
Abstract
Purpose of Review
The main goal of this article is to discuss the role of the epithelial sodium channel (ENaC) in extracellular fluid and blood pressure regulation.
Recent Findings
Besides its role in sodium handling in the kidney, recent studies have found that ENaC expressed in other cells including immune cells can influence blood pressure via extra-renal mechanisms. Dendritic cells (DCs) are activated and contribute to salt-sensitive hypertension in an ENaC-dependent manner. We discuss recent studies on how ENaC is regulated in both the kidney and other sites including the vascular smooth muscles, endothelial cells, and immune cells. We also discuss how this extra-renal ENaC can play a role in salt-sensitive hypertension and its promise as a novel therapeutic target.
Summary
The role of ENaC in blood pressure regulation in the kidney has been well studied. Recent human gene sequencing efforts have identified thousands of variants among the genes encoding ENaC, and research efforts to determine if these variants and their expression in extra-renal tissue play a role in hypertension will advance our understanding of the pathogenesis of ENaC-mediated cardiovascular disease and lead to novel therapeutic targets.
Similar content being viewed by others

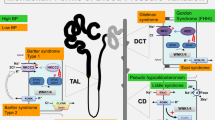

Introduction
The balance between salt and body fluid volume is necessary for regulating blood pressure. High blood pressure is the leading cause of morbidity and mortality due to cardiovascular-related diseases, such as stroke, heart failure, myocardial infarction, and chronic kidney disease [1]. Reducing dietary intake of sodium (Na+) decreases both hypertension and rate of morbidity and mortality associated with cardiovascular events [2]. A meta-analysis investigating the long-term effects of dietary salt intake on blood pressure showed that a reduction of salt for 4 weeks or more results in a significant reduction in blood pressure regardless of sex or ethnic group. A major problem with excess salt consumption is that approximately 25% in the general population and nearly half of the hypertensive population are salt-sensitive [3]. Salt-sensitivity is defined by the hyperresponsive increase and decrease in blood pressure to salt loading and salt depletion, and is an independent predictor of death and cardiovascular events [4, 5]. In contrast to salt-resistant individuals, those who are salt-sensitive experience abnormal changes in blood pressure in response to even minor changes in plasma salt levels [6].
Recently, a paradigm-shift in the understanding of Na+ handling has elucidated a role of extra-renal interstitial space [7, 8]. Studies using 23Na MRI and mathematical modeling demonstrated high Na+ content in the skin and muscle junctional zones positively correlates with blood pressure in humans [9,10,11]. These observations regarding tissue Na+ have relevance to immune cell activation which contributes to hypertension since monocytes can enter and re-emerge from tissues with minimal or no differentiation [12]. There is strong evidence that monocytes contribute to both blood pressure elevation and end-organ damage associated with hypertension. Deletion of monocytes markedly reduces experimental hypertension [13]. Cells derived from monocytes, including macrophages and DCs have also been implicated in hypertension [14,15,, 15, 16••]. Despite the extensive studies, the mechanisms mediating tissue sodium-induced activation of immune cells and hypertension are still not well understood.
The epithelial Na+ channel (ENaC) plays a critical role in body fluid volume and Na+ homeostasis which underly the pathogenesis of salt-sensitive hypertension [17, 18]. This channel has been extensively studied in the kidney where it plays a role in controlling Na+ and K+ handling (Fig. 1). However, it is expressed in other tissues such as the endothelium, vascular smooth muscle, tongue, colon, and immune cells and has been found to influence blood pressure via extra-renal mechanisms. Here, we review the mechanisms of the ENaC-mediated Na+ balance and its relationship to salt-sensitive hypertension and inflammation. Understanding the relationship between salt and the predisposition for high blood pressure could provide valuable insight in drug development for the prevention and treatment of hypertension.

ENaC regulation in the kidney, vasculature, and immune system. ENaC expression and activation in renal distal tubule epithelial cells is regulated by hormonal factors, proteases, lipids, and select ions promoting hypertension, renal injury and inflammation, hypokalemia, and metabolic alkalosis. In vasculature, inhibition of ENaC leads to increased nitric oxide production mediating vascular tone and myogenic response. Activation of ENaC in innate immune cells stimulates ROS production, pro-inflammatory cytokine secretion, and antigen presentation resulting in inflammation and salt-sensitive hypertension
ENaC: Kidney and Beyond
ENaC belongs to the ENaC/degenerin family of ion channels which are sensitive to extracellular factors. ENaC is typically a heterotrimer consisting of three homologous subunits: α, β, and γ [19,20,21]. A fourth subunit, δ, is functionally similar to the α-subunit and is found in various epithelial and non-epithelial tissues of humans such as in the pancreas, lung, and brain [22,23,24,25]. Unlike other subunits, the α-subunit is able to form a homo-trimeric channel that is able to conduct Na+. Co-expression of all three subunits (α, β, and γ; or δ, β, and γ) is required to attain full channel activity [20]. αβγ and δβγ have different functional properties. For example, αβγ channels are inhibited by extracellular Na+, and full activation requires furin-mediated proteolytic processing in the trans-Golgi network and at the cell surface by specific proteases, referred to as channel activating proteases (CAPs) [26,27,28]. In contrast, human δβγ channels are largely insensitive to extracellular Na+ and are not activated by proteases [22, 29].
