
Abstract
A genetic transformation system has been developed for callus cells of Crataegus aronia using Agrobacterium tumefaciens. Callus culture was established from internodal stem segments incubated on Murashige and Skoog (MS) medium supplemented with 5 mg l−1 Indole-3-butyric acid (IBA) and 0.5 mg l−1 6-benzyladenine (BA). In order to optimize the callus culture system with respect to callus growth and coloration, different types and concentrations of plant growth regulators were tested. Results indicated that the best average fresh weight of red colored callus was obtained on MS medium supplemented with 2 mg l−1 2,4-dichlorophenoxyacetic acid (2,4-D) and 1.5 mg l−1 kinetin (Kin) (callus maintenance medium). Callus cells were co-cultivated with Agrobacterium harboring the binary plasmid pCAMBIA1302 carrying the mgfp5 and hygromycin phosphotransferase (hptII) genes conferring green fluorescent protein (GFP) activity and hygromycin resistance, respectively. Putative transgenic calli were obtained 4 weeks after incubation of the co-cultivated explants onto maintenance medium supplemented with 50 mg l−1 hygromycin. Molecular analysis confirmed the integration of the transgenes in transformed callus. To our knowledge, this is the first time to report an Agrobacterium-mediated transformation system in Crataegus aronia.
Similar content being viewed by others

Introduction
Hawthorn is a common name of all plants species in the genus Crataegus that belongs to the Rosaceae family. It is a thorny shrub or small tree that has bright green leaves, white flowers and bright red berries (Chang et al. 2002). It has around 280 species distributed across different temperate regions, including the Mediterranean region, North Africa, Europe and Central Asia and eastern North America (Ozcan et al. 2005). Crataegus aronia (synonym for Crataegus azarolus L.) is the main hawthorn species found in the eastern Mediterranean region and it is distributed mainly on dry hillsides and in mountainous regions (Ljubuncic et al. 2005).
Many hawthorn species are grown for their edible fruits in Asia, Central America, and various Mediterranean countries, and they are commonly used as ornamental plants or as frost-resistant rootstocks for pear and apple (Ercisli 2004; Rigelsky and Sweet 2002). In addition, hawthorn species are listed as herbal drugs in pharmacopoeias, due to their high contents of flavonoids, oligomeric proanthocyaninidins, and ethanobotanical and ethanopharmacological compounds (Chang et al. 2005). Hawthorn constituents are predicted to be good antioxidants, responsible for free radical removal activity in living cells (Bahorun et al. 2003), and they play an important role in the prevention and treatment of cardiovascular diseases (Chang et al. 2002, 2005; Rigelsky and Sweet 2002).
Hawthorn callus and cell suspension cultures are valuable systems commonly used in the production of secondary metabolites (Bahorun et al. 2002). The first callus culture of hawthorn was reported by Schrall and Becker (1977). Crataegus monogyna callus and cell suspension cultures initiated from floral buds were used for the analysis of polyphenols constituents, their production levels and their antioxidant activity (Bahorun et al. 1994, 2003; Valls et al. 2007). Hawthorn callus produces mainly proanthocyanidins, B2 dimer, epicatechin, chlorogenic acid and major flavonoid hyperoside.
In hawthorn, biotechnological approaches to increase the production of polyphenols are needed. The development of an efficient method for genetic transformation of hawthorn callus is important for boosting the production of medicinal metabolites. Agrobacterium-mediated transformation has been commonly applied to engineer many plant species for desirable traits (Gelvin 2009). This system is favored over other methods, such as particle bombardment and electroporation, due to the integration of low numbers of the transgene into the genome, the stability in the host cells and the high transformation efficiency (Dai et al. 2001; Travella et al. 2005). Such a system has been commonly used to enhance the production of secondary metabolites in many plant species (reviewed in Matkowski 2008). For instance, larger amounts of rosmarinic acid accumulated in transformed lavender callus cells when compared with non-transformed cultures (Bauer et al. 2004).
