
Abstract
Tea (Camellia sinensis L.) is considered as to be one of the most consumed beverages globally and a reservoir of phytochemicals with immense health benefits. Despite numerous advantages, tea compounds lack a robust multi-disease target study. In this work, we presented a unique in silico approach consisting of molecular docking, multivariate statistics, pharmacophore analysis, and network pharmacology approaches. Eight tea phytochemicals were identified through literature mining, namely gallic acid, catechin, epigallocatechin gallate, epicatechin, epicatechin gallate (ECG), quercetin, kaempferol, and ellagic acid, based on their richness in tea leaves. Further, exploration of databases revealed 30 target proteins related to the pharmacological properties of tea compounds and multiple associated diseases. Molecular docking experiment with eight tea compounds and all 30 proteins revealed that except gallic acid all other seven phytochemicals had potential inhibitory activities against these targets. The docking experiment was validated by comparing the binding affinities (Kcal mol−1) of the compounds with known drug molecules for the respective proteins. Further, with the aid of the application of statistical tools (principal component analysis and clustering), we identified two major clusters of phytochemicals based on their chemical properties and docking scores (Kcal mol−1). Pharmacophore analysis of these clusters revealed the functional descriptors of phytochemicals, related to the ligand–protein docking interactions. Tripartite network was constructed based on the docking scores, and it consisted of seven tea phytochemicals (gallic acid was excluded) targeting five proteins and ten associated diseases. Epicatechin gallate (ECG)-hepatocyte growth factor receptor (PDB id 1FYR) complex was found to be highest in docking performance (10 kcal mol−1). Finally, molecular dynamic simulation showed that ECG-1FYR could make a stable complex in the near-native physiological condition.
Graphical abstract

Similar content being viewed by others

Introduction
Over the last few years, the pharmaceutical industry faced several challenges owing to the ever-rising increase in the complexities of diseases worldwide. This significant obstacle that the medical community has been facing has instigated a much-needed shift in the treatment strategies that are currently being employed. A dramatic switch that favors multi-target therapeutics and herbo-synthetic combinations as complementary or sometimes even alternative therapy is being explored [1]. There is enough literature to support the claim that secondary metabolites derived from several plants are pharmacologically active and have been quite successful in treating a wide range of diseases [2, 3]. Plant-based medication is not only readily available and affordable, but it also causes the least number of side effects. In the hunt for therapeutically beneficial novel medications, the medicinal plants that have been in use for more than a thousand years are being re-examined [4, 5]. Most of these plants and their phytochemical extracts are being used globally with little or no awareness about their benefits. One such plant extract that we consume daily is the tea prepared with the young leaves, buds, and stalks of Camellia sinensis (L.) Kuntze (Family: Theaceae) [6].
This plant is commonly cultivated commercially in tropical and subtropical regions, primarily in the southern parts of Asia and its leaf infusion, as tea is the most consumed beverage in the world. The pharmacological activity of C. sinensis extracts has been proven to help treat common ailments like hypertension, diabetes, and obesity. It also possesses anticancer, anti-inflammatory, antioxidant, and cardiopulmonary-protective properties [7, 8]. Recently, tea was reported to have beneficial role in the modulation of human gut microbes [9], also. Both black and green teas are rich in antioxidant polyphenols such as catechins [10]. These polyphenolic antioxidants are known to protect cells from intracellular ROS production, hence preventing the oxidative destruction cycle, which includes cell membrane rupture, DNA damage, and subsequently necrosis, mutagenesis, and cell death [11, 12]. This information can be exploited to computationally map the diseases that can be cured using these phytochemicals.
Lately, computational methods have been critical in the design and discovery of novel medicines for various illnesses [13]. The in silico technique of molecular docking is currently regarded as one of the most dependable, cost-effective, time-saving, and efficient ways for screening potential medicines. The use of this approach is extensively advocated and accepted, given the abundance of phytochemicals with potential pharmacological effects [14]. Recently, in silico biology witnessed its extensive applications for search and screening of plants drugs against various conditions such as acetylcholinesterase [15], microbial infections [16] and COVID-19 [17, 18]. Pharmacophore approach is often integrated with the molecular docking technique to understand the chemistry of protein ligand binding interaction. Pharmacophores explain the functional groups that are involved in such docking interaction. Recently, Oluyori et al. [19] identified SARS-CoV-2 inhibitors from bitter cola by using pharmacophore modeling. Molecular docking coupled with statistical methodology may be considered as to obtain accurate and valid results. In our previous studies, we successfully applied multivariate statistical models to validate our findings [20, 21]. The limiting factor of the conventional molecular docking method is that it targets a single protein rather than multiple proteins. Network pharmacology, also known as polypharmacology method, was developed to target multiple proteins and corresponding diseases [22]. Choudhary and Singh [23] utilized network pharmacology approach for targeting various proteins by Piper longum phytochemicals. Li et al. [24] found that the phytochemicals of Shenlian extract could target 37 proteins by using a similar approach. Recently, Wu et al. [25] used this tool for three flower teas (Rosa rugosa, Chrysanthemum morifolium, and Citrus aurantium) targeting nonalcoholic steatohepatitis [25]. Overall, the network pharmacology approach is witnessing an extensive application in the field of computational biology; however, such an approach is scarce for conventional tea based on C. sinensis leaves. Therefore, we believe that the innumerable health benefits of tea warrant further study on its phytochemicals and the diseases it can be used to treat. Our study is claimed to give a new insight on the numerous health benefits of tea by combining a revised approach consisting of a combination of statistics, pharmacophore alignment, virtual library screening and molecular docking.
In the present study, eight important tea compounds (Epicatechin, epicatechin gallate, epigallocatechin gallate, catechin, quercetin, kaempferol, ellagic acid and gallic acid) were docked with thirty target proteins as obtained from the virtual databases. Finally, tripartite network consisting of phytochemicals, proteins and associated diseases was formed with the aid of multivariate principal component analysis (PCA) and pharmacophore-based experimentations.
Materials and methods
Data collection
Tea phytochemicals, target proteins, and related illnesses were selected after extensive literature mining. Databases such as IMPPAT and PubChem were utilized to screen out phytochemicals from Camilla chinensis. IMPPAT or Indian Medicinal Plants, Phytochemistry, and Therapeutics (https://cb.imsc.res.in/imppat/home) is a curated database with material from more than 50 specialized books, and 7000 research article abstracts on Indian traditional plants of ethno botanical value [26]. The structures of the compounds can be downloaded in the sdf, mol, pdb, or pdbqt formats both two-dimensionally and three-dimensionally from this database. PubChem (https://pubchem.ncbi.nlm.nih.gov/) is the world’s largest open access repository of publicly available chemical data at the National Institutes of Health (NIH) [27]. The phytochemicals identified via these servers were further analyzed to find interacting proteins and their corresponding diseases using the PCIDB (https://www.genome.jp/db/pcidb) web server. The Phytochemical Interactions Database, or PCIDB, is an open-source web server with over 100,000 records, most of them from plants and linked to different protein and disease databases [28]. The proteins that interacted with the shortlisted phytochemicals were found using the UniProt database (https://www.uniprot.org/), which houses freely accessible information about protein sequences and their respective functions [29]. Once these proteins were found, the associated diseases were identified using KEGG DISEASE (https://www.genome.jp/kegg/disease/) and OMIM (https://www.ncbi.nlm.nih.gov/omim) databases. The Kyoto Encyclopedia of Genes and Genomes DISEASE Database (KEGG) [30] and Online Mendelian Inheritance in Man (OMIM) [31] are both open-access databases that provide data regarding disease pathways and other gene-related information. Finally, the drugs designated to target the selected proteins were taken from the DrugBank database (https://go.drugbank.com/) [32].
Preparation of ligands
The Avogadro software was primarily used for the preparation of ligand molecules before docking. This software serves as a cross-platform molecule editor and visualizer for computational chemistry, molecular modeling, bioinformatics, materials science, and related fields [33]. The three-dimensional structures of the phytochemicals selected for the study, namely gallic acid, catechin, epigallocatechin gallate, epicatechin, epicatechin gallate, quercetin, kaempferol and ellagic acid, were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in the.sdf format and converted to their respective.pdb formats using the Avogadro software. Three-dimensional structures of the control drug molecules were downloaded from the DrugBank database (https://go.drugbank.com/) in the.pdb formats and were subjected to energy minimization and structural optimization by using Avogadro.