Mechanisms of ENaC Regulation
Na+ Self-Inhibition
Elevated extracellular Na+ inhibits ENaC through two different mechanisms. First, extracellular Na+ binds ENaC at a defined site in the α-subunit and drives an allosteric change reducing the ENaC open probability, which is referred to as Na+ self-inhibition [30, 31]. This self-inhibition is rapid, low-affinity, and cation-selective. Not only can Na+ inhibit ENaC activity, but Li+ is also able to have an inhibitory effect while K+ only has a minimal inhibitory effect [26, 32, 33]. Second, increased intracellular Na+ slowly inhibits the channel over time [34, 35]. In cultured cells, increases in intracellular Na+ renders channels insensitive to cleavage and activation by proteases [36, 37]. ENaC regulation by Na+ self-inhibition (NaSI) enables the distal nephron to control Na+ reabsorption based on fluctuating urinary Na+ concentrations. Numerous studies have found that many of the human ENaC nonsynonymous single nucleotide variants affect channel function by altering the NaSI response [38,39,40,41,42].
ENaC Regulation by Post-Translational Proteolytic Cleavage
Another mechanism by which ENaC is regulated is through post-translational proteolytic cleavage at defined sites in their extracellular domains with release of imbedded inhibitory tracts leading to activation of the channel through an increase in channel open probability [28, 39, 43]. The ENaC subunits αβγ are assembled and processed in Golgi and post-Golgi compartments and are cleaved by furin [28, 43, 44]. The α-subunit is cleaved twice by furin, releasing an inhibitory tract and transitioning channel from a low to an intermediate activity state. Furin cleaves the γ-subunit once. Its cleavage by other proteases including prostasin, matriptase, cathepsin B, elastase, kallikrein, urokinase, and plasmin at sites distal to its inhibitory tract releases the tract and transition channels to a high activity state [43, 45,46,47,48,49,50,51,52,53]. Other key regulatory factors interface with ENaC subunit proteolysis to determine channel open probability [27•].
ENaC Regulation by Lipids
Lipid signaling molecules including phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol (3,4,5)-trisphosphate have been found to enhance ENaC open probability by binding to cationic sequences within ENaC subunits [54,55,56]. In contrast, the CYP-epoxygenase metabolite 11,12-epoxyeicosatrienoic acid (EET) inhibits ENaC by reducing its channel open probability [57, 58]. Post-translational modification of specific cytoplasmic Cys residues on the β- and γ-subunits by palmitoylation is another mechanism by which ENaC is activated by lipids [59, 60].
ENaC Regulation by Aldosterone
Aldosterone regulates ENaC activity through a variety of mechanisms, including an increase in expression and, in concert with other hormones, activation of a serum-and-glucocorticoid-induced kinase (SGK1) [61, 62]. Aldosterone is secreted from the adrenal gland in response to salt loss and volume depletion, and in response to an elevated serum [K+] [63, 64]. The binding of aldosterone to the mineralocorticoid receptor in renal epithelial cells activates SGK1, which in turn phosphorylates NEDD4-2, a ubiquitin ligase that interacts with ENaC at the cell surface through carboxyl-terminal Pro-Tyr motifs on channel subunits. Ubiquitination of ENaC subunits targets the channel for internalization and degradation. Phosphorylation of NEDD4-2 recruits a 14-3-3 protein and prevents the interaction of NEDD4-2 with ENaC, contributing to the accumulation and enhanced expression of ENaC at the plasma membrane, as well as an increase in channel open [65,66,67].
ENaC Function in the Kidney
Classically, ENaC has been demonstrated to be involved in reabsorption of filtered Na+ in the distal nephron, including the aldosterone-sensitive distal nephron (ADSN) and the collecting duct [68,69,70,71,72,73]. In the kidney, ENaC acts as the final and rate-limiting step in determining transepithelial Na+ reabsorption, net total body Na+ content, fluid volume, and blood pressure in the collecting duct of the nephron. ENaC is primarily expressed in the apical membrane of late distal tubule epithelial cells, which is also known as the ASDN. In addition to its role in Na+ reabsorption, ENaC also plays a critical role in the secretion of K+ in the ASDN [31, 74, 75]. The discovery of mutations in the human ENaC channel confirmed the role of ENaC in regulating blood pressure homeostasis. These ENaC mutations lead to Mendelian forms of hypertension or hypotension, Liddle syndrome, and pseudohypoaldosteronism type 1 (PHA1), respectively. Liddle’s syndrome occurs through a gain-of-function mutation in the cytoplasmic C-terminus of either the β- or γ-subunit of ENaC resulting in an autosomal-dominant form of salt-sensitive hypertension, hypokalemia, and metabolic alkalosis through constitutively active ENaC [76]. In this setting, hypokalemia is predicted to activate the Na/Cl co-transporter, which also contributes to the hypertension in Liddle syndrome [77]. Conversely, inherited loss-of-function mutations in ENaC result in PHA1 and present physiologically as severe hypotension, renal salt wasting, metabolic acidosis, and hyperkalemia [78,79,80,81]. A recent study has shown that treatment of hyperaldosteronism with low-dose amiloride, a pharmacological inhibitor of ENaC activity, normalized previously elevated blood pressure within 1–4 weeks of starting the amiloride treatment and was maintained for 14–28 years [82]. In cases of uncontrolled hypertension, it has been demonstrated that mineralocorticoid receptor blockade with spironolactone was sufficient in reducing systolic blood pressure regardless of their levels of aldosterone. In many cases where ENaC activity is constitutively active, the Na+ channel can be inhibited by the K+-sparing diuretic, amiloride [83,84,85]. For instance, a population of African-Americans was resistant to the blood pressure–lowering effects of spironolactone but had a significant reduction when treated with amiloride [86]. This evidence suggests a therapeutic role for targeting hyperactive ENaC activity in hypertensive patients.