In this study, we describe the establishment of a callus culture system for Crataegus aronia. An Agrobacterium-mediated transformation system suitable for Crataegus aronia callus cells was also developed. The genetic transformation system developed successfully transferred a T-DNA containing the mgfp5 and hptII genes into hawthorn callus cells. Furthermore, molecular evidence for the integration of the T-DNA in the transgenic hawthorn culture is also presented.
Materials and methods
Plant material
One-year-old stem cuttings (30–50 cm in length) of Crataegus aronia were collected from a mature tree planted at the University of Jordan campus during September 2008. The stem cuttings were cut by sterile pruner into single internodal stem segments (1–2 cm in length) that were surface sterilized by soaking in 100% ethanol for 1 min and then for 20 min in 3.5% sodium hypochlorite solution containing a few drops of Tween® 20. After sterilization, the internodal cuttings were rinsed five times with sterile distilled water, dried on sterile paper, and used as explants.
Callus induction and maintenance
Preliminary experiments revealed that no callus was induced with explants cultured onto basal MS (Murashige and Skoog 1962) medium without any plant growth regulators or using plant growth regulators combinations described in previous studies (Bahorun et al. 1994). Therefore, series of experiments were performed to induce callus formation using different levels of individual cytokinins and auxins. For this purpose, MS media supplemented with 100 mg l−1 myo-inositol, 100 mg l−1 citric acid, 30 g l−1 sucrose and different concentrations of either Indol-3-acetic acid (IAA) or IBA or 1-naphtaleneacetic acid (NAA) (0, 0.5, 5, or 10 mg l−1) as auxin sources or BA or Kin or Zeatin (0, 0.5, 5 or 10 mg l−1) as cytokinin sources were prepared. The pH of the media was adjusted to 5.8 before the addition of 6 g l−1 agar and then autoclaved for 15 min at 121°C and 1.06 kg cm−2. The sterilized explants were placed horizontally on the media and incubated for 6 weeks at 25 ± 2°C under 16 h photoperiod provided by an irradiance of 56 μmol m−2 s−1 supplied by cool white fluorescent lamps. The results obtained indicated that only IBA at a concentration of 5 mg l−1 was successful in producing small size calli with a callusing percentage of 10% (data not shown).
In order to further improve callus induction, various concentrations of either BA or Kin (0.5, 5 or 10 mg l−1) were added to the 5 mg l−1 IBA MS medium. Each treatment was replicated ten times and each replicate consisted of one Petri dish that contained five internodal stem segments inserted horizontally on the surface of the corresponding medium. The cultures were incubated for 6 weeks under the same conditions described above. After determining the best callus induction medium, pieces (~250 mg in weight) of 45-day-old induced calli were separated from the internodal explants and then subcultured onto fresh callus induction medium. The callus culture was maintained for a 6-month period on the best callus induction medium with a subculture interval of 4 weeks.
To determine the best medium for callus growth and development, 4-week-old callus pieces (250 mg in weight) were cultured onto fresh MS medium without any plant growth regulators for 1 week. The calli were then transferred onto MS media supplemented with 100 mg l−1 myo-inositol, 30 g l−1 sucrose and different concentrations of either NAA or 2,4-D (0, 0.5, 1 or 2 mg l−1) in combination with different concentrations of either BA or Kin (0, 1.5 or 3 mg l−1). Each treatment was replicated ten times and each replicate consisted of one Petri dish that contained five callus pieces (250 mg in weight) placed on the surface of the corresponding medium. The best callus maintenance medium was determined after 4 weeks of culture based on best callus growth, in terms of increase in fresh weight and morphological changes. This medium was named as callus maintenance medium and was used thereafter in maintaining the callus culture with a subculture event every 4 weeks.
