
Abstract
Ultra-performance liquid chromatographic technique was applied for the first time for determination of radiochemical and chemical purity of [11C]methionine with measurement time less than 2 min. The pH and eluent strength of mobile phase were optimized to achieve a gradient elution for separation of non-radioactive impurities as well as radioactive ingredients. The developed method was validated according to the following parameters: linearity, repeatability, recovery, limit of quantitation. The novel chromatographic procedure with reduced measurement time could be uniquely applied for the quality control of [11C]methionine to considerably shorten the release time and avoid radioactivity loss of radiopharmaceutical.
Similar content being viewed by others

Introduction
Methionine has an important role in rapid synthesis of different proteins in brain tumors due to increased expression of amino acid transporters. Labeling the molecule with 11C isotope makes methionine a useful radiotracer for imaging oncological lesions with positron emission tomography (PET). Malignant tumor cell is characterized with high uptake of [11C]methionine. On the other hand, the accumulation of the radiolabeled amino acid in normal tissue is negligible. Interestingly, the extent of uptake is proportional with tumor grade. [11C]methionine is applied primarily in examination of gliomas to delineate uptake areas for surgery, radiotherapy and to guide biopsies [1].
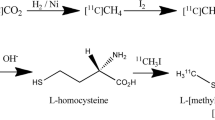
In most cases, the radiosynthesis of [11C]methionine is performed by methylation of L–homocysteine or L–homocysteine thiolactone with [11C]methyl iodide choosing various technical solutions. Remotely controlled production is a convenient method to obtain [11C]methionine with high radiochemical yield and purity [2, 3]. Avoiding preparative high performance liquid chromatographic (HPLC) purification step [4] the synthesis could be accomplished using solid phase extraction (SPE) cartridges which may also serve as a scene for the labeling procedure [5, 6]. Even “Loop” method could be applied successfully for routine production of [11C]methionine [7]. Ultimately, optimized radiosynthetic procedures provide reproducible production of [11C]methionine batches for routine PET examinations [8].
The quality control of [11C]methionine covers the determination of radiochemical and chemical purity. According to the European Pharmacopeia (Ph. Eur.) reversed phase liquid chromatography could be applied in order to identify the main component as well as radioactive and non-radioactive impurities [9]. This methodology was successfully adapted for quantitative analysis of homocysteine, homocysteine thiolactone and methionine by several authors [2,3,4,5,6]. Radiochemical components, namely [11C]methionine, [11C]methionine sulfone, [11C]methionine sulfoxide and [11C]methanol could be measured simultaneously [10,11,12]. On the other hand, additional impurities could be found in the final product not mentioned in Ph. Eur. monograph. Pascali et al. [13] detected homocystine and iodide ion by a reversed phase HPLC method. [11C]methyl iodide could be also identified as a radiochemical impurity [11, 12]. Giglio et al. [12] developed a gradient method for examination of the entire impurity profile of [11C]methionine with measurement time of 16 min. However, in order to avoid considerable loss of radioactivity it is very important to have a fast QC protocol to shorten the release time of the radiopharmaceutical [14]. Hence, the aim of this work was to develop a liquid chromatographic method applying the previously not used UPLC technique to separate radiochemical as well as chemical compounds of [11C]methionine injection with reduced measurement time. A fast chromatographic procedure could be achieved by simultaneous analysis of 11C-labeled and non-radioactive ingredients on a stationary phase with particle size lower than 2 μm. Due to UPLC conditions the examination time could be decreased even by an order of magnitude in comparison with conventional HPLC methods. Consequently, the quality control protocol could be shortened and the release time of [11C]methionine could be considerably reduced which is a critical issue for radiopharmaceuticals with short shelf lives.
Experimental
Reagents for synthesis of [11C]methionine and [11C]methyl iodide as well as for chromatographic analysis were purchased from Sigma and used without further purification. GE PETtrace cyclotron was applied for production of isotope according to nuclear reaction of 14N(p,α)11C at 16.4 MeV using N2 (0.2%, O2) gas mixture under 10 bar pressure. The synthesis of [11C]methyl iodide was carried out on a GE PETtrace MeI Microlab module via “gas phase” method. [11C]methionine was obtained by the reaction of [11C]methyl iodide with 2 mg L–homocysteine absorbed on 20 mg Al2O3 in 1 mL ethanol according to method proposed by Schmitz et al. [15]. The purification of crude reaction mixture was performed on Sep–Pak C18 Plus and Sep–Pak Alumina N light cartridges (Waters) by elution with 9 mL of saline. The final solution was pushed through Millex GS 0.22 μm sterile filter (Millipore).