ENaC Beyond the Kidney
Vascular Smooth Muscle Cells
In addition to the ASDN and collecting duct, certain subunits of ENaC have been shown to mediate vascular tone through their expression in vascular smooth muscle cells. It has been demonstrated that all ENaC subunits (α, β, and γ) are expressed in mesenteric, cerebral, and renal arteries [23, 87,88,89]. Perez et al. showed that all ENaC subunits are expressed in rat mesenteric resistance arteries and elegantly showed that inhibition of ENaC using either benzamil or amiloride increases the ratio of phosphorylated epithelial nitric oxide synthase (p-eNOS)/total eNOS through a phosphoinositide 3-kinase (P13K)/Akt-dependent mechanism [87]. Moreover, within the cerebral vasculature, Drummond et al. demonstrated by mRNA and protein expression that both β- and γ-subunits of ENaC are expressed in cerebral resistance arteries and that inhibition of ENaC with amiloride or benzamil prevented pressure-induced vasoconstriction [23]. Within the kidney, Guan et al. demonstrated that vascular smooth muscle cells from afferent arterioles express α, β, and γ ENaC subunits. They went on to demonstrate that micromolar doses of amiloride or benzamil, which do not affect l-type calcium channels, inhibited afferent arteriole myogenic response [88]. To investigate which subunit of ENaC contributes to the myogenic response in resistance vessels, Ge et al. used a mouse model of reduced βENaC expression (βENaC m/m). They showed that reduction of βENaC led to the impairment of whole kidney renal autoregulatory capability [89]. Taken together, these studies suggest that ENaC plays a fundamental physiological role in vascular smooth muscle cell function and regulation of blood flow through resistance vessels by modulating the myogenic response.
Vascular Endothelium
All 4 subunits of ENaC (α, β, δ, and γ) have been demonstrated to be expressed in the vascular endothelium by mRNA transcription and/or protein expression by immunofluorescence [90,91,92,93,94,95,96,97]. Interestingly, there is evidence that endothelial ENaC can be regulated much likely in the kidney. For example, Oberleithner and colleagues showed that nanomolar concentrations of aldosterone increases the expression of ENaC subunits by approximately 36% and total cellular ENaC by 91% in human umbilical vein cells (HUVECs) [94]. Moreover, co-administration of amiloride and aldosterone leads to an 84% reduction in total ENaC in HUVECs, suggesting a regulation mechanism similar to the kidney. However, the way that endothelial ENaC handles extracellular Na+ differs from the renal epithelium. For instance, increases in extracellular Na+ content leads to downregulation of ENaC within the renal epithelium. In contrast, in endothelial cells, aldosterone plus high extracellular Na+ (beyond 140 mM) increases ENaC protein expression within minutes [91]. The precise mechanism by which this occurs is currently unknown, although it is hypothesized that in endothelial cells, aldosterone and high extracellular Na+ environment activates SGK1 which in turn phosphorylates NEDD4-2 which renders it inactive. Thus, ENaC plays a critical role in both intra- and extra-renal vascular function.
ENaC Activity and Regulation in Immune Cells
Activation of the adaptive immune system in hypertension may occur through the loss of self-tolerance, suggesting it may be an autoimmune disease. Antigen-presenting cells (APCs), including macrophages, DCs, and B cells, are critical initiators of the immune response. Of these APCs, DCs are the most proficient classical antigen presenters and play an important role in the discrimination between self and non-self-antigens. In 1973, Ralph Steinman first discovered and described DCs, and since their discovery, they have been extensively studied and well characterized as potent stimulators of T cell activation [98]. APCs present antigens that are then recognized by T cell receptors, stimulating T cell proliferation and activation. Various hypertensive stimuli including angiotensin II (Ang II) and norepinephrine and dietary salt stimulate the infiltration of monocytes, macrophages, DCs, and T lymphocytes into the vasculature and kidney to promote Na+ retention, blood pressure elevation, vasoconstriction, and end-organ damage [14, 99,100,101,102,103]. Until now, salt sensitivity studies have focused on the roles of the vasculature, kidney, and sympathetic activity; however, the contribution of immune cells remains largely unknown.
