
Abstract
Background
Antibody-drug conjugates (ADCs) are gaining widespread use in the treatment of breast cancer, although toxicity remains an underexplored issue in the real-world clinical setting. Individual case safety reports collected in large pharmacovigilance databases can advance our knowledge on their safety profile in routine clinical practice.
Objective
We prioritized adverse events (AEs) reported with ADCs approved for breast cancer using the Food and Drug Administration Adverse Event Reporting System (FAERS).
Methods
We assessed clinical priority of AEs reported in FAERS (February 2013–March 2022) for trastuzumab emtansine (T-DM1), trastuzumab deruxtecan (T-DXd), and sacituzumab govitecan (SG) by attributing a score to each AE disproportionally reported with ADCs. Four criteria were assessed: clinical relevance, reporting rate, reported case fatality rate, and stability of disproportionality signals (consistency of the reporting odds ratio across multiple analyses using three different comparators).
Results
We retained 6589 reports (77.4% referring to T-DM1 as suspect), and 572 AEs generated a disproportionality signal in at least one analysis. The majority of these AEs (62%) were classified as moderate clinical priorities (e.g., interstitial lung disease with T-DXd, thrombocytopenia, peripheral neuropathy with T-DM1, febrile neutropenia, and large intestine perforation with SG). Three AEs emerged as high clinical priorities (6 points): septic shock and neutropenic colitis with SG (N = 8 and 13, with median onset 13 and 10 days, respectively), without co-reported immunosuppressive agents; and pulmonary embolism with T-DM1 (N = 31, median onset 109 days, 52% with reported metastasis).
Conclusion
The heterogeneous spectrum of post-marketing toxicities for ADCs used in breast cancer, as emerging from the FAERS, is largely in line with preapproval evidence. Although causality cannot be proved, we call for increased awareness by oncologists on potential serious unexpected reactions, including early onset of septic shock and neutropenic colitis with SG, and late emergence of pulmonary embolism with T-DM1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The heterogeneous spectrum of post-marketing toxicities for antibody-drug conjugates is largely predictable from preapproval evidence. |
The analysis of individual case safety reports from pharmacovigilance databases supports post-marketing surveillance of adverse events of clinical interest. |
Septic shock and neutropenic colitis with sacituzumab-govitecan, and pulmonary embolism with trastuzumab-emtansine, deserve prioritization by clinicians. |
1 Introduction
Breast cancer is the most common cancer worldwide, with large heterogeneity in terms of biology and molecular pathology. These hallmarks, including immunohistochemical, genomic, and immunological markers, pose the basis for a personalized pharmacotherapy, comprising chemotherapy (e.g., anthracyclines, taxanes), targeted therapy (e.g., CDK4/6 inhibitors, PARP inhibitors, antiestrogens), and immunotherapy (immune checkpoint inhibitors) [1].
Antibody-drug conjugates (ADCs) combine the selectivity of monoclonal antibodies (mAbs) with the potency of cytotoxic agents (known as payload) [2]. To date, 13 ADCs have received marketing approval worldwide in hematological malignancies, and seven in solid tumors. Among ADCs approved for solid tumors, those targeting HER2 are the most widely prescribed. The encouraging results obtained by ADCs in breast cancer have stimulated research, and clinical trials have shown promising results for some new HER2-ADCs [3].
The anti-HER2 trastuzumab emtansine (T-DM1, trastuzumab conjugated with a microtubule inhibitor) was the first marketed ADC in solid tumors, and is approved for late-stage breast cancer [22 February 2013 by the Food and Drug Administration (FDA)] and as adjuvant treatment for patients with early stage HER2-positive breast cancer (3 May 2019). These approvals were based on the phase III EMILIA trial, which demonstrated an improvement in progression-free survival (PFS) and overall survival (OS) with T-DM as a second line treatment compared with standard therapy at that time (capecitabine and lapatinib) [4]. The results of the KATHERINE trial led to approval of TDM-1 for patients with HER2 positive disease who had not achieved a pathological complete response (pCR) after neoadjuvant treatment with trastuzumab and taxane therapy [5].
On 20 December 2019, the FDA approved trastuzumab-deruxtecan (T-DXd, conjugated with a topoisomerase I inhibitor) for HER2-positive unresectable or metastatic breast cancer following two or more prior anti-HER2-based regimens, and later extended the indications to include tumors with low HER2 expression as well as certain tumors from other anatomic sites. The favorable results of the phase III DESTINY-03 trial are positioning T-DXd as the standard of care for second-line therapy of HER2-positive metastatic breast cancer [6], while DESTINY-04 confirmed the role of T-DXd in HER2 low breast cancer [7, 8].
Sacituzumab govitecan (SG) targets the human trophoblast cell-surface antigen 2 (Trop-2) expressed on the majority of breast cancer cells [9], and is conjugated with SN-38 (topoisomerase I inhibitor). It was authorized by the FDA on 22 April 2020 for relapsed or refractory metastatic triple negative breast cancer (TNBC) that has received at least two prior therapies for metastatic disease, on the basis oif the phase III ASCENT study documenting a numerically higher median PFS (primary endpoint), OS, and objective response rate compared with physician therapeutic choice [10]. On 3 February 2023, SG also received FDA approval for unresectable locally advanced or metastatic hormone receptor-positive, HER2-negative breast cancer that has received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting, on the basis of statistically significant and clinically meaningful PFS and OS data from the phase 3 TROPiCS-02 study [11].