Evaluation of drug-likeness
The SwissADME server (http://www.swissadme.ch/) was employed to assess the drug-likeness of the selected phytochemicals. This tool allows us to compute physicochemical descriptors and estimate ADME parameters, pharmacokinetic characteristics, drug-like nature, and medicinal chemistry friendliness of one or more small compounds. Some of the characteristics that were taken into account to predict the drug-likeness of the compounds were the topological polar surface area (TPSA, important for the estimation of brain permeability and gastrointestinal absorption of the potential drugs), gastrointestinal absorption, PGP substrate, lipophilicity (XLOGP3), and water solubility (Log S) [34]. The Lipinski’s rule of 5 was also used to predict the drug-likeness of the molecules. The rule of 5 indicates that poor absorption or penetration is more probable when there are more than 5 H-bond donors and 10 H-bond acceptors, while molecular weight (MWT) is more than 500, and computed Log P (CLogP) is greater than 5 [35].
Preparation of receptors
The three-dimensional structures of the protein targets were downloaded from the RSCB PDB database (https://www.rcsb.org/). This is a database that stores three-dimensional structural data for big biological entities including proteins and nucleic acids [36]. Out of the thirty proteins that were selected for the study, the native structures of 28 were retrieved from the PDB database itself. These proteins include the carbonic anhydrase 12 [PDB ID: 1JCZ, 1.55 Å, X Ray Diffraction], Dihydrofolate reductase [PDB ID: 1BOZ, 2.10 Å, X Ray Diffraction], Epidermal Growth Factor and Receptor [PDB ID: 1IVO, 3.30 Å, X Ray Diffraction], Prothrombin [PDB ID: 1A2C, 2.10 Å, X Ray Diffraction], Carbonic anhydrase 2 [PDB ID: 12CA, 2.40 Å, X Ray Diffraction], Receptor tyrosine protein kinase erbB-2 [PDB ID: 1MFG, 1.25 Å, X Ray Diffraction], Protein kinase C gamma type (PRKCG) [PDB ID:2E73, Solution NMR], Neutrophil elastase [PDB ID: 1BOF, 2.20 Å, X Ray Diffraction], 72 kDa type IV collagenase [PDB ID: 1CK7, 2.80 Å, X Ray Diffraction], Hepatocyte growth factor receptor [PDB ID: 1FYR, 2.40 Å, X Ray Diffraction], Lysosomal alpha-glucosidase [PDB ID: 5KZW, 2.00 Å, X Ray Diffraction], Cytochrome P450 2D6 [PDB ID: 2F9Q, 3.00 Å, X Ray Diffraction], Cyclin-dependent kinase 4 [PDB ID: 2W96, 2.30 Å, X Ray Diffraction], Angiotensin-converting enzyme [PDB ID: 1O86, 2.00 Å, X Ray Diffraction], Matrix metalloproteinase-9 [PDB ID: 1GKC, 2.30 Å, X Ray Diffraction], P-selectin [PDB ID: 1G1Q, 2.40 Å, X Ray Diffraction], Acetylcholinesterase [PDB ID: 1B41, 2.76 Å, X Ray Diffraction], Carbonic anhydrase 4 [PDB ID: 1ZNC, 2.80 Å, X Ray Diffraction], G1/S-specific cyclin-D1 [PDB ID: 2W96, 2.30 Å, X Ray Diffraction], Dipeptidyl peptidase 4 [PDB ID: 1J2E, 2.60 Å, X Ray Diffraction], Prostaglandin G/H synthase 2 [PDB ID: 5F19, 2.04 Å, X Ray Diffraction], Receptor-type tyrosine-protein kinase FLT3 [PDB ID: 1RJB, 2.10 Å, X Ray Diffraction], PI3-kinase subunit alpha [PDB ID: 2RD0, 3.05 Å, X Ray Diffraction], Xanthine dehydrogenase/oxidase [PDB ID: 2CKZ, 3.20 Å, X Ray Diffraction], Cyclin-dependent kinase 6 [PDB ID: 1BI7, 3.40 Å, X Ray Diffraction], Protein kinase C theta type [PDB ID: 1XJD, 2.00 Å, X Ray Diffraction], Potassium voltage-gated channel subfamily H member 2 or hERG-1 [PDB ID: 1BYM, Solution NMR], and Serine-protein kinase ATM [PDB ID: 5NP1, 5.70 Å, Electron microscopy]. Nonpolar H was removed, and polar H was added to the receptor proteins by using AutoDock tool maintaining physiological pH.
The structures of the remaining two proteins UDP-glucuronosyltransferase 1–1 and UDP-glucuronosyltransferase 1–4 were unavailable, and hence, protein structure prediction was performed by homology modeling. The SWISS-MODEL server (https://swissmodel.expasy.org/) was used for homology modeling [37]. UCSF Chimera software was used to further optimize all 3D protein structures for docking [38]. The quality assessment of the modeled proteins was performed by SwissADME MolProbity tool and UCLA-DOE LAB SAVES v6.0: PROCHECK tool (https://saves.mbi.ucla.edu/).
Active site prediction, molecular docking and docking validation
The active sites present in the target proteins were predicted by CASTp (Computed Atlas of Surface Topography of proteins) server. CASTp (http://sts.bioe.uic.edu/castp/index.html?3trg) is a freely accessible server, and the sites predicted by the server were used to set the dimensions of the grid box while performing docking [39]. The binding grid co-ordinates of the thirty proteins are shown in Supplementary Table S3. Protein–ligand molecular was performed by AutoDock Vina software. AutoDock Vina relies on a sophisticated gradient optimization method, followed by the prediction of the best binding orientation of the ligand within the protein cavity [40]. The eight tea phytochemicals were docked with all thirty proteins in the present study. The phytochemical–protein docking results were compared with that of the respective control drugs. Finally, the top result for each ligand was superimposed with the respective control by using BioVia Discovery Studio software, and the similarity of docking poses was examined.
Statistical analysis
Principal component analysis
Minitab 18 statistical software was used to perform principal component analysis or PCA to group phytochemicals based on the docking scores (binding affinity in Kcal mol−1). This statistical tool allows us to reduce the difficulty that accompanies the interpretation of larger data sets. It decreases the dimensionality of such datasets, therefore improving interpretability, while minimizing data loss. By reducing the overall distance between the data and their projection onto the PC (principal components), the first PC (PC1) maximizes variance. The second and following PCs are unrelated to the first, although chosen in the same way [41, 42].
Heatmap generation and clustering analysis
A second confirmatory statistical test for clustering of docking scores was performed by TBtools software along with heat map generation. Pearson correlation methodology was used for the statistical clustering. TBtools (a Toolkit for Biologists integrating various biological data-handling tools) is an open access and stand-alone software suite, with various integrated tools. This software is based on simple IOS logic (input, output and start) [43].
Structural classification and phytochemical alignment
Structural classification of eight (8) phytochemicals of tea was performed by the ClassyFire server (http://classyfire.wishartlab.com/). Classyfire performs automated structural classification of the input molecules. It relies on hierarchical chemical classification principle and provides structural information as created by ChemOnt [44].
PharmaGist, an open-source server, was used to perform pharmacophore analysis of the eight tea compounds. Pharmacophore screening is helpful to identify the common descriptors or functional groups. Based on the clustering of multivariate statistical PCA modeling (Minitab), each of the clusters was subjected to pharmacophore analysis. PharmaGist analysis is based on the DUD (directory of useful decoys) data set, which contains 2950 active ligands for 40 different receptors, with 36 decoy compounds for each of the active ligands [45]. Three-dimensional structural rendering of the output files was performed by PyMol software to represent common descriptors.
Tripartite network construction
The tool Cytoscape (https://cytoscape.org/) allows for global datasets and functional annotations to be projected and integrated, creating strong visual mappings that span these datasets [46]. To understand the interaction among phytochemicals, protein targets and associated diseases, a tripartite network was constructed using Cytoscape v3.7.2.