Several studies have identified multiple neoantigens, or modified endogenous molecules no longer identified as “self,” for their potential to trigger the adaptive immune system. It is thought that these molecules are modified by post-translational modification, adduct formation, oxidation, and nitrosylation. One particular molecule that has been intensely studied for over 40 years for its potential role in hypertension and transport and delivery of antigenic peptides is heat shock protein 70 (HSP70) [104]. Only recently has HSP70 been suggested to induce an autoimmune reaction leading to T cell activation and polarization of CD4+ into regulatory T cells in salt-sensitive hypertension [105, 106]. Additionally, the Toll-like receptor 9 (TLR9) expressed in the endoplasmic reticulum of immune cells recognize mitochondrial DNA-derived cell-free unmethylated CpG dinucleotides, which are upregulated in patients with essential hypertension [107]. Internalization of these CpG motifs activates the TLR9 signaling cascade through pro-inflammatory transcriptional factors NF-κB and AP-1 [108]. The ability for TLR9 to discriminate between methylated and unmethylated DNA is critical in preventing an autoimmune reaction. Moreover, studies in our laboratory have found that a new neoantigen in APCs contributes to the development of hypertension, its associated inflammation, and end-organ damage [16••]. We established a critical role of reactive oxygen species (ROS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in activating DCs in hypertension and in the modulation of gene expression and immunogenicity. In response to a hypertensive stimuli including Ang II, DOCA-salt, or N-nitro-l-arginine methyl ester (l-NAME/high-salt feeding), gamma-ketoaldehydes known as isolevuglandins isoLGs are formed in DCs [16••]. Importantly, scavenging of IsoLG-adducts attenuates blood pressure, inflammation, and vascular stiffness [109].
Recently, we investigated the signaling mechanisms of Na+-dependent ENaC activation in DCs [14]. We demonstrated that an increase in extracellular Na+ concentrations leads to an ENaC-dependent activation of the NADPH-oxidase and subsequent superoxide production leading to formation of the highly immunoreactive IsoLGs. Moreover, in monocyte-derived DCs, the production of IsoLG-adducted proteins can lead to loss of immune tolerance in DOCA-salt hypertension [14, 16]. This ENaC-dependent increased formation of IsoLG-adducted proteins in DCs after exposure to high salt correlates with an increase in surface expression of B7 ligands CD80 and CD86 indicating DC maturation and is essential for the pathogenesis of hypertension [16••]. These studies suggest a potential relationship between innate immunity, ENaC, and hypertension.
One important intracellular enzyme induced by Na+ is SGK-1. The role of SGK-1 in modulating blood pressure has predominantly been studied in the distal convoluted tubule where it regulates ENaC expression. Recent work in our lab has demonstrated that in APCs, the salt-sensing kinase SGK-1 mediates salt-sensitive hypertension by regulating increased expression of ENaC α- and γ-subunits, which leads to IsoLG-adduct formation, interleukin-1β (IL-1β) production, and T cell activation [110•]. Studies by Kleinewiefeld et al. and Wu et al. showed that when exposed to elevated Na+ concentration, there is a marked induction of Th17 polarization in naïve T cells [111, 112]. Inhibiting SGK-1 prevented activation of Forkhead box protein O1 and subsequent differentiation to the Th17 phenotype. In addition, SGK-1 signaling inhibits FOXP3+ regulatory T cells [113, 114], and both Th17 and regulatory T cells play a role in autoimmune tolerance and the genesis of hypertension [115, 116].
IL-1β and ENaC: New Mechanism for Salt-Sensitive Hypertension?
In recent years, significant research progress has been made to better understand the relationship between inflammation and the pathogenesis of hypertension [117, 118]. Both animal and human studies suggest that cytokines such as IL-1β induce a pro-inflammatory state potentiating blood pressure elevation through the alteration of renal, endothelial, and immune responses [99]. In mice, targeting IL-1β activity has been shown to decrease blood pressure through pharmacological inhibition, IL-1β targeted antibody treatment, and genetic deletion [119,120,121]. In a recent article by Rothman et al., secondary analysis of the CANTOS trial suggested that while IL-1β inhibition with canakinumab reduced cardiovascular event rates, this benefit may not be related to incident hypertension and raises the question of the importance of inflammation in hypertension and development of cardiovascular disease [122•]. Moreover, there is a connection between high-salt environments and inflammation. Prior work by Shapiro and Dinarello showed that high salt concentrations drive peripheral blood mononuclear cells to produce the pro-inflammatory cytokine IL-1β [123]. Additionally, high salt increased ENaC-dependent production of IL-1β in DCs to mediate salt-sensitive hypertension by priming and polarizing T cells to produce interleukin 17-A (IL-17A) [14, 101, 124].
Although it is known that there are increased levels of circulating IL-1β in hypertension, only recently has the inflammasome activation been suggested to play a role in its production. Consisting of the sensing domain NOD-like receptor family, pyrin domain containing (NLRP3) and adaptor protein apoptosis-associated speck-like protein containing a carboxy-terminal caspase recruitment domain (ASC), the stimulated complex forms to recruit and proteolytically cleave pro-caspase-1 into the bioactive caspase-1. Caspase-1 activation results in the subsequent maturation and secretion of IL-1β [125, 126]. Hypertensive stimuli, including elevated Na+ and Ang II, are linked to ROS production which has been extensively studied for its role in inflammation, and recent evidence has shown that multiple sources of intracellular ROS are responsible for the activation of the NLRP3 inflammasome [127]. In a recent study by Krishnan et al., apoptosis-ASC−/− mice were protected from DOCA-salt-induced elevated blood as well as renal inflammation and fibrosis [128]. Additionally, they demonstrated that pharmacological inhibition of the NLRP3 inflammasome abolishes DOCA-salt hypertension. In humans, elevated circulating levels of IL-1β and increased inflammasome gene expression have been correlated with age-related hypertension [129,130,131]. Moreover, mutations in the non-coding region of NLRP3 gene in humans were associated with susceptibility to developing hypertension [132]. Recently, ENaC-mediated Na+ influx was responsible for NLRP3 inflammasome activation in PBMCs of cystic fibrosis patients [133]. This suggests a possible link between increased Na+ content and IL-1β production in the pathogenesis of salt-sensitive hypertension. Current studies examining the inflammasome, its components, relationship to ENaC activity, and downstream effector cytokines provide promising insight into the role of salt and inflammation in the development of hypertension.