Most ADCs result in off-target toxicities largely due to the cytotoxic payload as well as on-target toxicities and other poorly understood and potentially life-threatening adverse effects. Increasing evidence suggests that new-generation ADCs shed circulating payload, which may contribute to both anti-tumor activity and toxicity observed in studies analyzing ultralow levels of the antibody target, such as DAISY [12].
Of note, these agents are associated with treatment-related adverse events in 91% of patients (including grade ≥ 3 events in 46%) [13], with an overall incidence of treatment‐related discontinuation of 3.2% on the basis of a meta-analysis of 169 clinical trials [14]. Given the rapid expansion of the clinical utilization of ADCs, safety is a major issue for prescribers, and a number of strategies are under investigation to optimize tolerability, thus maximizing their therapeutic value.
Considering the limitations of clinical trials in fully capturing the safety profile of drugs, especially in the detection of rare AEs, individual case safety reports (ICSRs) databases represent a valuable source for post-marketing surveillance and better characterization of AEs occurring in the real-life multifaceted scenario of patients with comorbidities and poly-pharmacotherapy receiving complex anticancer regimens [15]. This is especially the case with ADCs receiving accelerated approval, fast-track/breakthrough designation, and priority review, thus requiring stringent post-marketing surveillance.
The aim of this study was to globally appraise the safety profile of ADCs approved for breast cancer, describing the spectrum of toxicities reported in the post-marketing surveillance and prioritizing AEs of clinical interest.
2 Methods
2.1 Study Design
This study is an observational, retrospective pharmacovigilance analysis of the FDA Adverse Event Reporting System (FAERS) database. It was designed with a multimodal stepwise approach, combining different criteria toward clinical prioritization of AEs, an approach recently proposed in pharmacovigilance [16, 17]. To this aim, descriptive and multiple disproportionality analyses were implemented. Finally, a case-by-case assessment was carried out to further characterize AEs with high clinical priority. The analyses were performed through the open-source R software (version 4.0.2; 22 June 2022).
2.2 Data Source
The FAERS archive is a publicly available post-marketing surveillance system collecting more than 20 million anonymized ICSRs that were submitted to the FDA by the pharmaceutical industry, healthcare providers, lawyers, and consumers. The FAERS, mainly representative of the USA, also gathers serious AEs from the rest of the world. This publicly available archive has attracted considerable interest among clinicians for safety assessment of anticancer drugs and relevant post-marketing characterization of AEs of special interest and is particularly suited for the detection of rare but serious AEs, which may escape detection and/or reporting within clinical trials [18, 19].
Of note, FAERS offers a unique opportunity to publicly access data and can be accessed through (a) a user-friendly public dashboard (https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/fda-adverse-event-reporting-system-faers-public-dashboard), containing many duplicates and limited information and (b) raw quarterly data downloadable as ASCII or XML files (https://fis.fda.gov/extensions/FPD-QDE-FAERS/FPD-QDE-FAERS.html), which need to be pre-processed but allow for more reliable and customized analyses.
We downloaded the FAERS quarterly data [20] since February 2013 up to the first quarter (January–March) of 2022. The AEs were codified through the Medical Dictionary for Regulatory Activities (MedDRA) terminology at the preferred terms (PTs) level. The drugs are recorded as free text and need a thorough standardization before any analysis. To this end, we used a specifically developed dictionary, known as the DiAna dictionary, to standardize drug names into active ingredients [21]. Duplicates were identified and removed on the basis of the presence of the overlapping data in six key variables (sex, age, reporter country, list of drugs, list of events, event date).
2.3 Drugs of Interest, Cases and Exposure Definition
Three ADCs were selected as study drugs: T-DM1, T-DXd, SG (see Supplementary Material 1 for details on the relevant codification).
Cases of interest were reports where at least one ADC of interest was recorded with different exclusion criteria applied. Considering the FDA approval of the trastuzumab emtansine, we selected only reports submitted since February 2013 to obtain a more homogeneous reference group and minimize the existence of pre-marketing reports. AEs associated with cancer (e.g., metastasis, breast cancer) and lack of efficacy (e.g., disease progression) were removed to minimize the existence of a “reverse causality bias” known as indication bias in pharmacovigilance, for which the indication for the prescribed drug is reported as an AE, a frequent phenomenon especially in oncology [22]. Finally, AEs related to medication errors were excluded.
In FAERS, the exposure to a given drug is classified by the reporter as primary suspect (PS), secondary suspect (SS), concomitant (C), or interacting (I). To avoid potential misclassification of AEs and increase the accuracy of analyses, we removed those reports recording one of the investigated ADCs as C or I. In other words, only reports where ADCs of interest were recorded as PS or SS were retained.
