
Abstract
Neurological symptoms are prevalent in both the acute and post-acute phases of coronavirus disease 2019 (COVID-19), and they are becoming a major concern for the prognosis of COVID-19 patients. Accumulation evidence has suggested that metal ion disorders occur in the central nervous system (CNS) of COVID-19 patients. Metal ions participate in the development, metabolism, redox and neurotransmitter transmission in the CNS and are tightly regulated by metal ion channels. COVID-19 infection causes neurological metal disorders and metal ion channels abnormal switching, subsequently resulting in neuroinflammation, oxidative stress, excitotoxicity, neuronal cell death, and eventually eliciting a series of COVID-19-induced neurological symptoms. Therefore, metal homeostasis-related signaling pathways are emerging as promising therapeutic targets for mitigating COVID-19-induced neurological symptoms. This review provides a summary for the latest advances in research related to the physiological and pathophysiological functions of metal ions and metal ion channels, as well as their role in COVID-19-induced neurological symptoms. In addition, currently available modulators of metal ions and their channels are also discussed. Collectively, the current work offers a few recommendations according to published reports and in-depth reflections to ameliorate COVID-19-induced neurological symptoms. Further studies need to focus on the crosstalk and interactions between different metal ions and their channels. Simultaneous pharmacological intervention of two or more metal signaling pathway disorders may provide clinical advantages in treating COVID-19-induced neurological symptoms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was identified for the first time in December 2019, which was responsible for the devastating infection of 2019 coronavirus disease (COVID-19) pandemic (Zhu et al. 2020). Early studies have focused on the respiratory system of COVID-19 patients, whose pulmonary symptoms have been well described (Guan and Zhong 2020; Huang et al. 2020). However, emerging evidence suggests that patients with COVID-19 have neurological symptoms and complications as well (Ellul et al. 2020; Helms et al. 2020; Jaywant et al. 2021; Méndez et al. 2022). Thus, understanding the mechanisms underlying COVID-19 neuropathology is critical for preventing neurological complications for COVID-19 patients.
Metal ions are widely distributed in the brain and play a pivotal role in the central nervous system (CNS) and neuronal function. It is well known that two types of metal ions are present in human: essential ions (such as sodium, potassium, calcium, iron and zinc) and nonessential ions (such as aluminum). Metal ions are involved in the development, metabolism, redox, and neurotransmitter delivery of the CNS with a strictly regulated metal absorption, efflux, distribution and storage (Black 1998; D'Ambrosi and Rossi 2015; Kauer and Gibson 2009; Masaldan et al. 2019; McDaid et al. 2020; Scheiber et al. 2014; Thirupathi and Chang 2019). In these processes, metal ions perform as the structural, catalytic, or regulatory ingredients of proteins including transcription factors, enzymes, transporter proteins and receptors. Because of their important functions, metal ions are enormously valuable to the brain. Lack of metals is associated with various degrees of damage to the CNS.
Although metals have physiological functions to the body, excessive amount of them are particularly neurotoxic to healthy nerve cells and tissues, which impairs nerve cell physiological activities and even causes cell death (Kawabata 2022; Sun et al. 2022). However, various pathological factors, such as SARS-CoV-2 infection and ischemia/hypoxia, may lead to excessive intake, uncontrolled release and metabolic disorders in metals, which are responsible for the pathogenesis of neurological symptoms due to COVID-19 infection and ischemic stroke (Alim et al. 2019; Almutairi et al. 2019; Danta 2020a; Pulido Perez et al. 2022; Vinceti et al. 2022). Therefore, metal homeostasis is important to neuro-homeostasis, which demands a precise balance among metal absorption, circulation, and storage.
Metal transport relies on various metal ion channels, which control the influx and efflux of metal ions. To date, diverse types of neurologically associated metal ion channels have been identified including glutamate receptors, transient receptor potential (TRP) channels, iron transporter-related proteins and zinc transporters. Under physiological conditions, these channels keep a dynamic homeostasis to guarantee the functions of the CNS. Under pathological conditions (such as ischemia and hypoxia), transporter proteins are increased or decreased and become aberrantly activated or inactivated, causing metal overload or deficiency and affecting many cellular signaling. Generally, oxidative stress, neuroinflammation and excitotoxicity are well-established factors contributing to neurological disorders, and various studies have shown that abnormalities in transporter proteins usually cause and exacerbate neurological disorders (Denechaud et al. 2022; Wang et al. 2023; Zhang et al. 2022a). Considering the key roles of the metal ions and metal ion channels in neurological symptoms associated with COVID-19, big efforts are devoted to understand the regulatory mechanisms for transporter proteins and their correlation with intracellular metal homeostasis, especially to the downstream signaling pathways causing COVID-19-induced neurological symptoms. In this review, we summarized the latest studies regarding the metal ions, metal ion channels, and relevant pathogenesis in COVID-19-induced neurological symptoms. In addition, the strategies in targeting metal ions and their transport proteins are also discussed. Maintenance in metal homeostasis may illuminate potential novel therapeutic targets for COVID-19-induced neurological symptoms (Fig. 1).