Conclusion
In an effort to determine therapeutic targets for salt-induced hypertension, human gene sequencing efforts have identified several ENaC gain- and loss-of-function mutations that have been described in Mendelian disorders characterized by either hypertension or hypotension [134,135,136,137,138,139,140,141,142,143,144]. It is not known if individuals with gain-of-function ENaC variants have increased risk for salt-sensitive hypertension. Inhibition of ENaC using inhibitors such as amiloride is not a routinely used approach for treatment of hypertension given their low efficacy when compared other diuretics. However, a meta-analysis by Hebert et al. found that treatment of elderly hypertensive patients with ENaC inhibitors combined with a thiazide diuretic reduces coronary mortality and sudden cardiac death [145]. To date, most of the studies on ENaC have focused on its role in regulating renal Na+ and K+ handling. The recent seminal discoveries of the existence and functioning of extra-renal ENaC including immune cells may illuminate additional therapeutic targets for ENaC in salt-induced cardiovascular disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Muntner P, Carey RM, Gidding S, Jones DW, Taler SJ, Wright JT Jr, et al. Potential US population impact of the 2017 ACC/AHA High Blood Pressure Guideline. Circulation. 2018;137(2):109–18.
Whelton PK, He J. Health effects of sodium and potassium in humans. Curr Opin Lipidol. 2014;25(1):75–9.
Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986;8(6 Pt 2):II127–34.
Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, et al. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997;350(9093):1734–7.
Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37(2 Pt 2):429–32.
de Wardener HE, He FJ, MacGregor GA. Plasma sodium and hypertension. Kidney Int. 2004;66(6):2454–66.
Wiig H, Luft FC, Titze JM. The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol (Oxf). 2018;222(3).
Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T, et al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest. 2017;127(5):1944–59.
Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Muller DN, et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61(3):635–40.
Linz P, Santoro D, Renz W, Rieger J, Ruehle A, Ruff J, et al. Skin sodium measured with (2)(3)Na MRI at 7.0 T. NMR Biomed. 2015;28(1):54–62.
Hofmeister LH, Perisic S, Titze J. Tissue sodium storage: evidence for kidney-like extrarenal countercurrent systems? Pflugers Arch. 2015;467(3):551–8.
Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610.
Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124(12):1370–81.
Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21(4):1009–20.
Kirabo A. A new paradigm of sodium regulation in inflammation and hypertension. Am J Physiol Regul Integr Comp Physiol. 2017;313(6):R706–R10.
•• Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124(10):4642–56. This article demonstrates a novel role for ENaC in immune cells and its contribution to salt-induced inflammation and hypertension.
Greene AS, Yu ZY, Roman RJ, Cowley AW Jr. Role of blood volume expansion in Dahl rat model of hypertension. Am J Phys. 1990;258(2 Pt 2):H508–14.
Hall JE. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation. 2016;133(9):894–906.
Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009;460(7255):599–604.
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367(6462):463–7.
Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife. 2018;7.
Ji HL, Su XF, Kedar S, Li J, Barbry P, Smith PR, et al. Delta-subunit confers novel biophysical features to alpha beta gamma-human epithelial sodium channel (ENaC) via a physical interaction. J Biol Chem. 2006;281(12):8233–41.
Drummond HA, Gebremedhin D, Harder DR. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension. 2004;44(5):643–8.
Giraldez T, Afonso-Oramas D, Cruz-Muros I, Garcia-Marin V, Pagel P, Gonzalez-Hernandez T, et al. Cloning and functional expression of a new epithelial sodium channel delta subunit isoform differentially expressed in neurons of the human and monkey telencephalon. J Neurochem. 2007;102(4):1304–15.
Giraldez T, Dominguez J, Alvarez de la Rosa D. ENaC in the brain--future perspectives and pharmacological implications. Curr Mol Pharmacol. 2013;6(1):44–9.
Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol. 2009;71:361–79.
• Kleyman TR, Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol. 2020;318(1):C150–C62. This review outlines the currently known critical regulatory factors of ENaC, both by extracellular Na+ and intracellular proteolytic activation factors.
Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem. 2009;284(31):20447–51.
Wichmann L, Vowinkel KS, Perniss A, Manzini I, Althaus M. Incorporation of the delta-subunit into the epithelial sodium channel (ENaC) generates protease-resistant ENaCs in Xenopus laevis. J Biol Chem. 2018;293(18):6647–58.
Kashlan OB, Blobner BM, Zuzek Z, Tolino M, Kleyman TR. Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J Biol Chem. 2015;290(1):568–76.
Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na(+) channel regulation by extracellular and intracellular factors. Annu Rev Physiol. 2018;80:263–81.
Bize V, Horisberger JD. Sodium self-inhibition of human epithelial sodium channel: selectivity and affinity of the extracellular sodium sensing site. Am J Physiol Renal Physiol. 2007;293(4):F1137–46.