Agrobacteriumtumefaciens strain and binary plasmid
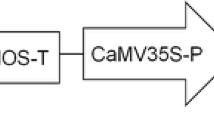
The binary plasmid pCAMBIA1302 was provided by CAMBIA (http://www.cambia.org.au; GenBank accession number: AF234298.1) Canberra, Australia. The pCAMBIA1302 harbors the mgfp5 gene as a reporter gene expressing the GFP and the hptII gene conferring hygromycin resistance as a selectable marker (Fig. 1). The binary plasmid was transferred into Agrobacterium tumefaciens strain GV3101 by electroporation. A single colony of the transformed Agrobacterium was used to inoculate liquid Luria broth (LB) medium supplemented with the proper antibiotics (50 mg l−1 rifampicin, 25 mg l−1 gentamicin and 50 mg l−1 kanamycin) and 100 μM acetosyringone and the culture was incubated in an orbital shaker at 30°C and 150 rpm for 24 h. The bacterial cells were harvested by centrifugation at 5,000g for 10 min and the pellet was resuspended in MS liquid medium supplemented with 20 g l−1 sucrose and 100 μM acetosyringone to an OD600 of 0.5 for the callus inoculation.

Schematic representation of the T-DNA region from the pCAMBIA1302 binary plasmid used for genetic transformation. LB Left border, RB right border, 35S CaMV 35S promoter, NOS nopaline synthase 3′ UTR, 35S-T CaMV 3′ UTR. Positions of the HindIII site (arrow) and hptII probe (hatched box) used in the Southern blot are indicated
Agrobacterium-mediated transformation and selection
Before starting the transformation experiments, the sensitivity of the callus cells to the antibiotic hygromycin was tested. For this purpose, hawthorn calli were cultured on callus maintenance media containing 0, 5, 10, 25 and 50 mg l−1 hygromycin. Callus growth was inhibited when cultured on MS media supplemented with hygromycin concentrations equal to or higher than 10 mg l−1 (data not shown). However, hygromycin concentration of 50 mg l−1 completely inhibited callus growth when compared with lower concentrations where some resistant nontransgenic callus escape was observed. Although the escapes were completely inhibited at a concentration of 50 mg l−1 hygromycin, a slow growth rate of the callus was observed when compared to hygromycin concentration of 10 mg l−1. Therefore, a concentration of 50 mg l−1 hygromycin was used in the transformation experiments to avoid escape, while a concentration of 10 mg l−1 hygromycin was used to maintain high growth rates of the putative transgenic callus (see below).
The transformation experiment was performed using 4-week-old callus, which was previously cultured on the best callus maintenance medium as described above. Callus pieces (~250 mg in weight) were immersed in the Agrobacterium suspension for 30 min. The infected calli were dried on sterile filter paper to remove excess bacteria. The infected calli were co-cultivated with the Agrobacterium on the callus maintenance medium supplemented with 100 μM acetosyringone for 2 days at room temperature in the dark. After the co-cultivation, the infected calli were rinsed three times in distilled water containing 500 mg l−1 cefotaxime followed with a final wash in distilled water without cefotaxime. The infected calli were transferred to callus maintenance medium supplemented with 50 mg l−1 hygromycin and 500 mg l−1 cefotaxime and incubated for 4 weeks under the same culture conditions described above. After 4 weeks, newly emerging hygromycin resistant calli were separated from the infected explants and subcultured onto callus maintenance medium supplemented with 10 mg l−1 hygromycin. The hygromycin resistant calli were maintained by subculturing at 4-week intervals onto the callus maintenance medium supplemented with 10 mg l−1 hygromycin. The transformation experiment was repeated twice with 120 callus pieces per experiment. The transformation efficiency was calculated as the number of hygromycin resistant calli events recovered per total number of infected explants.
PCR analysis
Total genomic DNA was isolated from 4-week-old transformed and non-transformed (negative control) calli cultured on the maintenance medium as described above using the NucleoSpin® Plant II kit (Macherey-Nagel, Diiren, Germany) following the supplier’s instructions. The isolated genomic DNA was used as a template in a polymerase chain reaction (PCR) to amplify a 449-bp fragment from the hptII gene in the putative transgenic callus. The primers used to amplify the corresponding DNA fragment were Hyg-F (5′-CTATTTCTTTGCCCTCGGACG-3′) and Hyg-R (5′-ACCTCGTGCACGCGGATTTC-3′). The reactions were performed in 25 μl volume containing 100 ng genomic DNA, 2.5 μl of dNTPs (100 μM), 5 μl of 5× PCR, 0.5 μM of each primer and 0.25 μl of 5 U μl−1 GoTaq DNA polymerase (Promega, Madison, Wisconsin). The PCR conditions were 94°C for 5 min, followed by 30–40 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min, and a final 10 min extension at 72°C. The PCR products were separated on 1% agarose gels and visualized by ethidium bromide staining.