Molecular dynamic (MD) simulation
MD simulation was performed as described in our previous study [20]. Briefly, GROMACS-2019.2 [47]-based bio-molecular package was used to perform the molecular docking simulation of Epicatechin gallate (ECG)-hepatocyte growth factor receptor (PDB id 1FYR) as facilitated by the Simlab, the University of Arkansas for Medical Sciences (UAMS), Little Rock, USA [48]. GROMOS96 43a1 force field was used for the simulation, and a ligand topology file was generated by PRODRG software [49]. A triclinic grid box was defined for the protein–ligand complex. The molecular dynamic simulation was performed in SPC water and 0.15 M counter ions (Na+/Cl−) environment. The system was supported with NVT/NPT ensemble temperature 300 K and 1 bar atmospheric pressure. The pressure and temperature were maintained with Parrinello-Rahmanbarostat and Parrinello-Danadio-Bussithermostat, respectively [50]. The resultant model was energy minimized by 5000 steepest descent integrator, and run time was fixed for 100 ns. Parameters, namely root-mean-square deviation (RMSD), radius of gyration (Rg), root-mean-square flexibility (RMSF) and ligand-H bonds, were evaluated for the protein–ligand complex. A comparative study was performed with above-mentioned parameters for ligand-bound and ligand-free proteins.
Free energy analysis by MM-PBSA calculation
The free energies of epicatechin gallate (ECG)-hepatocyte growth factor receptor (PDB id 1FYR) complex (ΔG_Vander Waal, ΔG_Electrostatic, ΔG_Polar, ΔG_Non-Polar and ΔG_Binding) were estimated by molecular mechanics-Poisson–Boltzmann solvent-accessible surface area (MM-PBSA) method using g-mmbsa package [51].
The following equation is performed for calculating ΔG_Bind (KJ mol−1):
where ΔG_Comp = the energy of protein–ligand complex, G_Prot and G_Lig = individual energy of protein and ligand, respectively. The MMPBSA calculation was performed for 5-ns trajectory.
Results and discussion
Data collections
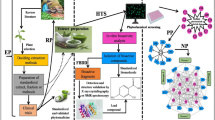
Tea (C. sinensis) leaves are rich in compounds such as catechin, epicatechin, epicatechin gallate and epigallocatechin gallate [3]. Catechin and its derivatives generally constituted of 30% dry weight of black tea leaves [52]. Epigallocatechin gallate was also reported to be the principal constituent of green tea [53]. Furthermore, phytochemicals such as quercetin, kaempferol, ellagic acid and gallic acid were also reported in tea [54, 55]. Considering previously reported health benefits, richness and dominance of above-mentioned phytochemicals in the tea leaves, these eight compounds were selected for this study. For instance, catechin–gallic acid esters epicatechin, epicatechin gallate and epigallocatechin gallate were reported to have antidiabetic and antioxidant properties [6]. Epigallocatechin gallate, in particular, also served as a hepatoprotective agent by suppressing cytotoxin-induced cell death [56]. Ellagic acid had antioxidant and anti-inflammatory properties. Gallic acid and kaempferol displayed anticancer and anti-inflammatory activities, while Quercetin had been deemed beneficial in cases of hypertension [3]. Based on these findings, eight tea phytochemicals, namely epicatechin, epicatechin gallate, epigallocatechin gallate, catechin, quercetin, kaempferol, ellagic acid and gallic acid, were selected for this study (Table 1). The schematic presentation of the work is represented in Fig. 1.

Schematic representation of the study
Furthermore, we identified 30 proteins and their associated target diseases though database scouting as mentioned in the Materials and Methods. In Table 2, we tabulated target proteins, associated diseases and control drugs of the respective proteins.
Drug-likeness of the compounds
SwissADME could effectively predict the drug-like properties of eight tea phytochemicals. However, it was noted in the literature that the drug metabolism should not be assessed in one parameter. Hou et al. [83] reported that the values obtained from TPSA (total polar surface area) did not correlate with another drug-likeliness parameter. In general, TPSA represents the bioavailability of drug candidates, and the recommended range is 20 to 140 Å2 [82]. Except two compounds, other phytochemicals satisfied the criteria (Table 3). Water solubility of the drug-like molecules is considered to have a significant impact on ADME (absorption, distribution, metabolism and excretion) properties, i.e., on the bioavailability of the compounds. The parameter Log S represents the intrinsic solubility of the candidate drugs in the water. The log value range within − 1 to − 5 shows the balancing between the solubility in water and lipids [83]. In our study, all the phytochemicals are found well within the range of log S (− 1.64 to − 3.70). Further, the transcellular passive diffusion is one of the major parameters for permeability and absorption of the drug-like compounds, and crossing the barrier of the gastrointestinal (GI) tract represents such property. Our study showed that except epicatechin gallate and epigallocatechin gallate, all six phytochemicals had high GI absorption capacity. XlogP3 is an automatic method and determinant of lipophilicity of drug-like molecules. The recommended range of XlogP3 is 1.48–6.19, and all the compounds passed the criteria. p-Glycoprotein substrate (PGP) is a representative of active transporter and generally removes the drug molecules from the cells, when positive. Except two compounds, all other six were found to be PGP negative [84]. Finally, we observed minimum Lipinski's violations (0–2). Overall, all the compounds were found to be passing one or other drug-likeness parameters.
Quality analysis of the homology modeled proteins
The homology modeling of two proteins (UDP-glucuronosyltransferase 1–1: UniProt id P22309 and UDP-glucuronosyltransferase 1–4: UniProt id P22310) were performed by SWISS-Model server, based on the templates of PDB id 6KVJ.1.A and 6O86.1.A. The quality analysis was done by the parameters MolProbity score, QMEAN and GMQE (Global Model Quality Estimate) Z scores. While QMEAN represents the degree of nativeness of the modeled structure, GMQE (Global Model Quality Estimation) shows expected accuracy of the structured models [85]. The results of these quality estimates are shown in supplementary tables S1 and S2. Finally, Ramachandran plots revealed > 85% favorable regions for both the modeled structures (Fig. S1 and S2). Overall, despite the unavailability of accurate templates for our modeled proteins, we were able to obtain moderate quality models.