Collier DM, Snyder PM. Extracellular chloride regulates the epithelial sodium channel. J Biol Chem. 2009;284(43):29320–5.
Anantharam A, Tian Y, Palmer LG. Open probability of the epithelial sodium channel is regulated by intracellular sodium. J Physiol. 2006;574(Pt 2):333–47.
Kellenberger S, Gautschi I, Rossier BC, Schild L. Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest. 1998;101(12):2741–50.
Heidrich E, Carattino MD, Hughey RP, Pilewski JM, Kleyman TR, Myerburg MM. Intracellular Na+ regulates epithelial Na+ channel maturation. J Biol Chem. 2015;290(18):11569–77.
Knight KK, Wentzlaff DM, Snyder PM. Intracellular sodium regulates proteolytic activation of the epithelial sodium channel. J Biol Chem. 2008;283(41):27477–82.
Sheng S, Bruns JB, Kleyman TR. Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J Biol Chem. 2004;279(11):9743–9.
Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol. 2006;290(6):F1488–96.
Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem. 2007;282(28):20180–90.
Shi S, Blobner BM, Kashlan OB, Kleyman TR. Extracellular finger domain modulates the response of the epithelial sodium channel to shear stress. J Biol Chem. 2012;287(19):15439–44.
Shi S, Kleyman TR. Gamma subunit second transmembrane domain contributes to epithelial sodium channel gating and amiloride block. American journal of physiology Renal physiology. 2013;305(11):F1585–92.
Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, et al. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J Biol Chem. 2007;282(9):6153–60.
Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, et al. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004;279(18):18111–4.
Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L813–9.
Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. A segment of gamma ENaC mediates elastase activation of Na+ transport. J Gen Physiol. 2007;130(6):611–29.
Widder JD, Guzik TJ, Mueller CF, Clempus RE, Schmidt HH, Dikalov SI, et al. Role of the multidrug resistance protein-1 in hypertension and vascular dysfunction caused by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27(4):762–8.
Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus Oocytes. J Gen Physiol. 2002;120(2):191–201.
Passero CJ, Mueller GM, Myerburg MM, Carattino MD, Hughey RP, Kleyman TR. TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the gamma-subunit distal to the furin cleavage site. American journal of physiology Renal physiology. 2012;302(1):F1–8.
Tan CD, Hobbs C, Sameni M, Sloane BF, Stutts MJ, Tarran R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J Physiol. 2014;592(Pt 23):5251–68.
Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol. 2012;303(4):F540–50.
Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20(2):299–310.
Ji HL, Zhao R, Komissarov AA, Chang Y, Liu Y, Matthay MA. Proteolytic regulation of epithelial sodium channels by urokinase plasminogen activator: cutting edge and cleavage sites. J Biol Chem. 2015;290(9):5241–55.
Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol. 2005;16(11):3182–7.
Helms MN, Liu L, Liang YY, Al-Khalili O, Vandewalle A, Saxena S, et al. Phosphatidylinositol 3,4,5-trisphosphate mediates aldosterone stimulation of epithelial sodium channel (ENaC) and interacts with gamma-ENaC. J Biol Chem. 2005;280(49):40885–91.
Pochynyuk O, Bugaj V, Vandewalle A, Stockand JD. Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am J Physiol Renal Physiol. 2008;294(1):F38–46.
Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, et al. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124(6):719–27.
Sun P, Antoun J, Lin DH, Yue P, Gotlinger KH, Capdevila J, et al. Cyp2c44 epoxygenase is essential for preventing the renal sodium absorption during increasing dietary potassium intake. Hypertension. 2012;59(2):339–47.
Mueller GM, Maarouf AB, Kinlough CL, Sheng N, Kashlan OB, Okumura S, et al. Cys palmitoylation of the beta subunit modulates gating of the epithelial sodium channel. J Biol Chem. 2010;285(40):30453–62.
Mukherjee A, Mueller GM, Kinlough CL, Sheng N, Wang Z, Mustafa SA, et al. Cysteine palmitoylation of the gamma subunit has a dominant role in modulating activity of the epithelial sodium channel. J Biol Chem. 2014;289(20):14351–9.
Lang F, Pearce D. Regulation of the epithelial Na+ channel by the mTORC2/SGK1 pathway. Nephrol Dial Transplant. 2016;31(2):200–5.
Gleason CE, Oses-Prieto JA, Li KH, Saha B, Situ G, Burlingame AL, et al. Phosphorylation at distinct subcellular locations underlies specificity in mTORC2-mediated activation of SGK1 and Akt. J Cell Sci. 2019;132(7).
Okubo S, Niimura F, Nishimura H, Takemoto F, Fogo A, Matsusaka T, et al. Angiotensin-independent mechanism for aldosterone synthesis during chronic extracellular fluid volume depletion. J Clin Invest. 1997;99(5):855–60.
Makhanova N, Sequeira-Lopez ML, Gomez RA, Kim HS, Smithies O. Disturbed homeostasis in sodium-restricted mice heterozygous and homozygous for aldosterone synthase gene disruption. Hypertension. 2006;48(6):1151–9.
Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86(4):1151–78.
Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, et al. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest. 2007;117(3):773–83.
Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol. 2015;10(1):135–46.
Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol. 2008;19(10):1845–54.