Chromatographic measurements were performed on Waters Acquity I Class UPLC system. UV–Vis detector was used for detection of non-radioactive ingredients. Radioactive components were identified by radioactive detector (NaI (Tl)/PMT, Hamamatsu). Elution was carried out at 0.6 mL min−1 and 225 nm detection wavelength with 1 μL injected volume. Components were separated on Acquity BEH C18 column (1.7 μm, 2.1 × 100 mm, 130 Å, 18% carbon load). Column oven was adjusted to 25 °C.
The following samples were used for the method development. Stock solutions of the examined ingredients were prepared by dissolution of appropriate mass of solid material in water. The concentration of reference components in test solutions was adjusted to 120% of the allowable concentration of non-radioactive ingredients, namely to 250 μg mL−1 for homocysteine, homocystine and methionine, 10 μg mL−1 for iodide ion and 80 μg mL−1 for homocysteine thiolactone unless otherwise stated. The concentration of non-radioactive analogs of radiochemical components, namely methionine sulfone, methionine sulfoxide, methanol, methyl iodide and methionine, were adjusted to 250 μg mL−1. The radioactive concentration of [11C]methionine batches was up to 500 MBq mL−1. Samples of [11C]methyl iodide were prepared with radioactivity of up to 2 GBq. All the examined solutions were filtered through 0.22 μm Millex-GV PVDF cartridge (Merck). The analysis of samples was repeated three times and average values were used for the evaluation of results.
The validation of the developed analytical method was performed according to ICH Q2(R1) guideline [16]. The linearity measurement of the chemical purity test was performed in the range of LOQ to at least 120% of the allowable concentration of non-radioactive ingredients. Accordingly, samples were prepared separately and analyzed with concentrations as follows: 0.07–0.5–0.7–1.8–3.5–5.3–7.1–17.7–35.4–53.1–70.8–88.5 μg mL−1 for homocysteine thiolactone, 0.5–1.5–2.0–4.9–9.8–14.8–19.7–49.2–98.4–147.6–196.8–246.0 μg mL−1 for homocysteine, 0.5–1.5–1.9–4.8–9.7–14.5–19.4–48.4–96.8–145.2–193.6–242.0 μg mL−1 for homocystine and 0.5–1.5–2.0–4.9–9.8–14.7–19.6–48.9–97.8–146.7–195.6–244.5 μg mL−1 for methionine. Accuracy and precision were determined by consecutive analysis of a sample with maximum allowable concentration of ingredients, namely, 200 μg mL−1 for homocysteine, homocystine and methionine as well as 60 μg mL−1 for homocysteine thiolactone. The measurement was repeated six times. Relative standard deviation (RSD) and recovery values were calculated from the obtained concentrations of the ingredients. LOQ was determined based on the Standard Deviation of the Response and the Slope. The validation of radiochemical purity test was performed using samples spiked with [11C]methionine and [11C]methyl iodide. The radioactivity ratio of [11C]methionine to [11C]methyl iodide was 91.5% The injected radioactivity values were adjusted by analyzing samples at different times. The radioactivity linear range was examined from 1.2 kBq to 499.8 kBq for [11C]methionine and from 3.7 kBq to 45.4 kBq for [11C]methyl iodide. Linearity was determined using the following injected radioactivities: 1.2–10.4–20.6–30.5–40.3–48.8–69.1–95.6–132.3–161.7–205.8–242.6–294.0–408.7–499.8 kBq for [11C]methionine and 3.7–4.4–6.3–8.7–12.0–14.7–18.3–22.1–27.2–37.2–45.4 kBq for [11C]methyl iodide. Accuracy and precision of the radiochemical purity test were determined by evaluation of measurements of linearity test. Recovery and RSD values were calculated from the obtained radioactivity ratio of [11C]methionine to [11C]methyl iodide.