Molecular docking
Molecular docking is the preferred tool for initial screening of drug molecules. It eliminates the need for tedious, time-consuming and expensive techniques that were previously used for drug discovery. AutoDock Vina is one of most established and robust open-source molecular docking software. This software is considered one of the top ranking programs consistent with the screening benchmark, known as directory of useful decoys by Watowich group [86]. Gaillard 2018 [87] compared the performance of AutoDock and AutoDock Vina based on a comparative assessment of scoring functions (CASF-2013) and reported superior function of AutoDock Vina in all aspects. In our previous study, we successfully deployed AutoDock Vina to identify piperine as the inhibitor of Dengue and Ebola virus enzymes [21]. In this work, all seven phytochemicals, except gallic acid, showed substantial inhibitory potential against target proteins. Out of all the phytochemicals, epigallocatechin gallate, epicatechin gallate, and quercetin have shown maximum coverage, targeting 22, 21 and 20 proteins, respectively, where the binding scores were better than those of the respective controls (Table 4). In the molecular docking experiment, we identified the best target protein for each of the phytochemicals in terms of binding affinity (Kcal mol−1). Target proteins, namely hepatocyte growth factor receptor (PDB id 1FYR), dihydrofolate reductase (PDB id 1BOZ), prostaglandin G/H synthase 2 (PDB id 5F19) and angiotensin-converting enzyme (PDB id 1O86), on an average was found to be most potential targets by the phytochemicals. Epigallocatechin gallate could inhibit the progression of tumor genes by blocking DNA methyltransferases, proteases, and dihydrofolate reductase (DHFR) activities [88]. In a recent study, Zong et al. [21] showed that this phytochemical prevented aminoglycosides-induced ototoxicity in Zebra fish, by blocking several enzymes including DHFR [89]. In our work, epigallocatechin gallate scored considerably well (− 9.8 kcal mol−1) against DHFR compared with other target proteins. Tea phytochemicals were earlier reported to have strong inhibiting potentials against angiotensin-converting enzyme (ACE) protein (Fauzi et al., 2018) [90]. Epicatechin and kaempferol were found to have substantial binding potentials with matrix metalloproteinase (MMP)-9 (PDB id 1GKC) compared to their respective controls, both having similar scores − 9 kcal mol−1. Kanbarkar and Mishra (2021) [91] recently in their findings showed that tea phytochemicals could inhibit MMP protein. Kaempferol could modulate the activity of MMP-2 and 9 proteins [92]. Anticancer activities of tea polyphenols were vastly reported in the literature [93, 94]. In their review, Cheng et al. (2020) [95] showed the modulating effect of Catechin and its derivatives against various types of cancer. The flavonoids quercetin and rutin could suppress the expressions of multiple oncogenes, including prostaglandin synthase 2 and showed antiglioma effects [96]. Consistent with these findings, among all 30 target proteins, prostaglandin G/H synthase 2 (PDB id 5F19) was the best targeted by the phytochemicals catechin and quercetin with the binding affinities − 9.2 and − 9.4 kcal mol−1, respectively. Recently, Wang et al. (2021) [97] showed that the tea phytochemical catechin had strong binding affinity with ACE protein. The IC50 value of ellagic acid was shown as 2 mM against ACE protein indicating its strong inhibitory potential [98]. In consistent with these findings, among all other proteins ellagic acid showed the highest binding potential to ACE protein (− 8.9 kcal mol−1) compared with control. Epicatechin gallate scored the highest (− 10 kcal mol−1) against hepatocyte growth factor receptor, among all target proteins as well as phytochemicals. Hepatocyte growth factor is one of the major elements for the development of hepatocellular carcinoma [99], and epicatechin gallate is the one of the most prominent phytochemicals in tea leaves [100, 101]. Elsewhere, it was shown that green tea and epicatechin gallate effectively prevented and managed nonalcoholic fatty liver diseases (NAFLD) [102]. Further, catechin derivatives were reported to be very effective against hepatocellular carcinoma [103]. Overall, all these findings were consistent with various wet laboratory-based studies, in turn validating the accuracy and robustness of our outcomes.
Interaction analysis and docking validation
For amino acid–ligand interaction analysis and docking pose validation, the best protein–ligand complexes, namely Epicatechin/Kaempferol-1GKC (UniProt P14780), Epicatechin gallate-1FYR (UniProt P08581), Epigallocatechin gallate-1BOZ (UniProt P00374), Catechin/Quercetin-5F19 (UniProt P35354), and Ellagic acid-1O86 (UniProt P12821), were selected. Control drugs were re-docked with the respective proteins. Overlaid 3D diagrams of phytochemical-control drug pockets are represented in Fig. 2. Epicatechin and kaempferol along with the control drug marimastat interacted with the common amino acids, namely LEU288A, ALA189A, TYR423A and HIS401A, of the protein matrix metalloproteinase-9 (PDB id 1GKC) through conventional H, Pi-H and other bonds. For the target protein hepatocyte growth factor receptor (PDB id 1FYR), control drug crizotinib, interacted with the amino acid VAL123A through conventional H bond similar to epicatechin gallate. Methotrexate and epigallocatechin gallate both interacted with the protein dihydrofolate reductase (PDB id 1BOZ), for the amino acids THR56A, SER59A, AL115A, ALA9A and LEU22A. Control drug Icosapent shared the similar binding pocket (HIS207B, HIS386B, and HIS388B) of the protein Prostaglandin G/H synthase 2 (PDB id 5F19) with two phytocompounds, namely catechin and quercetin. Finally, ellagic acid similar to its corresponding control drug perindopril stabilized its interaction with the target angiotensin-converting enzyme through the common amino acids GLU123A, ARG124A, TYR135A and SER517A (Table 5).

Superimposed three-dimensional docking interaction between control drugs and phytochemicals to target proteins (Uniprot id), along with H bond interactions; catechin (CAT), epigallocatechin gallate (EGCG), epicatechin (EC), epicatechin gallate (ECG), quercetin (QUE), kaempferol (KAE), and ellagic acid (EA)
Statistical analysis
Application of advanced statistical tool and molecular docking methodology could give insightful information on the underlying mechanism of protein–ligand biding affinities. Recently, PCA was successfully deployed for understanding the molecular interaction of Mur enzymes and gallomyricitrin [104]. In our previous studies, we used PCA tool to categorize phytochemical groups and correlated their chemical classes with their docking scores [20, 105]. Docking outputs from 8 ligands and 30 proteins along with the control docking results were taken as inputs for PCA, and the results are presented in Fig. 3a. The first principal component (PC1) and the second component (PC2) explained approximately 61.50 and 1.78% of the variance. Each of the principal components shows different dimensions of the measured dataset, and all these components are transformed into uncorrelated datasets. We observed four distinct clusters in the analysis. While cluster 4 consisted only of hydroxybenzoic acid derivative gallic acid, cluster 2 had control drugs. Ellagic acid and catechin gallate esters epigallocatechin gallate and epicatechin gallate were grouped into cluster 1, and other flavonoids quercetin, catechin, epicatechin and kaempferol were found in the cluster 3. Placement of gallic acid and control drugs in the separate cluster was indicative of comparatively weaker biding affinities to target proteins compared to other compounds.

Statistical analysis of binding affinities (kcal mol−1), a principal component analysis and b heat map with clustering
Further, to validate the finding from PCA analysis, we performed heat map generation and clustering analysis based on the Pearson correlation algorithm (Fig. 3b). All clustering algorithms rely on the grouping principle based on the input data. Clustering principle is commonly used in gene expression experiments; however, we used this statistical methodology earlier in molecular docking study [21]. Correlation-based clustering method has an advantage over the distance-based matrices in terms of scaling irrelevance and primary focus on the relativity of output data [106]. In this study, we observed a clustering pattern similar to the PCA results.
Molecular alignment and pharmacophore analysis
Pharmacophore approach is being extensively used in the computer-aided drug discovery. It is defined as the ‘ensemble of various steric and electronic features’ for a particular molecule. Pharmacophores of ligand entities represent functional descriptors (groups) such as aromatic, acceptor, and donor that are responsible for interaction with target proteins. Recently, we aligned three molecules, namely curcumin, piperine and chloroquine, to understand the common interacting descriptors with COVID 19 spike protein as a target [20]. In a similar study, Oluyori et al., 2022 [19] showed that standard aromatic rings of garcinia biflavonoid I, garcinia biflavonoid II, kolaflavone and amentoflavone were responsible for their interaction with the main protease and RNA-dependent-RNA polymerase of protein of SARS-CoV-2. In this study, based on the PCA/clustering outputs, we performed molecular alignment to identify functional pharmacophores of the molecules. For PCA cluster 1 (ellagic acid, epigallocatechin gallate and epicatechin gallate), three H bond acceptors, two H bond donors and one aromatic group were identified as common descriptors. On the other hand, PCA cluster 3 (quercetin, catechin, epicatechin and kaempferol) had three H bond donors, one H bond acceptor and two aromatic groups as common functional groups (Fig. 4). Further, we observed alignment scores for PCA cluster and 2 as 26.638 and 33.237, respectively (Figs. S1 and S2). Alignment scores represent the overall alignment performance of the input molecules. Two-dimensional interaction diagrams of the best ranked ligands with their respective target proteins showed the identified descriptors as functional pharmacophores (Fig. 5).