Butterworth MB, Edinger RS, Frizzell RA, Johnson JP. Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol. 2009;296(1):F10–24.
Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77(2):359–96.
Hamm LL, Feng Z, Hering-Smith KS. Regulation of sodium transport by ENaC in the kidney. Curr Opin Nephrol Hypertens. 2010;19(1):98–105.
Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch. 2009;458(1):111–35.
Grunder S, Muller A, Ruppersberg JP. Developmental and cellular expression pattern of epithelial sodium channel alpha, beta and gamma subunits in the inner ear of the rat. Eur J Neurosci. 2001;13(4):641–8.
Kleyman TR, Satlin LM, Hallows KR. Opening lines of communication in the distal nephron. J Clin Invest. 2013;123(10):4139–41.
Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, et al. The Na+−dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest. 2010;120(5):1627–35.
Furuhashi M, Kitamura K, Adachi M, Miyoshi T, Wakida N, Ura N, et al. Liddle’s syndrome caused by a novel mutation in the proline-rich PY motif of the epithelial sodium channel beta-subunit. J Clin Endocrinol Metab. 2005;90(1):340–4.
Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016;89(1):127–34.
Bonny O, Hummler E. Dysfunction of epithelial sodium transport: from human to mouse. Kidney Int. 2000;57(4):1313–8.
Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, et al. A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci U S A. 1997;94(21):11710–5.
Hummler E, Horisberger JD. Genetic disorders of membrane transport. V. the epithelial sodium channel and its implication in human diseases. Am J Phys. 1999;276(3):G567–71.
Boiko N, Kucher V, Stockand JD. Pseudohypoaldosteronism type 1 and Liddle’s syndrome mutations that affect the single-channel properties of the epithelial Na+ channel. Physiol Rep. 2015;3(11).
Izzo JL Jr, Hong M, Hussain T, Osmond PJ. Maintenance of long-term blood pressure control and vascular health by low-dose amiloride-based therapy in hyperaldosteronism. J Clin Hypertens (Greenwich). 2019;21(8):1183–90.
Bubien JK, Watson B, Khan MA, Langloh AL, Fuller CM, Berdiev B, et al. Expression and regulation of normal and polymorphic epithelial sodium channel by human lymphocytes. J Biol Chem. 2001;276(11):8557–66.
Ismailov II, Berdiev BK, Fuller CM, Bradford AL, Lifton RP, Warnock DG, et al. Peptide block of constitutively activated Na+ channels in Liddle’s disease. Am J Phys. 1996;270(1 Pt 1):C214–23.
Zhou ZH, Bubien JK. Nongenomic regulation of ENaC by aldosterone. Am J Physiol Cell Physiol. 2001;281(4):C1118–30.
Laffer CL, Elijovich F, Eckert GJ, Tu W, Pratt JH, Brown NJ. Genetic variation in CYP4A11 and blood pressure response to mineralocorticoid receptor antagonism or ENaC inhibition: an exploratory pilot study in African Americans. J Am Soc Hypertens. 2014;8(7):475–80.
Perez FR, Venegas F, Gonzalez M, Andres S, Vallejos C, Riquelme G, et al. Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3-kinase/Akt in small-diameter mesenteric arteries. Hypertension. 2009;53(6):1000–7.
Guan Z, Pollock JS, Cook AK, Hobbs JL, Inscho EW. Effect of epithelial sodium channel blockade on the myogenic response of rat juxtamedullary afferent arterioles. Hypertension. 2009;54(5):1062–9.
Ge Y, Gannon K, Gousset M, Liu R, Murphey B, Drummond HA. Impaired myogenic constriction of the renal afferent arteriole in a mouse model of reduced betaENaC expression. Am J Physiol Renal Physiol. 2012;302(11):F1486–93.
Golestaneh N, Klein C, Valamanesh F, Suarez G, Agarwal MK, Mirshahi M. Mineralocorticoid receptor-mediated signaling regulates the ion gated sodium channel in vascular endothelial cells and requires an intact cytoskeleton. Biochem Biophys Res Commun. 2001;280(5):1300–6.
Korte S, Wiesinger A, Straeter AS, Peters W, Oberleithner H, Kusche-Vihrog K. Firewall function of the endothelial glycocalyx in the regulation of sodium homeostasis. Pflugers Arch. 2012;463(2):269–78.
Chen W, Valamanesh F, Mirshahi T, Soria J, Tang R, Agarwal MK, et al. Aldosterone signaling modifies capillary formation by human bone marrow endothelial cells. Vasc Pharmacol. 2004;40(6):269–77.
Krueger B, Schlotzer-Schrehardt U, Haerteis S, Zenkel M, Chankiewitz VE, Amann KU, et al. Four subunits (alphabetagammadelta) of the epithelial sodium channel (ENaC) are expressed in the human eye in various locations. Invest Ophthalmol Vis Sci. 2012;53(2):596–604.
Kusche-Vihrog K, Sobczak K, Bangel N, Wilhelmi M, Nechyporuk-Zloy V, Schwab A, et al. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Arch. 2008;455(5):849–57.
Mirshahi M, Nicolas C, Mirshahi S, Golestaneh N, d’Hermies F, Agarwal MK. Immunochemical analysis of the sodium channel in rodent and human eye. Exp Eye Res. 1999;69(1):21–32.