2.4 Descriptive Analysis
Descriptive analysis was carried out to explore and compare characteristics of ADC reports in terms of patient demographics (sex, age, country, type of reporter) and outcomes (seriousness). The reference group included only oncological reports, identified as any report recoding at least one drug listed within L01, L02BA, L02BG, and L02AE classes of the Anatomical Therapeutic Chemical (ATC) classification system, not including T-DXd, TDM-1, and SG records. Descriptive percentages refer to the proportion of reports recording a specific value, and cannot be interpreted as risk estimates. Data were reported as counts and relevant percentages for categorical variables, and as median values and interquartile ranges (IQRs) for continuous variables. Fisher’s exact test and χ2 test were adopted to compare categorical variables, while continuous variables were assessed using Kruskal–Wallis test; results were deemed significant for p < 0.05.
2.5 Disproportionality Analysis
Disproportionality analysis is a consolidated approach used to generate hypotheses on a possible drug-event association by comparing the proportion of reports recording a specific AE for a single drug or pharmacological class with the proportion of reports recording the same AE for a reference group [23]. If the proportion of AEs is greater in patients exposed to a specific drug (cases) than in patients not exposed to the same drug (reference group), a higher-than-expected reporting is detected, and a disproportionality signal can be claimed. Through this so-called case/non-case design, an association can be hypothesized between the specific drug and the event and can inform clinical practice for relevant monitoring or targeted preventive strategies [24].
Different disproportionality measures are available, including Bayesian and frequentist approaches. Since no gold standard exists and the performance of the various measures is comparable, we calculated the frequentist reporting odds ratios (ROR) with relevant two-sided 95% confidence interval (CI). In fact, the ROR is relatively easy to understand, interpret, and compute (using the 2 × 2 contingency table), and allows ICSRs database as a data source for a case-control study [25]. We corrected the ROR for multiple comparison using the Bonferroni test, as recently argued to minimize false positive results [26]. We used common threshold to define a statistically significant disproportionality signal, i.e., a lower limit of 95% CI > 1, with at least three cases reported.
The selection of the most appropriate comparator is debated but crucial in pharmacovigilance to minimize confounders [27]. We conducted a stepwise disproportionality approach using three different comparators: (a) all other drugs recorded in FAERS (a traditional screening approach); (b) all anticancer drugs, identified through the ATC classification system (L01, L02BA, L02BG, L02AE), reported as PS and SS drugs—this strategy allowed to control for major confounders, including confounding by indication (i.e., cancer may be a risk factor per se for a given AE); and (c) anticancer drugs used in breast cancer. To this purpose, indications that specify other tumors and did not contain any reference to breast cancer, according to High Level Term of MedDRA terminology, were excluded. These latter approaches have been described as disproportionality by therapeutic area or active-comparator disproportionality analysis and can be useful to partially mitigate the so-called channeling bias (i.e., selective prescription toward more severe patients, notably for ADCs), thereby further minimizing false-positive results [28, 29], especially in the oncological area [30].
2.6 Classification and Prioritization of Relevant Disproportionality Signals
For each drug, AEs emerging with a significant association in at least one disproportionality analysis were ranked on the basis of a semiquantitative score assessing the following criteria (Table 1):
-
Clinical relevance: we used the lists of Important Medical Events (IMEs—serious events—version 26.0) and Designated Medical Events (DMEs—rare but serious events likely to be drug induced), provided by the European Medical Agency) [31].
-
Reporting rate: the proportion of the AE of interest as compared with other AEs (i.e., the ratio between cases and non-cases). To mirror clinical trials, the following traditional categories were used: very common (≥ 10%), common (1–10%), and uncommon (≤ 1%).
-
Signal stability: consistency/robustness of disproportionality signals across multiple analyses. Maximum score was awarded to full consistency (disproportionality signals in three out of three analyses on the basis of different comparators).
-
Reported case fatality rate: the proportion of reports where death was recorded, as compared with all AEs. Notably, the mortality rate in oncology is expected to be per se remarkably high regardless of the drug contribution, and it is challenging to discriminate between AEs due to the drug and AEs due to the natural disease progression. For these reasons, we adopted a conservative approach, and the highest score was assigned only when the case fatality rate was > 50%.
A score of 0–2, 3–5, and 6–8 identified, respectively, AEs with low (yellow light), moderate (orange light), or high (red light) priority.
2.7 Case-By-Case Evaluation of AEs with High Clinical Priority
A case-by-case analysis was carried out in the attempt to identify potential drug- and patient-related risk factors. Therefore, reports were further individually inspected to remove potential remaining duplicates (on the basis of high similarity on event date, age, sex, reporter country, co-reported PTs, and co-reported drugs), and to analyze the following clinical features: latency [i.e., time to onset expressed in days with IQR, calculated as the difference between the start of therapy and the date the event occurred], and concomitant drugs as plausible risk factors for AE occurrence or being a proxy of a disease associated with AE susceptibility or a proxy of a preexisting event.
3 Results
3.1 Descriptive Analysis
The characteristics of cases and non-cases are summarized in Table 2.