Molecular alignment of phytochemicals showing pharmacophores (ACC acceptor, DON donor, AR aromatic)

Protein–phytochemical interacting amino acids showing pharmacophores involved: catechin (CAT), epigallocatechin gallate (EGCG), epicatechin (EC), epicatechin gallate (ECG), quercetin (QUE), kaempferol (KAE), and ellagic acid (EA)
Phytochemical interaction with target proteins and respective diseases
Network pharmacology is an advanced technique, which enables researchers to identify multiple targets for their candidate compounds in the shortest time possible. This is an emerging methodology in drug discovery studies and is being extensively used in phytochemical-based research. In our earlier study, we successfully deployed network pharmacology approach to identify multiple disease targets of ginger phytochemicals [107]. Recently, Sarkar et al. [108] used this methodology to identify targets for mango constituent. Tea compounds were essentially reported to have multiple health benefits and diseases modulating role; however, the application of a network pharmacology approach for these tea molecules is scarce to the best of our knowledge. Nevertheless, a few such works were presented for non-Camellia sinensis tea [109, 110]. In this work, a tri-partite network consisting phytochemicals, proteins and associated diseases is constructed based on the top docking score (binding affinity Kcal mol−1) for each of the phytocompounds (Fig. 6). Target proteins and their related diseases are presented in Table 2. Gallic acid did not qualify due to overall weak binding affinities toward target proteins. While the result was analyzed, it was found that epicatechin and kaempferol both were connected with the common target (1GKC, matrix metalloproteinase-9). Matrix metalloproteinase-9 is associated with diseases such as like intervertebral disc disease and penile cancer.

Tripartite network of phytochemicals, proteins and associated diseases: catechin (CAT), epigallocatechin gallate (EGCG), epicatechin (EC), epicatechin gallate (ECG), quercetin (QUE), kaempferol (KAE), and ellagic acid (EA)
Molecular dynamic simulation
The computer-assisted molecular docking simulation tool is very useful in computational biology study to determine the accuracy and stability of the docked complex in a near-native physiological environment. Kushwaha et al. [111] recently exhibited the binding stability of quercetin-3-rutinoside-7-glucoside at the active site of SARS-Cov-2 Mpro by using MD simulation tool. Similarly, Islam et al. [112] use MD simulation tool to evaluate the docking potential of the selected phytochemicals against the main protease of SARS-CoV-2. In our study, epicatechin gallate (ECG)-hepatocyte growth factor receptor (PDB id 1FYR) complex scored highest (− 10 kcal mol−1) in terms of binding affinity and was analyzed for understanding the stability and conformational dynamics of the complex using MD simulation tool. The results obtained collectively from all the four parameters, namely RMSD, RMSF, gyration and ligand–protein H bonds, clearly showed that the complex formed by the phytochemical ECG was stable and could effectively bind with the target protein hepatocyte growth factor receptor. Root mean square deviation (RMSD) is indicative of inter-residual interactions within protein. We performed a 100-ns simulation for both apo and bound states of the selected protein. We observed a gradual increment of the RMSD in both the states up to 20 ns, and thereafter, a stable RMSD was observed throughout the remaining simulation time within the range of 0.25–0.35 nm on an average. Overall, marginally higher RMSD values were seen for the bound state protein (~ 0.31 nm) than the apo state (~ 0.25 nm), possibly due to the introduction of the ligand (ECG) into the structure (Fig. 7a). A similar observation was reported for radius of gyration (Rg). The compactness of the protein structure in the free and bound forms was analyzed in Rg analysis (Fig. 7b). Before beginning the simulation, the Rg values of apo and bound states were calculated as 1.7 and 1.64 nm, respectively. After initial fluctuation up to 30 ns, both the structures were stabilized at ~ 1.7 and ~ 1.5 nm of Rg for apo and bound states, respectively. In the MD simulation, initial fluctuations at the start of the trajectory were commonly cited in the literature. In addition, the marginal difference of output parameter values between bound and free-state of protein was also reported elsewhere to explain stable dynamics [113, 114]. Further, to study the residual mobility of ligand-bound and ligand-unbound forms, we analyzed the root-mean-square fluctuations (RMSF) of individual amino acid residues. As shown in Fig. 7c, at the bound state, we did not observe any remarkable change in the residual flexibility when compared to the apo form. In both cases, the difference in residual flexibility was minimal. Stable hydrogen bond formation between ligand and proteins is important for the structural stability of the complex. For interaction with the protein 1FYR, ECG showed maximum 5 H bonds. We observed at least 2 H to be long-lived throughout the simulation of 100 ns (Fig. 7d).

Molecular dynamic (MD) simulation of epicatechin gallate (ECG)-hepatocyte growth factor receptor (PDB id 1FYR) complex: a root-mean-square deviation (RMSD), b radius of gyration, c root-mean-square fluctuation (RMSF) and ligand–protein H bonds
Free energy analysis by MM-PBSA calculation
In a recent development, MM-PBSA method is considered as an advanced methodology for the estimation of free energy. Although this method increases the computational cost significantly, it can provide more accurate results than the conventional score-based molecular docking technique [115]. The results showed that epicatechin gallate (ECG) had a very high binding affinity (− 242.09 ± 10.97 kJ mol−1) toward the receptor protein hepatocyte growth factor receptor (PDB id 1FYR). The dynamic stability of a protein–ligand complex was further explained by other important energy terms, namely van der Waals, electrostatic, nonpolar and polar. Among these energy terms, polar or polar solvation energy had the opposite effect, which links binding energy to the unfavorable positive value [116]. In this study, we observed that van der Waals contributed more negative energy than its electrostatic counterpart (Table 6). In our study, the low contribution of polar solvation energy was important to stabilize the ECG-1FYR complex. Overall, a stable energy trajectory was observed, as demonstrated by representative 5-nm snapshots on five energy terms, namely, ΔG_Van der Waal, ΔG_Electrostatic, ΔG_Polar, ΔG_Non-Polar and ΔG_binding (Fig. 8).

Free energy terms of epicatechin gallate (ECG)-hepatocyte growth factor receptor (PDB id 1FYR) complex, a ΔG_Van der Waal, b ΔG_Electrostatic, c ΔG_Polar, d ΔG_Non-Polar and e ΔG_binding
Conclusion
In this study, we found that tea phytochemicals were capable of targeting multiple target proteins associated with various ailments. The results supported by multiple tools like statistics, pharmacophore analysis, network pharmacology and molecular docking showed that seven tea phytochemicals (gallic acid was excluded) could altogether target five proteins and ten associated diseases at a minimum. However, this was a minimum estimate due to stringent screening applied in the present work. Further, among eight phytochemicals studied, epicatechin gallate was found to bind strongly with the target protein hepatocyte growth factor receptor (PDB id 1FYR) with the affinity as 10 kcal mol−1. Epicatechin gallate formed a highly stable complex with the target protein in a physiological environment as found by MD simulation.