Southern blot analysis
Southern analysis was performed using 20 μg of genomic DNA isolated from 4-week-old transgenic callus lines and non-transformed (negative control) callus cells cultured on the maintenance medium as described above. The DNA was digested with the HindIII restriction enzyme (MBI, Fermentas, Lithuania) and then separated by electrophoresis in 1% w/v agarose gel. The digested DNA was transferred to a nylon membrane (MBI) and was fixed by UV irradiation. The 449-bp PCR fragment containing the hptII coding region (described above) was used as a probe after labelling with biotin using the Biotin DecaLabel™ DNA Labeling Kit (MBI) following the manufacturer’s instructions. The hybridization reaction was carried out for 24 h at 42°C. The membrane was washed twice at 42°C with 2× SSC + 1% SDS (10 min per wash at 42°C), followed by twice washing with 0.1× SSC + 0.5% SDS (30 min per wash at 65°C). Detection was performed using the Biotin Chromogenic Detection Kit (MBI). All steps were performed following the suppliers’ instructions.
Gene expression analysis
Reverse transcription-polymerase chain reaction (RT-PCR) was used to detect the presence of mgfp5 mRNAs and to determine gene expression levels in transgenic callus lines. For this purpose, total RNA was isolated from 4-week-old PCR positive callus lines and non-transformed (negative control) callus cells using the SV Total RNA Isolation System (Promega) following the supplier instructions. The RNA was used in the synthesis of cDNA using Oligo (dT)18 as a primer and the SuperScript II RNase H− Reverse Transcriptase system (Invitrogen, Carlsbad, CA) following the supplier’s instructions. One microlitre of the first-strand cDNA was used as a template in a PCR reaction using the GFP-F (5′-GGAGTTGTCCCAATTCTTGT-3′) and GFP-R (5′-ATGCCGTTCTTTTGCTTGTC-3′) primers as described above. The GFP-F and GFP-R primers set will amplify a 455-bp DNA fragment in the mgfp5 coding sequence. Hawthorn actin gene-specific primers (CaAct-F (5′-TGGAGCCACAACCTTGATCT-3′) and CaAct-R (5′-GCTTTGATGAAGATTCTCACTG-3′) were used as internal control. The CaAct-F and CaAct-R primers set will amplify a 435-bp DNA fragment in a hawthorn actin coding sequence. The RT-PCR experiments were repeated three times and the PCR products were separated in 1% agarose, stained with ethidium bromide and visualized under ultraviolet light.
For microscopic detection of GFP expression, 250 mg of hawthorn callus cells transformed with pCAMBIA1302 were resuspended in 2 ml of callus maintenance medium. Aliquots from the cell suspension were examined for GFP expression under a Leica DM2500 microscope equipped with a GFP filter set (480/40 nm excitement filter, 505 nm LP dichromatic beam splitter, and 510 nm LP emission filter). Photographs were taken with the Leica DFC340 FX digital camera.
Results and discussion
Establishment of callus culture
In vitro callus induction was performed successfully, producing friable calli with a maximum callus induction percentage of 70% on MS medium supplemented with 5 mg l−1 IBA and 0.5 mg l−1 BA (Fig. 2a). This medium was named as callus induction medium. All other tested plant growth combinations failed to induce any callus and the explants turned brown in color and subsequently died.