Overview of this review article. In the case of SARS-CoV-2 infection, metal ion disorders and their channels aberrations induced neuroinflammation, oxidative stress, excitotoxicity and neuronal cell death, ultimately leading to a series of severe neurological symptoms. Administration of metal ion modulators or their channel modulators restores the metal ion homeostasis, thereby ameliorating COVID-19-induced neurological symptoms
Role of the major metal ions in the central nervous system
Sodium (Na) and potassium (K)
The membrane translocation of metal ions is accountable for excitability and bioelectricity in nerves, muscles and other cells. Neurons are excitable cells whose physiological foundation is the transition between resting and action membrane potentials (Wicher et al. 2006). This transition is determined by the selective permeability of the plasma membrane to Na+ and K+ under different conditions (Khaliq and Raman 2006). In the condition of resting membrane potential, the plasma membrane is much more permeable to K+ than to Na+ in the excitable neurons, resulting in a resting membrane potential closer to the equilibrium potential of K+ (− 90 mV) than that of Na+ (+ 65 mV) (Wang et al. 1994). Normally, the activity of the transporter Na+/K+-ATPase (also known as the Na+ pump) is critical for sustaining this asymmetric membrane translocation of Na+ and K+ (Scuri et al. 2007). Upon cells excitation, the plasma membrane at the axon hillock depolarizes with the assistance of opened voltage-gated Na+ channels. Gradually, Na+ returns to the cell, further depolarizing the membrane and eliciting an action potential in a positive feedback manner (Donnelly 2013). Once neurons are depolarized, Na+ channels are closed, accompanied by the opening of voltage-gated K+ channels, inducing repolarization (Chow and Leung 2020). As K+ flows out of the cell, the membrane potential declines and reverts to near resting potential. To achieve appropriate ion homeostasis, Na+/K+-ATPase performs a fundamental role in facilitating the efflux of three Na+ and the entry of two K+ into the cell by sacrificing the energy generated by the hydrolysis of one ATP molecule to provide energy for ion exchange and substance transport (Pivovarov et al. 2018). Thus, the Na+/K+ gradient generated by Na+/K+-ATPase is the basis for trafficking of other ions, substrates and neurotransmitters between the intra- and extracellular compartments (Hernández 1992).
Calcium (Ca)
Ca2+ is an indispensable divalent cation that functions as a second messenger in modulating neurodevelopment, synaptic transmission, neuronal excitability, and neuronal morphology in the CNS (Chaudhuri et al. 2021; Lin et al. 2019). For instance, Ca2+ exerts a pivotal role in triggering long-term potentiation (LTP) and depression (LTD), as well as in synaptic information storage patterns that underlie memory formation and maintenance (Hell 2016; Neveu and Zucker 1996). Ca2+ is also able to activate protein kinase including calpain and calcium/calmodulin-dependent protein kinase type II (CAMK II), to initiate downstream pathways (Coultrap et al. 2011; Tao et al. 2021). The wide variety of Ca2+ functions in the brain is only available when the gradient of Ca2+ concentrations tightly conserved in the cells (Dixon et al. 2022; Golovina et al. 1996). The concentration of Ca2+ in the cytoplasm is at least 10,000-fold lower compared to extracellular compartments and some intracellular compartments such as the endoplasmic reticulum (ER) and mitochondrion. This huge concentration gap is governed by mechanisms involving calcium channels, pumps, binding proteins, and other metal ions such as magnesium, which is considered to be a calcium antagonist (Yamanaka et al. 2019).
Iron (Fe)
Iron, the most abundant essential element in humans, is widely spread in nearly all tissues and organs, such as the heart, liver, lungs, kidneys, spleen and brain. The absorption, circulation, storage, and regulation of iron cooperates intensively to uphold human iron homeostasis. Iron is present in several forms: functional iron that forms hemoglobin, myoglobin, enzymes, as well as functional proteins, and reserve iron in ferritin and hemosiderin (Thirupathi and Chang 2019). Being utilized by various key enzymes with its oxidation states and forming coordination bonds, iron is involved in the sustainment of normal physiological functions as an indispensable co-factor for proteins concerned with oxygen transport, cellular respiration, energy production, DNA synthesis and repair, as well as mitochondrial and immunological maintenance (Halcrow et al. 2021). Several studies have collectively confirmed that iron plays an important role in brain health due to iron-dependent enzymes and proteins, such as monoamine oxidase, tryptophan hydroxylase, and aldehyde oxidase, which are required for synapse development, myelination, and neurotransmitter transport (Bar-Am et al. 2015; Li et al. 2017; Specker et al. 2022). In addition, brain is energetically reliant on iron-dependent proteins associated with cellular respiration, as it consumes large amount of oxygen. Iron also exhibits strong redox activity and is frequently converted between divalent and trivalent states through reduction and oxidation by ferric reductase or Fenton reaction, respectively, to accommodate its absorption, transport and storage (Kapralov et al. 2020). For this reason, the distribution of iron in the brain is heterogeneous in time and space. The rate of iron accumulation in the brain varies during different stages of brain development and neurological disorders, which is coupled with oxidative stress, neuroinflammation, and cell death in the meantime (Feng et al. 2021b).