Oberleithner H, Peters W, Kusche-Vihrog K, Korte S, Schillers H, Kliche K, et al. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch. 2011;462(4):519–28.
Oberleithner H, Schneider SW, Albermann L, Hillebrand U, Ludwig T, Riethmuller C, et al. Endothelial cell swelling by aldosterone. J Membr Biol. 2003;196(3):163–72.
Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137(5):1142–62.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–40.
Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10(2):203–7.
Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–7.
Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295(2):F515–24.
Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, et al. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110(12):1604–17.
Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425.
Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, et al. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. Am J Physiol Renal Physiol. 2013;304(3):F289–99.
Wendling U, Paul L, van der Zee R, Prakken B, Singh M, van Eden W. A conserved mycobacterial heat shock protein (hsp) 70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross-react with the mammalian self-hsp70 homologue. J Immunol. 2000;164(5):2711–7.
Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9(4):293–307.
Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4(4):249–58.
Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126(4):1607.
• Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, et al. High salt activates CD11c(+) antigen-presenting cells via SGK (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. 2019;74(3):555–63 This article indicates a role for SGK1 in regulating ENaC expression and activity in CD11c+ antigen presenting cells to mediate salt-sensitive hypertension and renal inflammation.
Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–22.
Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–7.
Safa K, Ohori S, Borges TJ, Uehara M, Batal I, Shimizu T, et al. Salt accelerates allograft rejection through serum- and glucocorticoid-regulated kinase-1-dependent inhibition of regulatory T cells. Journal of the American Society of Nephrology : JASN. 2015;26(10):2341–7.
Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. 2015;125(11):4212–22.
Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57(3):469–76.
Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, et al. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight. 2017;2(13).
Solak Y, Afsar B, Vaziri ND, Aslan G, Yalcin CE, Covic A, et al. Hypertension as an autoimmune and inflammatory disease. Hypertens Res. 2016;39(8):567–73.
Dinh QN, Drummond GR, Sobey CG, Chrissobolis S. Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int. 2014;2014:406960.
Ling YH, Krishnan SM, Chan CT, Diep H, Ferens D, Chin-Dusting J, et al. Anakinra reduces blood pressure and renal fibrosis in one kidney/DOCA/salt-induced hypertension. Pharmacol Res. 2017;116:77–86.
Chamberlain J, Francis S, Brookes Z, Shaw G, Graham D, Alp NJ, et al. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS One. 2009;4(4):e5073.
Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein E-deficient mice. Atherosclerosis. 2011;216(2):313–20.
• Rothman AM, MacFadyen J, Thuren T, Webb A, Harrison DG, Guzik TJ, et al. Effects of interleukin-1beta inhibition on blood pressure, incident hypertension, and residual inflammatory risk: a secondary analysis of CANTOS. Hypertension. 2020;75(2):477–82. As a secondary analysis of the CANTOS trials, this article describes a role for IL-1β in the pathogenesis of cardiovascular events separate from essential hypertension.
Shapiro L, Dinarello CA. Hyperosmotic stress as a stimulant for proinflammatory cytokine production. Exp Cell Res. 1997;231(2):354–62.
Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97(4):696–704.
Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol. 2009;21(1):10–6.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5.
Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D, et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. 2016;173(4):752–65.
Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. 2017;23(2):174–84.
Fearon WF, Fearon DT. Inflammation and cardiovascular disease: role of the interleukin-1 receptor antagonist. Circulation. 2008;117(20):2577–9.
Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–61.
Omi T, Kumada M, Kamesaki T, Okuda H, Munkhtulga L, Yanagisawa Y, et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet. 2006;14(12):1295–305.
Scambler T, Jarosz-Griffiths HH, Lara-Reyna S, Pathak S, Wong C, Holbrook J, et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife. 2019;8.
Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79(3):407–14.
Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, et al. Mechanism by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995;83(6):969–78.
Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11(1):76–82.
Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci U S A. 1995;92(25):11495–9.
Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC. A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci U S A. 1995;92(12):5699–703.
Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. 1996;97(7):1780–4.
Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12(3):248–53.
Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996;13(2):248–50.
Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem. 2010;285(40):30363–9.
Sheng S, Hallows KR, Kleyman TR. Epithelial Na+ channels. In: Alpern RJ, Caplan MJ, Moe OW, editors. Seldin and Giebisch’s the kidney: physiology & pathophysiology fifth. Edition ed. New York, NY: Academic Press; 2012. p. 983–1017.
Rossier BC. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr Opin Pharmacol. 2014;15:33–46.
Hebert PR, Coffey CS, Byrne DW, Scott TA, Fagard RH, Rottman JN, et al. Treatment of elderly hypertensive patients with epithelial sodium channel inhibitors combined with a thiazide diuretic reduces coronary mortality and sudden cardiac death. Journal of the American Society of Hypertension : JASH. 2008;2(5):355–65.
Funding
This study was supported by the National Institutes of Health grants K01HL130497, R01HL147818, T32HL144446, F32HL142937, and P30DK079307.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Inflammation and Cardiovascular Diseases
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pitzer, A.L., Van Beusecum, J.P., Kleyman, T.R. et al. ENaC in Salt-Sensitive Hypertension: Kidney and Beyond. Curr Hypertens Rep 22, 69 (2020). https://doi.org/10.1007/s11906-020-01067-9
Published:
DOI: https://doi.org/10.1007/s11906-020-01067-9