Establishment and maintenance of callus culture of Crataegus aronia. a Callus development on Crataegus aronia internodal explant after 21 days of incubation on MS media supplemented with 5 mg l−1 IBA and 0.5 mg l−1 BA (callus induction media). b Callus proliferation on Crataegus aronia internodal explant after 45 days of incubation on callus induction media. c Callus growth after 30 days of subculturing on MS media supplemented with 2 mg l−1 2,4-D and 1.5 mg l−1 Kin (callus maintenance media). d Callus growth after 30 days of subculturing on MS media supplemented with 5 mg l−1 IBA and 0.5 mg l−1 BA (callus induction media)
In general, the incubation of Crataegus aronia internodal stem segments on the callus induction medium resulted in the inflation and swelling of the explants, and the first callus was observed 21 days after incubation (Fig. 2a). After 45 days of incubation on the callus induction medium, friable and variegated colored callus was apparent and it started to cover the explant (Fig. 2b). Bahorun et al. (1994) induced callus using floral buds of Crataegus monogyna cultured on modified B5 Gamborg medium supplemented with 2.0 mg l−1 2,4-D and 0.5 mg l−1 Kin. In comparison to the results of this study, callus induction of Crataegus aronia was achieved on MS media supplemented with 5 mg l−1 IBA and 0.5 mg l−1 BA. The discrepancy between the two results might be attributed to differences in explant type used. However, the callus induction in the current study is in general agreement with the previous finding by Daniele et al. (2008), where a combination of 0.5 mg l−1 IBA and 0.5 mg l−1 or 1.0 mg l−1 BA resulted in the best callus induction frequency of Grindelia robusta Nutt.
To improve the growth of the obtained callus, the effect of different plant growth regulators on callus growth and coloration was examined. The results showed that callus fresh weight varied between treatments depending on auxin or cytokinin sources, concentrations and their combinations (Table 1). For callus cultured on MS media supplemented with two different auxins in the absence of cytokinin, 2.4-D irrespective to its concentration in the media, gave the highest average callus fresh weight when compared to NAA (Table 1). MS media supplemented with different levels of 2,4-D produced deep red colored callus, while callus cultured on NAA was green in color (data not shown). On the other hand, callus cultured on medium supplemented with 3 mg l−1 BA in the absence of auxin source had the highest average callus fresh weight when compared with Kin (Table 1). Callus produced on MS media supplemented with BA gave light red color, while that produced on MS media supplemented with Kin gave green color (data not shown). For treatments with combinations of different types and concentrations of auxins and cytokinins, the best average callus fresh weights were obtained on MS media supplemented with combinations of 2 mg l−1 2,4-D with either 1.5 mg l−1 BA or 1.5 mg l−1 Kin (Table 1). In all combinations, treatments with 2,4-D, red coloration of callus was observed, although lighter red coloration was observed with BA (data not shown), while dark red color was observed with Kin (Fig. 2c).
Compared to callus induction medium, which produced an average callus fresh weight of 1.43 ± 0.11 g, the combination of 2 mg l−1 2,4-D with 1.5 mg l−1 Kin produced a significantly higher average callus fresh weight (3.45 ± 0.10 g) with a predominant red callus coloration indicating the presence of high levels of polyphenols (Fig. 2). Therefore, MS medium supplemented with 2 mg l−1 2,4-D in combination with 1.5 mg l−1 Kin was used subsequently for callus maintenance instead of the callus induction medium. In general agreement with the current study results, Bahorun et al. (1994) reported optimum callus growth and polyphenols production from Crataegus monogyna callus culture on Gamborg B5 medium supplemented with 0.5 mg l−1 Kin and 2.0 mg l−1 2,4-D. In another study, Ogita (2005) recommended the addition of 3.0 mg l−1 2,4-D to enhance Phyllostachys nigra callus proliferation. In contrast to the current study results, Schrall and Becker (1977) found that NAA had a better influence on total phenols production from Crataegus monogyna callus when compared with 2,4-D.