References
De B, Bhandari K, Katakam P, Goswami TK (2019) Development of a standardized combined plant extract containing nutraceutical formulation ameliorating metabolic syndrome components. SN Appl Sci 1:1–12. https://doi.org/10.1007/s42452-019-1518-9
Abu Bakar MF, Mohamed M, Rahmat A, Fry J (2009) Phytochemicals and antioxidant activity of different parts of bambangan (Mangifera pajang) and tarap (Artocarpus odoratissimus). Food Chem 113:479–483. https://doi.org/10.1016/j.foodchem.2008.07.081
Bag S, Mondal A, Majumder A, Banik A (2022) Tea and its phytochemicals: hidden health benefits & modulation of signaling cascade by phytochemicals. Food Chem 371:131098. https://doi.org/10.1016/j.foodchem.2021.131098
Siddiqui M, Shah N, Dur-re-shahwar M et al (2021) The phytochemical analysis of some medicinal plants. J Liaquat Univ Med Health Sci. https://doi.org/10.38106/LMRJ.2021.3.1-02
Badalamenti N, Sottile F, Bruno M (2022) Ethnobotany, phytochemistry, biological, and nutritional properties of genus crepis—a review. Plants 11:519. https://doi.org/10.3390/plants11040519
Samanta S (2020) Potential bioactive components and health promotional benefits of tea (camellia sinensis). J Am Coll Nutr. https://doi.org/10.1080/07315724.2020.1827082
Ferreira MCL, Lima LN, Cota LHT et al (2020) Effect of camellia sinensis teas on left ventricular hypertrophy and insulin resistance in dyslipidemic mice. Braz J Med Biol Res 53:1–6. https://doi.org/10.1590/1414-431x20209303
Sánchez M, González-Burgos E, Iglesias I et al (2020) The pharmacological activity of camellia sinensis (L.) kuntze on metabolic and endocrine disorders: a systematic review. Biomolecules 10:2–33. https://doi.org/10.3390/biom10040603
Khairudin MAS, Mhd Jalil AM, Hussin N (2021) Effects of polyphenols in tea (Camellia sinensis sp.) on the modulation of gut microbiota in human trials and animal studies. Gastroenterol Insights 12:202–216. https://doi.org/10.3390/gastroent12020018
Deka H, Barman T, Dutta J et al (2021) Catechin and caffeine content of tea (Camellia sinensis L.) leaf significantly differ with seasonal variation: a study on popular cultivars in North East India. J Food Compos Anal. https://doi.org/10.1016/j.jfca.2020.103684
do Carmo MAV, Pressete CG, Marques MJ et al (2018) Polyphenols as potential antiproliferative agents: scientific trends. Cur Opin Food Sci 24:26–35. https://doi.org/10.1016/j.cofs.2018.10.013
Zhao Y, Fang C, Jin C et al (2022) Catechin from green tea had the potential to decrease the chlorpyrifos induced oxidative stress in larval zebrafish (Danio rerio). Pestic Biochem Phys 182:105028. https://doi.org/10.1016/j.pestbp.2021.105028
Rampogu S, Baek A, Gajula RG et al (2018) Ginger ( Zingiber officinale ) phytochemicals — gingerenone - a and shogaol inhibit SaHPPK : molecular docking, molecular dynamics simulations and in vitro approaches. Ann Clin Microbiol. https://doi.org/10.1186/s12941-018-0266-9
Berg EL (2014) Systems biology in drug discovery and development. Drug Discov 19:113–125. https://doi.org/10.1016/j.drudis.2013.10.003
Chitra L, Penislusshiyan S, Soundariya M et al (2022) Anti-acetylcholinesterase activity of corallocarpus epigaeus tuber: in vitro kinetics, in silico docking and molecular dynamics analysis. J Mol Struct. https://doi.org/10.1016/j.molstruc.2022.132450
Nischitha R, Shivanna MB (2022) Diversity and in silico docking of antibacterial potent compounds in endophytic fungus chaetomium subaffine sergeeva and host heteropogon contortus (L.) P. Beauv Process Biochem 112:124–138. https://doi.org/10.1016/j.procbio.2021.11.013
Souid I, Korchef A, Souid S (2022) In silico evaluation of Vitis amurensis Rupr. polyphenol compounds for their inhibition potency against CoVID-19 main enzymes Mpro and RdRp. SPJ. https://doi.org/10.1016/j.jsps.2022.02.014
Devi A, Chaitanya NS (2021) Designing of peptide aptamer targeting the receptor-binding domain of spike protein of SARS-CoV-2: an in silico study. Mol Divers 26:157–169. https://doi.org/10.1007/s11030-020-10171-6
Oluyori AP, Olanipekun BE, Adeyemi OS et al (2022) Molecular docking, pharmacophore modelling, MD simulation and in silico ADMET study reveals bitter cola constituents as potential inhibitors of SARS-CoV-2 main protease and RNA dependent-RNA polymerase. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.2024883
Nag A, Paul S, Banerjee R, Kundu R (2021) In silico study of some selective phytochemicals against a hypothetical SARS-CoV-2 spike RBD using molecular docking tools. Comput Biol and Med 137:104818. https://doi.org/10.1016/j.compbiomed.2021.104818
Nag A, Chowdhury RR (2020) Piperine, an alkaloid of black pepper seeds can effectively inhibit the antiviral enzymes of dengue and ebola viruses, an in silico molecular docking study. Virusdisease 31:308–315. https://doi.org/10.1007/s13337-020-00619-6
Hopkins AL (2008) Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4:682–690. https://doi.org/10.1038/nchembio.118
Choudhary N, Singh V (2018) A census of P. longum’s phytochemicals and their network pharmacological evaluation for identifying novel drug-like molecules against various diseases, with a special focus on neurological disorders. PLoS ONE 13(1):e0191006. https://doi.org/10.1371/journal.pone.0191006
Li J, Wang C, Wang Y et al (2022) Network pharmacology analysis and experimental validation to explore the mechanism of Shenlian extract on myocardial ischemia. J Ethnopharmacol. https://doi.org/10.1016/j.jep.2022.114973
Wu P, Liang S, He Y et al (2022) Network pharmacology analysis to explore mechanism of three flower tea against nonalcoholic fatty liver disease with experimental support using high-fat diet-induced rats. Chin Herb Med. https://doi.org/10.1016/j.chmed.2022.03.002
Mohanraj K, Karthikeyan BS, Vivek-Ananth RP et al (2018) IMPPAT: a curated database of Indian medicinal plants, phytochemistry and therapeutics. Sci Rep 8:1–17. https://doi.org/10.1038/s41598-018-22631-z
Kim S, Thiessen PA, Bolton EE et al (2016) PubChem substance and compound databases. Nucleic Acids Res 44:D1202–D1213. https://doi.org/10.1093/nar/gkv951
Ali M, Therapy BC (2019) Olive Net Library presentation by : DR . MOHAMMED ALI ABD EL-HAMMED ABD ALLAH. https://doi.org/10.13140/RG.2.2.24146.91847
Bateman A, Martin MJ, O’Donovan C et al (2015) UniProt: a hub for protein information. Nucleic Acids Res 43:D204–D212. https://doi.org/10.1093/nar/gku989
Yi Y, Fang Y, Wu K et al (2020) Comprehensive gene and pathway analysis of cervical cancer progression. Oncol Lett 19:3316–3332. https://doi.org/10.3892/ol.2020.11439
Amberger JS, Bocchini CA, Schiettecatte F et al (2015) OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an Online catalog of human genes and genetic disorders. Nucleic Acids Res 43:D789–D798. https://doi.org/10.1093/nar/gku1205
Wishart DS, Knox C, Guo AC et al (2008) DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 36:901–906. https://doi.org/10.1093/nar/gkm958
López R (2014) Capillary surfaces with free boundary in a wedge. Adv Math 262:476–483. https://doi.org/10.1016/j.aim.2014.05.019
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:1–13. https://doi.org/10.1038/srep42717
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 64:4–17. https://doi.org/10.1016/j.addr.2012.09.019
Berman HM, Battistuz T, Bhat TN et al (2002) The protein data bank. Acta Crystallogr Sect D: Biol Crystallogr 58:899–907. https://doi.org/10.1107/S0907444902003451
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. https://doi.org/10.1093/nar/gky427
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Tian W, Chen C, Lei X et al (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46:W363–W367. https://doi.org/10.1093/nar/gky473
Allouche A (2012) Software news and updates gabedit — a graphical user interface for computational chemistry softwares. J Comput Chem 32:174–182. https://doi.org/10.1002/jcc
Jollife IT, Cadima J (2016) Principal component analysis: a review and recent developments. Philos Trans Royal Soc A PHILOS T R SOC A 374(2065):20150202. https://doi.org/10.1098/rsta.2015.0202
Lever J, Krzywinski M, Altman N (2017) Points of significance: principal component analysis. Nat Methods 14:641–642. https://doi.org/10.1038/nmeth.4346
