grants from NIH (RO1NS126227, U01NS120901), and has consulted for Janssen and Roche Pharmaceuticals without personal compensation. Dr. McKeon has patents for Septin-5-IgG and Septin-7-IgG licensed to Euroimmun, a patent for GFAP-IgG issued, a patent for MAP1B-IgG with royalties paid to himself and licensed to Ravo Diagnostika, and patents for KLCHL11-IgG and PDE10A-IgG pending.
Human and Animal Rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gilligan, M., McGuigan, C. & McKeon, A. Paraneoplastic Neurologic Disorders. Curr Neurol Neurosci Rep 23, 67–82 (2023). https://doi.org/10.1007/s11910-023-01250-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-023-01250-w