A total of 6589 cases were identified, with an increase since 2020, likely explained by extended therapeutic indications and marketing authorizations of T-DXd and SG. More than three-quarters of the reports (77.4%) refer to T-DM1, followed by T-DXd (12.8%) and SG (9.8%), likely reflecting the marketing age. Most cases have been reported in female patients (> 96%) across all the ADCs. Physicians and other healthcare professionals submitted about 80% of reports, a trend likely related to administration within a hospital setting of these molecules. Most of the cases (26.6%) have been reported in patients aged 50–64 years, with SG having similar percentages in the range 30–49 (37%) and 50–64 (38.3%) years. Most of the cases were from the USA for T-DXd (63.86%) and SG (61.73%), whereas TDM-1 had similar numbers of reports from the USA (37.42%) and Europe (37.77%). The difference in records between the USA and Europe could be due to different drug approval times from regulatory agencies rather than different reporting behaviors. Indeed, the difference is more evident with T-DXd and SG authorized in USA a year before than in Europe, compared with TDM-1, which was approved in 2013 and has a longer history of clinical use. Death was recorded in 17.1% of cases, with the highest percetage of fatal (33.95%) and life-threatening (4.48%) events for SG compared with T-DM1 (14.93% and 2.61%, respectively) and T-DXd (17.06% and 3.2%, respectively). The highest hospitalization rate was reported for T-DM1 (26.45%) compared with T-DXd (21.80%) and SG (16.20%). Between 2013 and 2019, reports were exclusively for TDM-1 and report rate was steady over this period, with a slight increase in the last 2 years. Since 2020, reports for T-DXd and SG were included in the FAERS database and both showed an increase in records between 2020 (315 for T-DXd and 90 for SG) and 2021 (410 for T-DXd and 297 for SG). Records in the first quarter of 2022 (118 for T-DXd, 190 for TDM-1, and 259 for SG) suggested a further upward trend, especially for SG.
3.2 Disproportionality Analysis
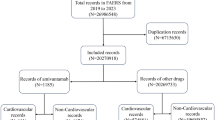
The flowchart (Fig. 1) describes key results of disproportionality analysis in terms of disproportionality signals. After excluding 105 PTs due to indication bias and medication errors, 53 PTs for T-DXd, 483 PTs for T-DM1, and 36 PTs for SG were finally retained as drug related and statistically significant. Most frequently reported AEs were nausea and fatigue for both T-DM1 and T-DXd, and diarrhea and neutropenia for SG.

Flowchart on the process of case selection, disproportionality analysis, and relevant prioritization.
3.3 Clinical Prioritization of Relevant Disproportionality Signals
In the flowchart (Fig. 1), the clinical prioritization is shown; most of the AEs (62.06%) had a moderate clinical priority: 25 PTs for T-DXd (47.17%), 166 PTs for T-DM1 (65.42%), and 14 for SG (38.89%). Among these, pneumonitis (score of 5) and interstitial lung disease (score of 4) with T-DXd; pneumonia (score of 5) and thrombocytopenia, peripheral neuropathy, and hepatotoxicity (score of 4 each) with T-DM1; and febrile neutropenia and large intestine perforation (score of 5 each) with SG.
Among the 572 PTs disproportionally associated with the ADCs, 177 (30.9%) were IMEs: 20 for T-DXd (37.7%), 141 (29%) for T-DM1, and 16 (44%) for SG; 4% were DME: 3 for T-DXd (5.7%), 4 for SG (11.1%), and 16 for T-DM1 (3.3%). Febrile neutropenia, hemolytic anemia, and pulmonary fibrosis, associated with T-DXd, are DMEs with moderate clinical priority.
Three AEs were classified as high clinical priorities: neutropenic colitis (DME) and septic shock (IME) for SG, and pulmonary embolism, included in IME list, for T-DM1, as presented in Table 3.
The full list of reported AEs, including all disproportionality analyses, sub-scores, and overall score, is provided in Supplementary Material 1.
3.4 Case-By-Case Assessment
Full details of the case-by-case evaluation is provided in Supplementary Material 2. After manual inspection to remove likely additional duplicates, 8/9 cases of septic shock (1 case removed as potential duplicate), 13/16 cases of neutropenic colitis (3 cases removed), and 31/58 cases of pulmonary embolism (27 cases removed) were finally retained. A shorter time to onset was highlighted for septic shock and neutropenic colitis (13 and 10 days, respectively) as compared with pulmonary embolism (109 days). Concomitant drug and disease-related risk factors were recorded in only a negligible proportion of cases of septic shock and neutropenic colitis, and no glucocorticoids with immunosuppressive effect were found. With regard to pulmonary embolism, antithrombotic drugs were found in 11 of 31 cases, but it is not possible to assert whether the antithrombotic agent was started before or after the pulmonary event. Antidepressants and antidiabetics were recorded as concomitant drugs in 19.3% of cases, whereas cardiovascular agents were found in 55% of cases. Hormone therapy was reported as concomitant in 7 out of 31 cases (22.6%). Metastasis (namely co-reported breast cancer metastatic, metastases to bone, and breast cancer stage IV) was specified in 31 cases (52%).
4 Discussion
This post-marketing surveillance study, through the analysis of ICSRs from FAERS database, describes the worldwide AE reporting of ADCs in breast cancer. Previous studies mainly focused on specific AEs such as cardiotoxicity, hepatotoxicity, myelotoxicity, and interstitial lung disease [32, 33]. Most importantly, we attempted to move a “conventional” pharmacovigilance study, aimed at detecting unexpected drug–event associations, or a new aspect of a known association, forward and proposed a score to prioritize AEs of special interest on the basis of accepted general criteria, adapted to the oncological area. This score was developed to highlight those toxicities of major clinical relevance and therefore of importance in clinical monitoring and the development of risk minimization strategies.