Chen C, Chen H, Zhang Y et al (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13:1194–1202. https://doi.org/10.1016/j.molp.2020.06.009
Djoumbou Feunang Y, Eisner R, Knox C et al (2016) ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J Cheminform. https://doi.org/10.1186/s13321-016-0174-y
Schneidman-Duhovny D, Dror O, Inbar Y et al (2008) PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Res 36:W223–W228. https://doi.org/10.1093/nar/gkn187
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (1971) Cytoscape: a software environment for integrated models. Genome Res 13:426
Kutzner C, Páll S, Fechner M et al (2019) More bang for your buck: improved use of GPU nodes for GROMACS 2018. J Comput Chem 40:2418–2431. https://doi.org/10.1002/jcc.26011
Bekker H, Berendsen HJC, Dijkstra EJ, et al (1993) Gromacs-a parallel computer for molecular-dynamics simulations. In: 4th International conference on computational physics (PC 92). World scientific publishing, pp 252–256
Schüttelkopf AW, Van Aalten DM (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D ACTA CRYSTALLOGR D 60:1355–1363. https://doi.org/10.1107/S0907444904011679
Huang J, Rauscher S, Nawrocki G et al (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Met hods 14:71–73. https://doi.org/10.1038/nmeth.4067
Kumari R, Kumar R, Consortium OSDD, Lynn A (2014) g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962
Mena P, Bresciani L, Brindani N et al (2019) Phenyl-γ-valerolactones and phenylvaleric acids, the main colonic metabolites of flavan-3-ols: synthesis, analysis, bioavailability, and bioactivity. Nat Prod Rep 36:714–752. https://doi.org/10.1039/C8NP00062J
da Silva PM (2013) Tea: a new perspective on health benefits. Food Res Intl 53:558–567. https://doi.org/10.1016/j.foodres.2013.01.038
Jeganathan B, Punyasiri PA, Kottawa-Arachchi JD et al (2016) Genetic variation of flavonols quercetin, myricetin, and kaempferol in the Sri Lankan tea (Camellia sinensis L.) and their health-promoting aspects. Intl J Food Sc 2016:1–9. https://doi.org/10.1155/2016/6057434
Jiang H, Yu F, Qin LI et al (2019) Dynamic change in amino acids, catechins, alkaloids, and gallic acid in six types of tea processed from the same batch of fresh tea (Camellia sinensis L.) leaves. J Food Compos Anal 77:28–38. https://doi.org/10.1016/j.jfca.2019.01.005
Aboulwafa MM, Youssef FS, Gad HA et al (2019) A comprehensive insight on the health benefits and phytoconstituents of Camellia sinensis and recent approaches for its quality control. Antioxidants 8:455. https://doi.org/10.3390/antiox8100455
Whittington DA, Waheed A, Ulmasov B et al (2001) Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. PNAS 98:9545–9550. https://doi.org/10.3390/antiox8100455
Gangjee A, Vidwans AP, Vasudevan A et al (1998) Structure-based design and synthesis of lipophilic 2, 4-diamino-6-substituted quinazolines and their evaluation as inhibitors of dihydrofolate reductases and potential antitumor agents. J Med Chem 41:3426–3434. https://doi.org/10.1021/jm980081y
Ogiso H, Ishitani R, Nureki O et al (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110:775–787. https://doi.org/10.1016/S0092-8674(02)00963-7
Rios Steiner JL, Murakami M, Tulinsky A (1998) Structure of thrombin inhibited by aeruginosin 298-A from a blue-green alga. J Am Chem Soc 120:597–598. https://doi.org/10.1016/S0092-8674(02)00963-7
Nair SK, Calderone TL, Christianson DW, Fierke CA (1991) Altering the mouth of a hydrophobic pocket. Structure and kinetics of human carbonic anhydrase II mutants at residue Val-121. J Biol Chem 266:17320–17325. https://doi.org/10.1016/S0021-9258(19)47376-6
Birrane G, Chung J, Ladias JA (2003) Novel mode of ligand recognition by the Erbin PDZ domain. J Biol Chem 278:1399–1402. https://doi.org/10.1074/jbc.C200571200
Cregge RJ, Durham SL, Farr RA et al (1998) Inhibition of human neutrophil elastase. 4. design, synthesis, x-ray crystallographic analysis, and structure- activity relationships for a series of P2-modified, orally active peptidyl pentafluoroethyl ketones. J Med Chem 41:2461–2480. https://doi.org/10.1021/jm970812e
Morgunova E, Tuuttila A, Bergmann U et al (1999) Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 284:1667–1670. https://doi.org/10.1126/science.284.5420.1667
Schiering N, Casale E, Caccia P et al (2000) Dimer formation through domain swapping in the crystal structure of the Grb2-SH2- Ac-pYVNV complex. Biochemistry 39:13376–13382. https://doi.org/10.1021/bi0012336
Rowland P, Blaney FE, Smyth MG et al (2006) Crystal structure of human cytochrome P450 2D6. J Biol Chem 281:7614–7622. https://doi.org/10.1074/jbc.M511232200
Day PJ, Cleasby A, Tickle IJ et al (2009) Crystal structure of human CDK4 in complex with a D-type cyclin. PNAS 106:4166–4170. https://doi.org/10.1073/pnas.0809645106
Natesh R, Schwager SL, Sturrock ED, Acharya KR (2003) Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 421:551–554. https://doi.org/10.1038/nature01370
Rowsell S, Hawtin P, Minshull CA et al (2002) Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J Mol Bio 319:173–181. https://doi.org/10.1016/S0022-2836(02)00262-0
Somers WS, Tang J, Shaw GD, Camphausen RT (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P-and E-selectin bound to SLeX and PSGL-1. Cell 103:467–479. https://doi.org/10.1016/S0092-8674(00)00138-0
Kryger G, Harel M, Giles K et al (2000) Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr D 56:1385–1394. https://doi.org/10.1107/S0907444900010659
Stams T, Nair SK, Okuyama T et al (1996) Crystal structure of the secretory form of membrane-associated human carbonic anhydrase IV at 2.8-Å resolution. PNAS 93:13589–13594. https://doi.org/10.1073/pnas.93.24.13589
Hiramatsu H, Kyono K, Higashiyama Y et al (2003) The structure and function of human dipeptidyl peptidase IV, possessing a unique eight-bladed β-propeller fold. Biochem Biophys Res Commun 302:849–854. https://doi.org/10.1016/S0006-291X(03)00258-4
Lucido MJ, Orlando BJ, Vecchio AJ, Malkowski MG (2016) Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry 55:1226–1238. https://doi.org/10.1021/acs.biochem.5b01378
Griffith J, Black J, Faerman C et al (2004) The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell 13:169–178. https://doi.org/10.1016/S1097-2765(03)00505-7
Huang C-H, Mandelker D, Schmidt-Kittler O et al (2007) The structure of a human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science 318:1744–1748. https://doi.org/10.1126/science.1150799
Jasiak AJ, Armache K-J, Martens B et al (2006) Structural biology of RNA polymerase III: subcomplex C17/25 X-ray structure and 11 subunit enzyme model. Mol Cell 23:71–81. https://doi.org/10.1016/j.molcel.2006.05.013
Russo AA, Tong L, Lee J-O et al (1998) Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395:237–243. https://doi.org/10.1038/26155
Xu Z-B, Chaudhary D, Olland S et al (2004) Catalytic domain crystal structure of protein kinase C-θ (PKC θ). J Biol Chem 279:50401–50409. https://doi.org/10.1074/jbc.M409216200
Cabral JHM, Lee A, Cohen SL et al (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95:649–655. https://doi.org/10.1016/S0092-8674(00)81635-9
Baretić D, Pollard HK, Fisher DI et al (2017) Structures of closed and open conformations of dimeric human ATM. Sci Adv 3:e1700933. https://doi.org/10.1126/sciadv.1700933
Ertl P, Rohde B, Selzer P (2000) Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 43:3714–3717. https://doi.org/10.1021/jm000942e
Hou T, Wang J (2008) Structure–ADME relationship: still a long way to go? Expert Opin Drug Metab Toxicol 4:759–770. https://doi.org/10.1517/17425255.4.6.759
Constantinides PP, Wasan KM (2007) Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: in vitro/in vivo case studies. J Pharm Sc 96:235–248. https://doi.org/10.1002/jps.20780
Adebiyi M, Olugbara O (2021) Binding site identification of COVID-19 main protease 3D structure by homology modeling. Indones J Electr Eng Comput Sci 21(3):1713. https://doi.org/10.11591/ijeecs.v21.i3.pp1713-1721
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Gaillard T (2018) Evaluation of AutoDock and AutoDock Vina on the CASF-2013 benchmark. J Chem Inf Model 58:1697–1706. https://doi.org/10.1021/acs.jcim.8b00312