Callus transformation
To develop a suitable transformation system for hawthorn callus cells, the GV3101 Agrobacterium strain harboring the plasmid pCAMBIA1302 containing the mgfp5 and hptII genes was used (Fig. 1). After 2 days of co-cultivating callus cells with the Agrobacterium, callus pieces were transferred directly onto the callus maintenance medium containing 50 mg l−1 hygromycin and 500 mg l−1 cefotaxime. After 1 week in culture under the described selection conditions, the co-cultivated calli started gradually to degenerate and turned brown (Fig. 3a). Four weeks after culturing, small clusters of hygromycin resistance cells started to appear on the surface of the brown degenerated explants (Fig. 3a). The newly growing hygromycin resistance calli were excised and subcultured onto callus maintenance medium supplemented with 10 mg l−1 hygromycin and 500 mg l−1 cefotaxime. The growth and development of the putative transgenic lines were visually compared with non-transformed callus that was subcultured onto the same plate (Fig. 3b). Positive transgenic callus lines were selected and maintained by subculturing at 4-week intervals on the callus maintenance medium supplemented with 10 mg l−1 hygromycin (Fig. 3b). In two different experiments, the transformation efficiency was 21.67% (26 putative transgenic calli out of 120 explants) for experiment 1 and 15.83% (19 putative transgenic calli out of 120 explants) for experiment 2. In total, 45 hygromycin resistance callus lines were obtained and an overall transformation efficiency of 18.8% was achieved.

Transformation of callus of Crataegus aronia by Agrobacterium containing the pCAMBIA1302 plasmid and the expression of GFP in transgenic lines. a Formation of hygromycin resistant calli from callus explants transformed with the pCAMBIA1302 plasmid after 30 days of incubation on maintenance media supplemented with 50 mg l−1hygromycin. b Growth of independent transgenic callus lines (1–6) and non-transformed callus (NT) after 30 days of incubation on maintenance media supplemented with 10 mg l−1 hygromycin. c Light microscopy of transgenic callus cells of Crataegus aronia. d GFP expression in transgenic callus cells of Crataegus aronia
Additionally, the putative transgenic callus lines were examined for GFP expression using fluorescence microscopy. In many instances, weak GFP expression was observed in hygromycin resistant transgenic callus lines (Fig. 3c), while no expression was detected in non-transformed callus (data not shown). Similar to our findings, Terakami et al. (2007) observed a weak GFP expression in transformed dwarf pomegranate calli that gradually disappeared after selection. Additionally, the production of pigments in transformed callus cells could interfere with GFP fluorescence as reported previously (Mercuri et al. 2001; Petri et al. 2008). For instance, no GFP fluorescence was detected in pigmented regions of petals of transformed Kalanchoe blossfeldiana plant when compared with non-pigmented region where green fluorescence was clearly visible (García-Sogo et al. 2010).
To detect the mgfp5 mRNA expression in transgenic lines, semi-quantitative RT-PCR analysis was performed. The mgfp5 transcript was detected in all tested transgenic lines, while no signal was detected in the non-transformed callus (Fig. 4). The RT-PCR DNA products obtained from different transgenic lines have similar expression levels of the mgfp5 transgene when compared to the actin expression levels (Fig. 4). These results indicate that mgfp5 is expressed in transgenic callus cells.

Semi-quantitative RT-PCR analysis of mgfp5 gene expression in putative transgenic callus lines of Crataegus aronia. a Agarose gel showing PCR products (455 bp) amplified from the mgfp5 gene with the primer pair GFP-F/GFP-R from cDNA libraries obtained from Crataegus aronia transgenic callus lines. b Agarose gel showing PCR products (435 bp) amplified from hawthorn actin gene with the primer pair CaAct-F/CaAct-F from cDNA libraries obtained from Crataegus aronia calli. The RT-PCR experiments were repeated three times and a representative result is shown. NT cDNA from non-transformed callus, lanes 1–6 cDNA from independent transgenic callus lines, M low range DNA marker (MBI, Fermentas, Lithuania)
The described genetic transformation system would be a valuable tool for the production of economically important secondary metabolites in hawthorn (Matkowski 2008). The recent developments in understanding molecular mechanisms regulating phenylpropanoid pathway in plants can be utilized to generate transgenic hawthorn callus lines manipulated to boost the production of certain polyphenolic compounds (Ikegami et al. 2007). For instance, the overexpression of VlmybA2 transcription factor gene from grape-induced anthocyanin production in transgenic tobacco callus (Geekiyanage et al. 2007). In addition, it would be interesting to develop an efficient regeneration protocol for the transgenic callus lines in order to produce transgenic hawthorn plants.