Results and discussion
Chemical purity
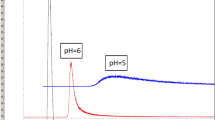
It is well known from the literature that [11C]methionine injection may contain the following non–radioactive impurities: homocysteine, homocysteine thiolactone, homocystine, methionine and iodide ion [9, 13]. However, homocysteine thiolactone can be only detected in the final product providing that it is used as a precursor for the radiosynthesis. On the other hand, [11C]methionine may contain homocysteine in every case, since it is used either directly as starting material for labeling procedure or obtained by basic hydrolysis of homocysteine thiolactone [12]. Although in this work homocysteine thiolactone was not used for precursor and therefore not expected to identify as non-radioactive impurity in analyzed drug batches, our goal was to develop a generic fast liquid chromatographic method for determination of the content of potential chemical impurities in [11C]methionine radiopharmaceutical produced by using either homocysteine thiolactone or homocysteine. For this purpose a reversed phase chromatography was applied which was also recommended by the Ph. Eur. monograph of [11C]methionine injection [9]. According to this guidance octadecylsilyl silica gel stationary phase and phosphate buffer eluent should be used. Adapting these recommendations to UPLC system an Acquity BEH C18 column (1.7 μm, 2.1 × 100 mm) was applied and eluted initially with 10 mM KH2PO4 solution. One of the main goals of this study was to examine the effect of pH of the mobile phase on the separation of non-radioactive ingredients. Hence, the pH of the eluent was adjusted either with NaOH or H3PO4 in the range of 3– 9 for monitoring the selectivity of the method. Initially, reference solutions of the components were examined separately for identification of impurities by retention time. Finally, mixture of ingredients was injected and peak resolutions were determined. It was found that at pH 9 only homocysteine, homocystine and methionine could be identified and separated with resolution greater than 1.5 (Fig. 1). At the same time iodide ion was co–eluted with the solvent front. Additionally, homocysteine thiolactone had greater retention time than 1 min. This might be in connection with its highest lipophilicity among examined impurities (logP = – 0.51). The pKa = 7.1 value of homocysteine thiolactone also indicated that under basic conditions the ion suppressed molecular form had increased lipophilicity which might have led to the increased retention. With decreasing the pH of the mobile phase to 8 only a slight decrease of retention times of the previously detected components could be observed and the quantitation of homocysteine thiolactone and iodide ion was still not possible. At neutral conditions only methionine could be identified as a pure peak with retention time of 0.606 min. At pH 7 the co–elution of homocysteine and homocystine was observed at 0.434 min.

Effect of pH of mobile phase in the range of 3–9 on the separation of non-radioactive components. (1: solvent front + iodide ion; 2: homocysteine thiolactone; 3: homocysteine; 4: homocystine; 5: methionine)
Increasing the acquisition time of measurements at pH 7–8–9 revealed that homocysteine thiolactone could be eluted with retention time from 1.514 to 2.024 min (Fig. 2). Decreasing the basicity of mobile phase from pH 9–7 led to decrease the retention time with 0.5 min.

Variation of retention time of homocysteine thiolactone at pH 7–8–9 of mobile phase. (2: homocysteine thiolactone)
Presumably further decrease of pH of the eluent would promote the elution of homocysteine thiolactone and ultimately the analysis could be performed in 1 min. Indeed, increasing the acidity of the mobile phase to pH 6 and 5 indicated a considerable change in the selectivity of the method (Fig. 1). Namely, beside methionine as well as the co–eluted homocysteine and homocystine pair, homocysteine thiolactone could be identified with retention time of 0.832 min and 0.551 min which was decreased further with decreasing the pH to 4. The co–elution of homocysteine and homocystine was also observed at pH 3 and the retention time of homocysteine thiolactone was decreased to 0.483 min. At the same time the retention time of methionine was increased to 0.645 min. The resolution between homocystine and homocysteine thiolactone as well as homocysteine thiolactone and methionine was 1.58 and 7.34, respectively. Finally, the pH 2 value of mobile phase was adjusted with 0.1% (m/m) H3PO4 solution instead of 10 mM KH2PO4 buffer. However, it was initiated a drastic effect on the selectivity of the method, since apart from iodide ion every component could be identified and separated with acceptable resolution (Rs > 1.5). The retention time of homocysteine thiolactone was decreased slightly in comparison with results obtained at pH 3 (Fig. 3). On the other hand, homocysteine and homocystine were resolved completely and interestingly its elution order was changed. The resolution between homocysteine thiolactone and homocysteine, homocysteine and homocystine as well as homocystine and methionine was 1.56, 1.91 and 9.36, respectively. The measurement time was only 1 min.