Overall, the main findings emerging from this large-scale contemporary pharmacovigilance analysis are: (1) we confirmed and enlightened the variegate spectrum of ADC-related toxicities, mainly driven by the type of monoclonal antibody and the payload (e.g., peripheral neuropathy with DM-1, diarrhea with SN-38); (2) we highlighted three serious unexpected AEs as high clinical priorities, namely septic shock and neutropenic colitis with SG and pulmonary embolism with T-DM1, which were further scrutinized through a case-by-case analysis as potential adverse drug reactions; (3) we found several AEs of moderate clinical priority, such as pneumonitis and interstitial lung disease with T-DXd, which should not be overlooked by oncologists. Therefore, this study, using the FAERS database, confirmed the irreplaceable and the complementary role of post-marketing surveillance for detection of rare but unpredictable AEs that may escape detection from pre-marketing pivotal trials, thus supporting clinicians in proactive monitoring and safer prescribing.
Septic shock emerged as a high clinical priority for SG, with a consistent disproportionality signal in all analyses and a reported case fatality rate higher than 77%. The case-by-case analysis did not highlight drug-related risk factors that may potentially increase patients’ susceptibility, such as co-reported immunosuppressive agents (corticosteroids), thus suggesting a possible drug-related component. A signal of sepsis was also recently found by Xia and colleagues [34], who performed a disproportionality analysis on FAERS to investigate the reporting of sepsis in patients with cancer treated with ADCs for solid and hematological cancers. Of note, in our FAERS analysis, sepsis and septic shock were classified as moderate priorities for T-DM1 and T-DXd, respectively, thus suggesting the potential existence of a class effect.
When comparing FAERS data with pivotal pre-marketing trials, one fatal case of septic shock caused by neutropenic colitis was determined to be related to SG in the latest TROPiCS-02 trial, whereas in the ASCENT trial the three deaths (one with sepsis) were judged to be unrelated to SG. Febrile neutropenia was reported in 5% of the patients in both trials, with granulocyte-colony stimulating factor (G-CSF) therapy initiated in almost 50% of the patients. Routine primary prophylaxis with G-CSF is not recommended, but secondary prophylaxis could be considered at the first occurrence of grade 4 neutropenia lasting 7 or more days, grade 3 febrile neutropenia, or grade 3–4 neutropenia that has delayed dosing by 2 or 3 weeks for recovery to grade ≤ 1 [35,36,37,38].
Neutropenic colitis also emerged as high clinical priority for SG; it was co-reported with diarrhea, neutropenia, and sepsis/septic shock (two cases), and was serious (hospitalization or death or life-threatening event recorded in 43.75%, 31.25%, and 18.75% of cases, respectively) without co-reported drugs that might increase patients’ susceptibility. Severe diarrhea (grade 3 or 4) is a well-known AE for SG and occurred in 10% of the patients in both trials and could be due to either early cholinergic syndrome (requiring atropine treatment) or delayed diarrhea due to SN-38. Of note, we found a median onset of 10 days for neutropenic colitis, presumably after day 8 infusion of SG. At the onset of delayed diarrhea, patients should be evaluated for infectious causes, and if ruled out, promptly initiate loperamide. Although in the TROPiCS-02 trial patients who were homozygous for UGT1A1 *28/*28 also had higher rates of grade 3 or 4 diarrhea and neutropenia than other subgroups, testing of UGT1A1 status is not required before treatment and detailed management strategy is lacking. Conversely, genotyping tests for UGT1A1 *6 and *28 are recommended by regulatory Agencies for irinotecan. Attention should, therefore, be paid to diarrhea, especially if ADCs are used in combination with immune checkpoint inhibitors, which are known to cause immune-related colitis.
For T-DM1, pulmonary embolism emerged as high clinical priority (reported case fatality rate of 45%), with a delayed latency (more than 3 months after start of therapy). Antithrombotic therapy with edoxaban or rivaroxaban was reported in 25% of patients, although it is not possible to determine whether antithrombotic treatment was started before or after the embolic event. The case-by-case evaluation detected seven cases (22.5%) in which concomitant hormone therapy with tamoxifen could be considered a drug-related risk factor. Among disease-related risk factors, metastasis was specified in more than half of the cases. Of note, in the final OS results of the EMILIA trial [39], only 1 case of pulmonary embolism (grade 1–2) was recorded in the T-DM1 arm, as compared with 14 cases (of which 9 were grade 3–4) in the capeticabine-lapatinib group. Oncologists should be aware that thrombosis may occur with T-DM1, although a contribution of the underlying metastatic setting cannot be ruled out.
A number of AEs were regarded as moderate clinical priorities; this is the case, for instance, with pneumonitis and interstitial lung disease for T-DXd, as well as hepatotoxicity, thrombocytopenia, and peripheral neuropathy for T-DM1. Although these AEs can be considered expected on the basis of pre-marketing and post-marketing evidence, clinicians should remain vigilant and be reminded as to the importance of proactive clinical monitoring and timely discontinuation as key risk minimization strategies [40].