Molecular analysis of transformants
PCR analysis of the putative transgenic callus lines was performed using primers specific for the hptII gene that will amplify a DNA product with an expected size of 449 bp, which corresponded to that of the plasmid control. The PCR results confirmed the presence of the transgene in the tested transgenic callus lines (Fig. 5a). No fragment was amplified in the non-transformed callus. Further PCR analysis confirmed the presence of the transgene in all 45 transgenic callus lines obtained in this study (data not shown).

Molecular analysis of putative transgenic callus lines of Crataegus aronia. a Agarose gel showing PCR products (449 bp) amplified from the hptII gene with the primer pair Hyg-F/Hyg-R from DNA extracts of putative transgenic callus. P pCAMBIA1302 DNA plasmid (positive control), NT DNA extracts from non-transformed callus, lanes 1–8 DNA extracts from independent transgenic callus lines, M Low range DNA marker (MBI, Fermentas, Lithuania). b Southern blot analysis of DNA isolated from putative transgenic lines. Genomic DNA (20 μg) was isolated from Crataegus aronia callus and digested with HindIII and probed with a biotin-labeled hptII gene sequence. NT DNA extracts from non-transformed callus; lanes 1–6 DNA extracts from putative transformants. The positions of Lambda EcoRI/HindIII molecular DNA markers are shown on the left
To confirm the integration of the transgene in the callus lines, Southern blot analysis was performed using six randomly selected transgenic callus lines. The HindIII restriction enzyme was used to digest the genomic DNA due to the presence of only one restriction site within the T-DNA region. Therefore, the presence of one band (expected to be above 2.5 kb) in the Southern blot analysis will be expected for each T-DNA integration event (Fig. 1). The results showed that all tested transgenic lines had 1–2 bands each of over 2.5 kb when compared to the non-transformed callus (Fig. 5b). Four of the six lines had a single band with different lengths and the other two lines had 2 bands with different lengths. No bands were detected in the HindIII-digested genomic DNA from the non-transformed control (Fig. 5b).
In conclusion, this is the first report of successful transformation in hawthorn callus cells using an Agrobacterium-mediated system. The transformation protocol offers a high transformation efficiency of up to 18.8%. Utilization of such system could be useful biotechnological approach to increase secondary metabolites levels in hawthorn callus cultures.
References
Bahorun T, Trotin F, Vasseur J (1994) Comparative polyphenolic productions in Crataegus monogyna callus culture. Phytochem 37:1273–1276
Bahorun T, Trotin F, Vasseur J (2002) Polyphenol production in Crataegus tissue cultures (Hawthorn). In: Bajaj YPS, Nagata T, Ebizuka Y (eds) Medicinal and aromatic plants. Springer, Netherlands, pp 24–30
Bahorun T, Aumjaud E, Ramphul H, Rycha M, Luximon-Ramma A, Trotin F, Aruoma O (2003) Phenolic constituents and antioxidant capacities of Crataegus monogyna (Hawthorn) callus extracts. Nahrung 47:191–198
Bauer N, Leljak-Levanic D, Jelaska S (2004) Rosmarinic acid synthesis in transformed callus culture of Coleus blumei Benth. Z Naturforsch 59:554–560
Chang Q, Zuo Z, Harrison F, Chow MS (2002) Hawthorn. J Clin Pharmacol 42:605–612
Chang WT, Dao J, Shao ZH (2005) Hawthorn: potential roles in cardiovascular disease. Am J Chin Med 33:1–10