Separation of chemical impurities using 0.1% (m/m) H3PO4 solution (pH 2) for mobile phase. (2: homocysteine thiolactone; 3: homocysteine; 4: homocystine; 5: methionine)
After the optimization of mobile phase composition, the developed UPLC method was validated. Linearity was verified in the concentration range of 0.07–88.5 μg mL−1 for homocysteine thiolactone, 0.5–246.0 μg mL−1 for homocysteine, 0.5–242.0 μg mL−1 for homocystine and 0.5–244.5 μg mL−1 for methionine. The regression coefficients (r2) were obtained between 0.9989 and 0.9994 (Supplementary Information, Fig. S1-S4). The repeatability was determined by injection of six replicates of reference solution with maximum allowable concentration of ingredients as follows: 60 μg mL−1 (homocysteine thiolactone), 200.0 μg mL−1 (homocysteine), 200.0 μg mL−1 (homocystine) and 200.0 μg mL−1 (methionine). The obtained RSD for homocysteine thiolactone, homocysteine, homocystine and methionine was 0.09%, 0.23%, 0.15% and 0.10%, respectively. The concentration recoveries were in the range from 100.8% to 102.2%. The quantitation limit based on the Standard Deviation of the Response and the Slope was 0.07 μg mL−1, 1.05 μg mL−1, 1.10 μg mL−1 and 0.73 μg mL−1 for homocysteine thiolactone, homocysteine, homocystine and methionine, respectively (Supplementary Information, Fig. S5-S8).
The developed UPLC method was applied for analysis of a crude radiosynthesis mixture to identify and quantify of non-radioactive impurities. According to the results among the previously examined ingredients homocysteine, homocystine and methionine could be detected with retention times of 0.516 min, 0.576 min and 0.814 min, respectively (Fig. 5). The concentration of homocysteine, homocystine and methionine in the crude mixture was 94.5 μg mL−1, 80.3 μg mL−1 and 4.1 μg mL−1, respectively. Homocysteine thiolactone was absent in the sample, since in our case it was not used as precursor for the radiosynthesis. The chemical purity of [11C]methionine batches from five consecutive productions was also determined by the novel UPLC method. It was found that the concentration of homocysteine varied from 80.1 to 128.4 μg mL−1. At the same time, the homocystine content was in the range of 91.4–137.2 μg mL−1 and methionine was detected with concentrations from 10.1 to 30.4 μg mL−1. The chemical purity of the examined batches fulfilled the Ph. Eur. requirements, since the concentrations of the non-radioactive impurities were below the allowable level of 200 μg mL−1.
Radiochemical purity
Beside the active ingredient of [11C]methionine other radioactive components could be identified in the final radiopharmaceutical solution [9, 12]. Namely, [11C]methionine sulfone, [11C]methionine sulfoxide, [11C]methanol and [11C]methyl iodide may effect considerably the radiochemical purity of the preparation. To determine the radiochemical purity [11C]methionine should be separated from other 11C-labeled impurities. In this work we aimed to perform the determination of chemical and radiochemical purity simultaneously. Hence, the previously developed method for chemical purity test was applied for the analysis of radiochemical compounds. Non–radioactive analogs were used as reference compounds during the development of radiochemical purity test for the sake of simplicity, since their chromatographic properties were identical with 11C-labeled ingredients. It was found that methionine sulfone, methionine sulfoxide and methanol was co–eluted with solvent front at 0.404 min. On the other hand, methionine was eluted with retention time of 0.798 min. The observed resolution between the identified peaks was 5.3. At the same time methyl iodide could not be eluted within 1 min due to its high lipophilicity. In order to simultaneously determine the chemical and radiochemical purity the elution should be started with 0.1% (m/m) H3PO4 solution. On the other hand, increased eluent strength should be applied for elution of methyl iodide with short retention time. It was achieved using acetonitrile during optimized gradient elution as follows (Table 1).