Finally, from a research perspective, sepsis, neutropenic colitis, and pulmonary embolism should also be considered as priorities for targeted analytical pharmaco-epidemiological research to investigate the role of other drug- and patient-related risk factors that cannot be captured by ICSR databases. This will help clarify whether these AEs are actual adverse drug reactions, especially for T-DM1, which is approved both in the adjuvant and metastatic settings.
We acknowledge limitations inherent to ICSRs and pharmacovigilance databases [41] that do not allow for a causal relationship to be inferred. Given the lack of a denominator and an expected under-reporting phenomenon, disproportionality measures such as ROR and its magnitude cannot quantify the real risk in clinical practice, but can only inform about a higher-than-expected AE reporting and not of AE occurrence. Consequently, incidence cannot be established. Moreover, verification of events through clinical features, including laboratory and instrumental tests, comorbidities and adjustment of therapeutic regimens, is limited. In this regard, the presence of missing data and incomplete information did not allow for the precise identification of the adjuvant and metastatic settings, which is highly relevant information for T-DM1.
However, FAERS is largely representative of the worldwide real-life use of drugs, which cannot be fully captured by clinical trials, thus supporting a good generalizability of results. We attempted to minimize the potential existence of a confounding by indication using different comparators, and there is no reason a priori to support the existence of notoriety bias (i.e., increased reporting following media attention or regulatory measures). However, residual channeling bias and confounders due to the underlying metastatic setting and comorbidities, as well as potential selective reporting toward serious AEs, cannot be ruled out. Our attempt to develop an original score to prioritize AEs of major clinical interest strengthened the clinical implications of these findings. Although the accuracy of this score cannot be determined (and was not the aim of the work), our approach was based on well-established criteria comprising qualitative items of unquestionable clinical relevance using conservative thresholds (i.e., reported case fatality rate) and multiple disproportionality analyses, thus increasing the accuracy and robustness of findings. This approach supports the potential role of the scoring system as a prioritization tool for pharmacovigilance of anticancer drugs.
5 Conclusion
The spectrum of toxicities for ADCs used in breast cancer is highly heterogeneous but largely predictable and mostly in line with preapproval evidence.
Although causality cannot be proved, we call oncologists to have increased awareness regarding three serious potential unexpected adverse reactions with high clinical priority: early onset of septic shock and neutropenic colitis with SG, and late emergence of pulmonary embolism with T-DM1.
In the absence of clear patient- and drug-related risk factors, stringent monitoring, timely discontinuation, and prompt treatment are key risk minimization strategies.
We support pharmacovigilance for the post-marketing prioritization of AEs of clinical interest, especially for the more recently approved T-DXd and SG, thus promoting real-time safer prescribing in oncology.
References
Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397:1750–69. https://doi.org/10.1016/S0140-6736(20)32381-3.
Dumontet C, Reichert JM, Senter PD, Lambert JM, Beck A. Antibody-drug conjugates come of age in oncology. Nat Rev Drug Discov. 2023;22:641–61. https://doi.org/10.1038/s41573-023-00709-2.
Rassy E, Rached L, Pistilli B. Antibody drug conjugates targeting HER2: clinical development in metastatic breast cancer. Breast. 2022;66:217–26. https://doi.org/10.1016/j.breast.2022.10.016.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. https://doi.org/10.1056/NEJMoa1209124.
von Minckwitz G, Huang CS, Mano MS, Loibl S, Mamounas EP, Untch M, et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med. 2019;380:617–28. https://doi.org/10.1056/NEJMoa1814017.
Cortés J, Kim SB, Chung WP, Im S, Park YH, Hegg R, et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N Engl J Med. 2022;386:1143–54. https://doi.org/10.1056/NEJMoa2115022.
Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N Engl J Med. 2022;387:9–20. https://doi.org/10.1056/NEJMoa2203690.
Schlam I, Tolaney SM, Tarantino P. How I treat HER2-low advanced breast cancer. Breast. 2023;67:116–23. https://doi.org/10.1016/j.breast.2023.01.005.
Ambrogi F, Fornili M, Boracchi P, Trerotola M, Relli V, SImeone P, et al. Trop-2 is a determinant of breast cancer survival. PLoS One. 2014;9: e96993. https://doi.org/10.1371/journal.pone.0096993.
Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384:1529–41. https://doi.org/10.1056/NEJMoa2028485.
Rugo HS, Bardia A, Marmé F, Cortés J, Schmid P, Loirat D, et al. Overall survival with sacituzumab govitecan in hormone receptor-positive and human epidermal growth factor receptor 2-negative metastatic breast cancer (TROPiCS-02): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2023;402(10411):1423-33. https://doi.org/10.1016/S0140-6736(23)01245-X.
Mosele F, Deluche E, Lusque A, Le Bescond L, FIlleron T, Pradat Y, et al. Trastuzumab deruxtecan in metastatic breast cancer with variable HER2 expression: the phase 2 DAISY trial. Nat Med. 2023;29:2110–20. https://doi.org/10.1038/s41591-023-02478-2.