Singh BN, Shankar S, Srivastava RK (2012) Intracellular signaling network as a prime chemotherapy target of green tea catechin, (–)-Epigallocatechin-3-gallate. Nutrition Diet and Cancer. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2923-0_15
Zong Y, Chen F, Li S, Zhang H (2021) (-)-Epigallocatechin-3-gallate (EGCG) prevents aminoglycosides-induced ototoxicity via anti-oxidative and anti-apoptotic pathways. Int J Pediatr Otorhinolaryngo 150:110920. https://doi.org/10.1016/j.ijporl.2021.110920
Fauzi NM, Kumolosasi E, Jasamai M, Azmi N (2019) Interaction between green tea and perindopril reduces inhibition of angiotensin-converting enzyme activity. Trop J Pharm Res 18:1185–1190. https://doi.org/10.4314/tjpr.v18i6.6
Kanbarkar N, Mishra S (2021) Matrix metalloproteinase inhibitors identified from Camellia sinensis for COVID-19 prophylaxis: an in silico approach. Adv Tradit Med 21:173–188. https://doi.org/10.1007/s13596-020-00508-9
Khan YH, Uttra AM, Qasim S et al (2021) Potential role of phytochemicals against matrix metalloproteinase induced breast cancer; an explanatory review. Front Chem. https://doi.org/10.3389/fchem.2020.592152
Miyata Y, Matsuo T, Araki K et al (2018) Anticancer effects of green tea and the underlying molecular mechanisms in bladder cancer. Medicines. https://doi.org/10.3390/medicines5030087
Rha C-S, Jeong HW, Park S et al (2019) Antioxidative, anti-inflammatory, and anticancer effects of purified flavonol glycosides and aglycones in green tea. Antioxidants 8:278. https://doi.org/10.3390/antiox8080278
Cheng Z, Zhang Z, Han Y et al (2020) A review on anti-cancer effect of green tea catechins. J Funct Foods 74:104172. https://doi.org/10.1016/j.jff.2020.104172
da Silva AB, Coelho PLC, das Neves Oliveira M et al (2020) The flavonoid rutin and its aglycone quercetin modulate the microglia inflammatory profile improving antiglioma activity. Brain Behav Immun 85:170–185. https://doi.org/10.1016/j.bbi.2019.05.003
Wang L, Ma L, Zhao Y et al (2021) Comprehensive understanding of the relationship between bioactive compounds of black tea and its angiotensin converting enzyme (ACE) inhibition and antioxidant activity. Plant Foods Hum Nutr. https://doi.org/10.1007/s11130-021-00896-6
Chen L, Wang L, Shu G, Li J (2021) antihypertensive potential of plant foods: research progress and prospect of plant-derived angiotensin-converting enzyme inhibition compounds. J Agric Food Chem 69:5297–5305. https://doi.org/10.1021/acs.jafc.1c02117
Yu J, Chen GG, Lai PB (2021) Targeting hepatocyte growth factor/c-mesenchymal–epithelial transition factor axis in hepatocellular carcinoma: Rationale and therapeutic strategies. Med Res Rev 41:507–524. https://doi.org/10.1002/med.21738
Liu Z, Bruins ME, de Bruijn WJ, Vincken J-P (2020) A comparison of the phenolic composition of old and young tea leaves reveals a decrease in flavanols and phenolic acids and an increase in flavonols upon tea leaf maturation. J Food Compos Anal 86:103385. https://doi.org/10.1016/j.jfca.2019.103385
Xu Y-Q, Gao Y, Granato D (2021) Effects of epigallocatechin gallate, epigallocatechin and epicatechin gallate on the chemical and cell-based antioxidant activity, sensory properties, and cytotoxicity of a catechin-free model beverage. Food Chem 339:128060. https://doi.org/10.1016/j.foodchem.2020.128060
Tang G, Xu Y, Zhang C et al (2021) Green tea and epigallocatechin gallate (EGCG) for the management of nonalcoholic fatty liver diseases (NAFLD): Insights into the role of oxidative stress and antioxidant mechanism. Antioxidants 10:1076. https://doi.org/10.3390/antiox10071076
Bimonte S, Albino V, Piccirillo M et al (2019) Epigallocatechin-3-gallate in the prevention and treatment of hepatocellular carcinoma: experimental findings and translational perspectives. Drug Des Devel Ther. https://doi.org/10.2147/DDDT.S180079
Kumari M, Singh R, Subbarao N (2021) Exploring the interaction mechanism between potential inhibitor and multi-target Mur enzymes of mycobacterium tuberculosis using molecular docking, molecular dynamics simulation, principal component analysis, free energy landscape, dynamic cross-correlation matrices, vector movements, and binding free energy calculation. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.1989040
Nag A, Banerjee R, Chowdhury RR, Venkatesh CK (2021) Phytochemicals as potential drug candidates for targeting SARS CoV 2 proteins, an in silico study. VirusDisease 32:98–107. https://doi.org/10.1007/s13337-021-00654-x
Kim T, Chen IR, Lin Y et al (2019) Impact of similarity metrics on single-cell RNA-seq data clustering. Brief Bioinform 20:2316–2326. https://doi.org/10.1093/bib/bby076
Nag A, Banerjee R (2021) Network pharmacological evaluation for identifying novel drug-like molecules from ginger (Zingiber officinale Rosc.) against multiple disease targets, a computational biotechnology approach. Netw Model Anal Health Inform Bioinforma 10:1–17. https://doi.org/10.1007/s13721-021-00330-6
Sarkar T, Bharadwaj KK, Salauddin M et al (2021) Phytochemical characterization, antioxidant, anti-inflammatory, anti-diabetic properties, molecular docking, pharmacokinetic profiling, and network pharmacology analysis of the major phytoconstituents of raw and differently dried mangifera indica (himsagar cultivar): an in vitro and in silico investigations. Appl Biochem Biotechnol. https://doi.org/10.1007/s12010-021-03669-8
Wei J, Zhang Y, Li D et al (2020) Integrating network pharmacology and component analysis study on anti-atherosclerotic mechanisms of total flavonoids of engelhardia roxburghiana leaves in mice. Chem Biodivers 17:e1900629. https://doi.org/10.1002/cbdv.201900629
Zhai L, Ning Z, Huang T et al (2018) Cyclocarya paliurus leaves tea improves dyslipidemia in diabetic mice: a lipidomics-based network pharmacology study. Front Pharmacol 9:973. https://doi.org/10.3389/fphar.2018.00973
Kushwaha PP, Singh AK, Prajapati KS et al (2021) Phytochemicals present in Indian ginseng possess potential to inhibit SARS-CoV-2 virulence: A molecular docking and MD simulation study. Microb Pathog. https://doi.org/10.1016/j.micpath.2021.104954
Islam R, Parves MR, Paul AS et al (2021) A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J Biomol Struct Dyn 39:3213–3224. https://doi.org/10.1080/07391102.2020.1761883
Aliebrahimi S, Montasser Kouhsari S, Ostad SN et al (2018) Identification of phytochemicals targeting c-Met kinase domain using consensus docking and molecular dynamics simulation studies. Cell Biochem Biophys 76:135–145. https://doi.org/10.1007/s12013-017-0821-6
Al-Shabib NA, Khan JM, Malik A et al (2018) Molecular insight into binding behavior of polyphenol (rutin) with beta lactoglobulin: Spectroscopic, molecular docking and MD simulation studies. J Mol Liq 269:511–520. https://doi.org/10.1016/j.molliq.2018.07.122
Ren W, Truong TM, Ai H (2015) Study of the binding energies between unnatural amino acids and engineered orthogonal tyrosyl-tRNA synthetases. Sci Rep 5:1–10. https://doi.org/10.1038/srep12632
He J-Y, Li C, Wu G (2014) Discovery of potential drugs for human-infecting H7N9 virus containing R294K mutation. Drug Des Devel Ther 8:2377. https://doi.org/10.2147/DDDT.S74061
Acknowledgements
The authors would like to acknowledge Centre for Research Project, CHRIST (Deemed to be University), Bangalore, for providing financial support toward a minor research project entitled “Effects of storage on polyphenols and sensory qualities of tea beverage” (MIRPDSC – 1904, Dt. 01/10/2019). We extend our acknowledgement to Centre for Academic and Professional Support (CAPS), CHRIST (Deemed to be University), Bangalore, for refining the manuscript as per professional language requirement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nag, A., Dhull, N. & Gupta, A. Evaluation of tea (Camellia sinensis L.) phytochemicals as multi-disease modulators, a multidimensional in silico strategy with the combinations of network pharmacology, pharmacophore analysis, statistics and molecular docking. Mol Divers 27, 487–509 (2023). https://doi.org/10.1007/s11030-022-10437-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10437-1