The initial eluent composition was 0.1% (m/m) H3PO4 (A eluent). Acetonitrile (B eluent) was added to mobile phase instantly after 0.3 min of the sample injection. The content of the organic modifier was adjusted to 90% (v/v). Due to the gradient delay time (ca. 0.7 min) the effect of B eluent could be observed only at about 1 min after the sample injection. Consequently, the chemical purity could be determined according to the previously optimized conditions. At the same time due to the effect of acetonitrile methyl iodide could be eluted with retention time of 1.351 min. The resolution between methionine and methyl iodide was 7.2 (Fig. 4). The measurement time was 2.0 min which was sufficient for conditioning the stationary phase with A eluent to the initial conditions. It is worth to note that a shift in retention times of a compound in non-radioactive and 11C-labeled form could be observed due to the connection of UV–Vis and radioactivity detector in sequence within the UPLC system. The difference of retention times was 0.2 ± 0.04 min at 0.6 mL min−1 flow rate. For instance, the retention time of [12C]methyl iodide and [11C]methyl iodide was 1.132 min and 1.352 min, respectively.

Analysis of non-radioactive analogues of radiochemical compounds by gradient elution. (6: solvent front; 7: methionine sulfone; 8: methionine sulfoxide; 9: methanol; 5: methionine; 10: solvent effect of the gradient elution; 11: methyl iodide)
The validation of the developed gradient elution method was accomplished using samples spiked with [11C]methionine and [11C]methyl iodide as radioactive reference components. The linearity was acceptable in the range of the injected radioactivity from 1.2 kBq to 499.8 kBq for [11C]methionine and from 3.7 kBq to 45.4 kBq for [11C]methyl iodide (Supplementary Information, Fig. S9-S10). The obtained r2 value was 0.9996 and 0.9993 for [11C]methionine and [11C]methyl iodide, respectively. The LOQ based on the Standard Deviation of the Response and the Slope was 6.1 kBq for [11C]methyl iodide. The obtained RSD and recovery values of radioactivity ratios of [11C]methionine to [11C]methyl iodide were 0.3% and 100.5%, respectively.
The crude radiosynthesis mixture was analyzed by the novel UPLC method to identify the radioactive components. From the obtained chromatogram only the peaks of [11C]methionine and [11C]methyl iodide could be identified (Fig. 5). [11C]methanol and the oxidation products of [11C]methionine, namely [11C]methionine sulfoxide and [11C]methionine sulfone could not be detected in the raw mixture. It was found that the 98.5% of the total radioactivity of the sample belonged to [11C]methionine. Additionally, the radiochemical purity of five [11C]methionine batches were determined. The content of [11C]methionine in samples was in the range of 98.0% to 98.9%. Interestingly, from the known radiochemical impurities only [11C]methyl iodide could be detected in the final samples during the shelf life of [11C]methionine batches.

Representative chromatograms from the analysis of a crude reaction mixture. (UV–Vis channel—3: homocysteine; 4: homocystine; 5: methionine. Radioactive channel—12: [11C]methionine; 13: [11C]methyl iodide)
Conclusions
The developed UPLC method is eligible for fast determination of radiochemical and chemical purity of [11C]methionine simultaneously. The measurement time is significantly shorter than that of proposed by European Pharmacopeia monograph. Despite of the gradient elution method the recondition of the system for repeated measurements could be achieved rapidly. The radiochemical purity could be determined accurately by complete separation of [11C]methionine from partly co–eluted radioactive impurities. Baseline separation of non–radioactive ingredients could be also achieved which is one of the main requirements for the determination of their concentration in the final product. Unfortunately, this UPLC method is not capable for quantitation of iodide ion. This parameter should be determined by another analytical procedure. Ion pair reversed phase chromatography might be applicable. However, UPLC method may be also promising if an analytical column with lower carbon load is used. In this case due to the greater number of silanol functional group of the stationary phase the increased polar selectivity may increase the retention of iodide ion.