Tarantino P, Ricciuti B, Pradhan SM, Tolaney SM. Optimizing the safety of antibody-drug conjugates for patients with solid tumours. Nat Rev Clin Oncol. 2023;20:558–76. https://doi.org/10.1038/s41571-023-00783-w.
Zhu Y, Liu K, Wang K, Zhu H. Treatment-related adverse events of antibody-drug conjugates in clinical trials: a systematic review and meta-analysis. Cancer. 2023;129:283–95. https://doi.org/10.1002/cncr.34507.
Raschi E, Gatti M, Gelsomino F, Ardizzoni A, Poluzzi E, De Ponti F. Lessons to be learnt from real-world studies on immune-related adverse events with checkpoint inhibitors: a clinical perspective from pharmacovigilance. Target Oncol. 2020;15:449–66. https://doi.org/10.1007/s11523-020-00738-6.
Gastaldon C, Schoretsanitis G, Arzenton E, Raschi E, Papola D, Ostuzzi G, et al. Withdrawal syndrome following discontinuation of 28 antidepressants: pharmacovigilance analysis of 31,688 reports from the WHO Spontaneous Reporting Database. Drug Saf. 2022;45:1539–49. https://doi.org/10.1007/s40264-022-01246-4.
Gatti M, Antonazzo IC, Diemberger I, De Ponti F, Raschi E. Adverse events with sacubitril/valsartan in the real world: emerging signals to target preventive strategies from the FDA adverse event reporting system. Eur J Prev Cardiol. 2021;28:983–9. https://doi.org/10.1177/2047487320915663.
Raschi E, Fusaroli M, Ardizzoni A, Poluzzi E, De Ponti F. Thromboembolic events with cyclin-dependent kinase 4/6 inhibitors in the FDA Adverse Event Reporting System. Cancers (Basel). 2021;13:1758. https://doi.org/10.3390/cancers13081758.
Raschi E, Fusaroli M, Ardizzoni A, Poluzzi E, De Ponti F. Cyclin-dependent kinase 4/6 inhibitors and interstitial lung disease in the FDA adverse event reporting system: a pharmacovigilance assessment. Breast Cancer Res Treat. 2021;186:219–27. https://doi.org/10.1007/s10549-020-06001-w.
FDA. FAERS Quarterly Data Extract Files. 2021. https://fis.fda.gov/extensions/FPD-QDE-FAERS/FPD-QDE-FAERS.html. Accessed 3 May 2021.
Fusaroli M, Giunchi V, Battini V, Puligheddu S, Khouri C, Carnovale C, et al. Standardization of drug names in the FDA adverse event reporting system: the DiAna dictionary. medRxiv. 2023. https://doi.org/10.1101/2023.06.07.23291076.
Hauben M, Hung E, Wood J, Soitkar A, Reshef D. The impact of database restriction on pharmacovigilance signal detection of selected cancer therapies. Ther Adv Drug Saf. 2017;8:145–56. https://doi.org/10.1177/2042098616685010.
Faillie JL. Case-non-case studies: principle, methods, bias and interpretation. Therapie. 2019;74:225–32. https://doi.org/10.1016/j.therap.2019.01.006.
Gatti M, Fusaroli M, Caraceni P, Poluzzi E, De Ponti F, Raschi E. Serious adverse events with tocilizumab: pharmacovigilance as an aid to prioritize monitoring in COVID-19. Br J Clin Pharmacol. 2021;87:1533–40. https://doi.org/10.1111/bcp.14459.
Poluzzi E, Raschi E, Piccinni C, De F. Data mining techniques in pharmacovigilance: analysis of the publicly accessible FDA Adverse Event Reporting System (AERS). Data Min Appl Eng Med InTech. 2012;12:265–302. https://doi.org/10.5772/50095.
Gaucher L, Sabatier P, Katsahian S, Jannot AS. Pharmacovigilance studies without a priori hypothesis systematic review highlights inappropriate multiple testing correction procedures. J Clin Epidemiol. 2023;162:127–34. https://doi.org/10.1016/j.jclinepi.2023.08.010.
Gravel CA, Douros A. Considerations on the use of different comparators in pharmacovigilance: a methodological review. Br J Clin Pharmacol. 2023;89:2671–6. https://doi.org/10.1111/bcp.15802.
Alkabbani W, Gamble JM. Active-comparator restricted disproportionality analysis for pharmacovigilance signal detection studies of chronic disease medications: an example using sodium/glucose cotransporter 2 inhibitors. Br J Clin Pharmacol. 2023;89:431–9. https://doi.org/10.1111/bcp.15178.
Grundmark B, Holmberg L, Garmo H, Zethelius B. Reducing the noise in signal detection of adverse drug reactions by standardizing the background: a pilot study on analyses of proportional reporting ratios-by-therapeutic area. Eur J Clin Pharmacol. 2014;70:627–35. https://doi.org/10.1007/s00228-014-1658-1.
Hauben M, Hung E, Wood J, Soitkar A, Reshef D. The impact of database restriction on pharmacovigilance signal detection of selected cancer therapies. Ther Adv Drug Saf. 2017;8:145–56. https://doi.org/10.1177/2042098616685010.
European Medicines Agency. Designated Medical Event (DME) list. 2020. https://www.ema.europa.eu/documents/other/designated-medical-event-dme-list_en.xls. Accessed 17 Mar 2022.