Dai SH, Zheng P, Marmey P, Zhang SP, Tian WZ, Chen SY, Beachy RN, Fauquet C (2001) Comparative analysis of transgenic rice plants obtained by Agrobacterium-mediated transformation and particle bombardment. Mol Breed 7:25–33
Daniele F, Laura G, Anahi B, Donata R (2008) In vitro plant regeneration from leaf callus of Grindelia robusta Nutt. Plant Biosyst 142:487–490
Ercisli S (2004) A short review of the fruit germplasm resources of Turkey. Genet Resour Crop Ev 51:419–435
García-Sogo B, Pineda B, Castelblanque L, Antón T, Medina M, Roque E, Torresi C, Beltrán JP, Moreno V, Cañas LA (2010) Efficient transformation of Kalanchoe blossfeldiana and production of male-sterile plants by engineered anther ablation. Plant Cell Rep 29:61–77
Geekiyanage S, Takase T, Ogura Y, Kiyosue T (2007) Anthocyanin production by over-expression of grape transcription factor gene VlmybA2 in transgenic tobacco and Arabidopsis. Plant Biotechnol Rep 1:11–18
Gelvin SB (2009) Agrobacterium in the genomics age. Plant Physiol 150:1665–1676
Ikegami A, Eguchi S, Kitajima A, Inoue K, Yonemori K (2007) Identification of genes involved in proanthocyanidin biosynthesis of persimmon (Diospyros kaki) fruit. Plant Sci 172:1037–1047
Ljubuncic P, Portnaya I, Cogan U, Azaizeh H, Bomzon A (2005) Antioxidant activity of Crataegus aronia aqueous extract used in traditional Arab medicine in Israel. J Ethnopharmacol 101:153–161
Matkowski A (2008) Plant in vitro culture for the production of antioxidants–a review. Biotechnol Adv 26:548–560
Mercuri A, Sacchetti A, De Benedetti L, Schiva T, Alberti S (2001) Green fluorescent flowers. Plant Sci 161:961–968
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with the tobacco tissue cultures. Physiol Plantarum 15:473–497
Ogita S (2005) Callus and cell suspension culture of bamboo plant, Phyllostachys nigra. Plant Biotechnol 22:119–125
Ozcan M, Haciseferogullari H, Marakoglu T, Arslan D (2005) Hawthorn (Crataegus spp.) fruit: some physical and chemical properties. J Food Eng 69:409–413
Petri C, Wang H, Alburquerque N, Faize M, Burgos L (2008) Agrobacterium-mediated transformation of apricot (Prunus armeniaca L.) leaf explants. Plant Cell Rep 27:1317–1324
Rigelsky JM, Sweet BV (2002) Hawthorn: pharmacology and therapeutic uses. Am J Health Syst Pharm 59:417–422
Schrall R, Becker H (1977) Production of catechins and oligomeric proanthocyanidins in tissue and suspension cultures of Crataegus monogyna, C. oxyacantha and Ginkgo biloba. Planta Med 32:297–307
Terakami S, Matsuta N, Yamamoto T, Sugaya S, Gemma H, Soejima J (2007) Agrobacterium-mediated transformation of the dwarf pomegranate (Punica granatum L. var. nana). Plant Cell Rep 26:1243–1251
Travella S, Ross SM, Harden J, Everett C, Snape JW, Harwood WA (2005) A comparison of transgenic barley lines produced by particle bombardment and Agrobacterium-mediated techniques. Plant Cell Rep 23:780–789
Valls J, Richard T, Trotin F, Monti JP, Merillon JM, Vitrac X (2007) Carbon-14 biolabeling of flavonols and chlorogenic acids in Crataegus monogyna cell suspension cultures. Food Chem 105:879–882
Acknowledgments
This work was supported in part by a grant from the Hamdi Mango Center for Scientific Research, University of Jordan, and in part by a grant from the Deanship of Scientific Research, University of Jordan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Al Abdallat, A.M., Sawwan, J.S. & Al Zoubi, B. Agrobacteriumtumefaciens-mediated transformation of callus cells of Crataegusaronia. Plant Cell Tiss Organ Cult 104, 31–39 (2011). https://doi.org/10.1007/s11240-010-9798-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-010-9798-1