References
Nanni C, Fantini L, Nicolini S, Fanti S (2010) Non FDG PET. Clin Radiol 65:536–548
Quincoces G, Penuelas I, Valero M, Serra P, Collantes M, Martí-Climent J, Arbizu J, García-Velloso MJ, Richter JÁ (2006) Simple automated system for simultaneous production of 11C–labeled tracers by solid supported methylation. Appl Radiat Isot 64:808–811
Cheung M, Ho C (2009) A simple, versatile, low–cost and remotely operated apparatus for [11C]acetate, [11C]choline, [11C]methionine and [11C]PIB synthesis. Appl Radiat Isot 67:581–589
Mitterhauser M, Wadsak W, Krcal A, Schmaljohann J, Eidherr H, Schmid A, Viernstein H, Dudczak R, Kletter K (2005) New aspects on the preparation of [11C]methionine—a simple and fast online approach without preparative HPLC. Appl Radiat Isot 62:441–445
Lodi F, Trespidi S, Di Pierro D, Marengo M, Farsad M, Fanti S, Franchi R, Boschi S (2007) A simple Tracerlab module modification for automated on-column [11C]metmehylation and [11C]carboxylation. Appl Radiat Isot 65:691–695
Lodi F, Rizzello A, Trespidi S, Di Pierro D, Marengo M, Farsad M, Fanti S, Al-Nahhasd A, Rubello D, Boschi S (2008) Reliability and reproducibility of N–[11C]methyl–choline and L–(S–methyl–[11C])methionine solid–phase synthesis: a useful and suitable method in clinical practice. Nucl Med Commun 29:736–740
Gomez V, Gispert JD, Amador V, Llop J (2008) New method for routine production of L–[methyl–11C]methionine: in loop synthesis. J Label Compd Radiopharm 51:83–86
Pichler V, Vraka C, Berroterán-Infante N, Krcal A, Eidherr H, Traub-Weidinger T, Hacker M, Mitterhauser M, Wadsak W (2018) L-[S-methyl-11C]methionine—an example of radiosynthetic optimization. Appl Radiat Isot 141:107–111
L–Methionine ([11C]methyl) injection 1617 (1/2008) Ph Eur 9.0, 1161–1163
Woods M, Leung L, Frantzen K, Garrick JG, Zhang Z, Zhang C, English W, Wilson D, Bénard B, Lin K-S (2017) Improving the stability of 11C-labeled l-methionine with ascorbate. EJNMMI Radiopharm Chem 2(13):1–9
Bogni A, Bombardieri E, Iwata R, Cadini L, Pascali C (2003) Stability of L–[S–methyl–11C]methionine solutions. J Radioanal Nucl Chem 256:199–203
Giglio J, Rosas G, Basso M, Boné A, Savio E, Engler H (2018) An alternative methodology for the determination of the radiochemical purity of 11C–methionine. EJNMMI Radiopharm Chem 3(15):1–12
Pascali C, Bogni A, Cucchi C, Laera L, Crispu O, Maiocchi G, Crippa F, Bombardieri E (2010) Detection of additional impurities in the UV-chromatogram of L–[S–methyl–11C]methionine. J Radioanal Nucl Chem 288:405–409
Nics L, Steiner B, Klebermass EM, Philippe C, Mitterhauser M, Hacker M, Wadsak W (2018) Speed matters to raise molar radioactivity: fast HPLC shortens the quality control of C–11 PET–tracers. Nucl Med Biol 57:28–33
Schmitz F, Plenevaux A, Del-fiore G, Lemaire C, Comar D, Luxen A (1995) Fast routine production of L–[11C–methyl]–methionine with Al2O3/KF. Appl Radiat Isot 46:893–897
ICH Guidance for Industry Q2(R1): validation of analytical procedures: text and methodology (ICH Q2(R1)). https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf
Acknowledgements
Open access funding provided by University of Debrecen (DE). This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. We would like to express our thanks to radiochemists at Division of Nuclear Medicine, University of Debrecen for their excellent assistance in radioisotope and [11C]methionine production.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rezes, R., Csoma, S.L., Németh, E. et al. Fast liquid chromatographic determination of radiochemical and chemical purity of [11C]methionine by UPLC technique. J Radioanal Nucl Chem 324, 1237–1244 (2020). https://doi.org/10.1007/s10967-020-07179-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07179-5