Li X, Chen G, Hu Y, Zhao B, Jiang J. Caution the arrhythmia association with antibody-dr]ug conjugates: a pharmacovigilance study. Anticancer Drug. 2022;33:226–34. https://doi.org/10.1097/CAD.0000000000001191.
Ma Z, Zhang Y, Zhu M, Feng L, Zhang Y, An Z. Interstitial lung disease associated with anti-HER2 anti-body drug conjugates: results from clinical trials and the WHO’s Pharmacovigilance database. Expert Rev Clin Pharmacol. 2022;15:1351–61. https://doi.org/10.1080/17512433.2022.2121705.
Xia S, Zhao Y, Guo L, Gong H, Wang Y, Ma R, et al. Do antibody-drug conjugates increase the risk of sepsis in cancer patients? A pharmacovigilance study. Front Pharmacol. 2022;13:967017. https://doi.org/10.3389/fphar.2022.967017.
Spring LM, Nakajima E, Hutchinson J, Viscosi E, Blouin G, Weekes C, et al. Sacituzumab govitecan for metastatic triple-negative breast cancer: clinical overview and management of potential toxicities. Oncologist. 2021;26:827–34. https://doi.org/10.1002/onco.13878.
Taplitz RA, Kennedy BE, Bow EJ, Crews J, Gleason C, Hawley DK, et al. Outpatient management of fever and neutropenia in adults treated for malignancy: American Society of Clinical Oncology and Infectious Diseases Society of America Clinical Practice Guideline Update. J Clin Oncol. 2018;36:1443–53. https://doi.org/10.1200/JCO.2017.77.6211.
Taplitz RA, Kennedy BE, Bow EJ, Crews J, Gleason C, Hawley DK, et al. Antimicrobial prophylaxis for adult patients with cancer-related immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. J Clin Oncol. 2018;36:3043–54. https://doi.org/10.1200/JCO.18.00374.
Zimmer AJ, Freifeld AG. Optimal management of neutropenic fever in patients with cancer. J Oncol Pract. 2019;15:19–24. https://doi.org/10.1200/JOP.18.00269.
Diéras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:732–42. https://doi.org/10.1016/S1470-2045(17)30312-1.
Lievano FA, Scarazzini LJ, Tyczynski JE, Schubert CM. Renz CL Risk minimization of antibody-drug conjugates in oncology: a review. Drug Saf. 2021;44:733–42. https://doi.org/10.1007/s40264-021-01069-9.
Raschi E, Poluzzi E, Salvo F, Pariente A, De Ponti F, Marchesini G, et al. Pharmacovigilance of sodium-glucose co-transporter-2 inhibitors: what a clinician should know on disproportionality analysis of spontaneous reporting systems. Nutr Metab Cardiovasc Dis. 2018;28:533–42. https://doi.org/10.1016/j.numecd.2018.02.014.
Acknowledgements
This work was supported by the Italian Ministry of Health (Ricerca Corrente) [no grant no. provided]. M.F. is enrolled in a PhD in General Medical and Services Sciences, University of Bologna, which supports his fellowship. E.R. and F.D.P. are supported by institutional research funds (Ricerca Fondamentale Orientata).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interest
C.Z. has received grants from Novartis, Roche, Eisai, AstraZeneca, Pfizer, PharmaMar, Tesaro, Pierre Fabre, Ist. Gentili, Teva, Seagen, Eli Lilly, Celgene, MSD, GSK, Amgen, and Daiichi; has received support for attending meetings and/or travel from Novartis, Roche, Pfizer, PharmaMar, Tesaro, Pierre Fabre, Ist. Gentili, and Celgene; and has participated in data safety monitoring or advisory boards for Novartis, Roche, Eisai, AstraZeneca, Pfizer, PharmaMar, Amgen, Tesaro, Quintiles IMS, Eli Lilly, Celgene, MSD, GSK, and Daiichi. L.G. reports serving as a consultant/advisor for Lilly, Novartis, AstraZeneca, GlaxoSmithKline, and Incyte. S.C., S.P., M.F., M.Y., F.D.P., and E.R. have no conflicts of interest that are directly relevant to the content of this article.
Availability of data and material
The datasets analyzed during the current study are available in the public domain: https://fis.fda.gov/extensions/FPD-QDE-FAERS/FPD-QDE-FAERS.html.
Code availability
Codes are available upon reasonable request to the authors.
Author contributions
S.C. conceived the study. E.R., M.F., S.P., and S.C. contributed to study design. S.P. and M.F. contributed to the acquisition and analysis of data. S.C. drafted the manuscript. All authors contributed to the interpretation of data, critically revised the manuscript, and provided final approval.
Ethics approval
This article does not contain any studies with human participants performed by any of the authors. This study is based on anonymous data that can be downloaded from a publicly available source. Therefore, no ethical approval is required.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cecco, S., Puligheddu, S., Fusaroli, M. et al. Emerging Toxicities of Antibody-Drug Conjugates for Breast Cancer: Clinical Prioritization of Adverse Events from the FDA Adverse Event Reporting System. Targ Oncol 19, 435–445 (2024). https://doi.org/10.1007/s11523-024-01058-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-024-01058-9