Zinc (Zn)
Zinc is the second most abundant trace element after Fe in the brain and is localized in the hippocampus, amygdala, cerebral cortex, thalamus, and olfactory cortex. Zinc performs a major role in DNA synthesis, brain development and neurotransmission (Sun et al. 2022). Growing evidence suggests that aberrant zinc levels are involved in many devastating diseases. For example, zinc deficiency can lead to mental health problems, growth retardation and immune disorders, while excess zinc distorts lymphocytes state and inhibit copper uptake (Chen et al. 2021; Ogawa et al. 2018; Park et al. 2020). There are three main forms of zinc in humans: free zinc, vesicular zinc, and protein-bound zinc. Most prevalent zinc in the brain is free zinc or chelatable zinc (Maret 2015, 2019). The chelatable zinc is mainly conserved in the presynaptic vesicles of specific excitatory glutamatergic neurons and is excreted into synaptic clefts with glutamate during neuronal excitation (Carrillo et al. 2020; Lavoie et al. 2011). Upon reaching the postsynaptic membrane, the released Zn2+ pairs with diverse receptors, such as ionotropic glutamate receptors, γ-amino butyric acid (GABA) receptors, glycine receptors, and P2-type purinergic receptors, functioning as a second messenger in signal transduction, neurotransduction, and neurogeneration (Chuang and Reddy 2019; Kovács et al. 2018). Increasing evidence demonstrates that Zn2+ in synaptic clefts moderates dendritic function in an activity-dependent manner via N-methyl-D-aspartate receptors (NMDARs) (Krall et al. 2020). In parallel, Zn2+ regulates α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptor (AMPARs) with a negative feedback (Kalappa et al. 2015). It has also been reported that liberated Zn2+ diffuses to heterologous synapses for transmission spatio-temporal neural information and regulation of synaptic plasticity (Tlili et al. 2011; Vogler et al. 2020). These data suggest that strictly controlled levels of zinc are critical in neurodevelopment and neurometabolism.
Copper (Cu)
Copper is the third most abundant trace element in the brain and it can be found in the thalamus, substantia nigra, striatum, and hippocampus. Copper is present in various redox enzymes such as cytochrome C oxidase (COX), Cu/Zn superoxide dismutase (Cu, Zn-SOD1), lysyl oxidase, uricase, dopamine hydroxylase and tyrosinase (Borisov and Forte 2021; Robinett et al. 2019). Possessing redox capability, copper facilitates the mitochondrial electron transport chain, neurotransmitter synthesis, myelination and clearance of reactive oxygen species (ROS) (Pezacki et al. 2022). The redox ability of copper is dependent on its capacity to operate as both electron “donor” and “acceptor”. Cuprous (Cu I) can redox into Cu II and Cu 0 through the single electron transfer charge-disproportionation between the “donor” and “acceptor” (Hatori and Lutsenko 2016; Liang et al. 2022a). Upon combining with proteins to form ceruloplasmin, Cu also converts Fe2+ to Fe3+, acting as a ferroxidase that contributes to Fe homeostasis (Hellman and Gitlin 2002). Another important function of copper is to serve as neurotransmitters (D'Ambrosi and Rossi 2015). Cu is also stored in presynaptic vesicles and released into synaptic clefts when a neuron is stimulated and subsequently connects to glutamate receptors and GABA receptors to modulate the neuronal excitability (D'Ambrosi and Rossi 2015; Tanaka and Kawahara 2017).
Selenium (Se)
Selenium, an essential trace element, is the active center of several selenium-dependent enzymes such as glutathione peroxidase (1, 2 and 4), iodothyronine deiodinases, methionine-R-sulfoxide reductase, thioredoxin reductase, and selenoproteins (Alshammari et al. 2022). Therefore, selenium plays an important role in scavenging ROS, managing immunity, inhibiting inflammation, ferroptosis and endoplasmic reticulum (ER) stress (Genchi et al. 2023). Selenium and selenium-containing proteins contribute to the human defense system as potent antioxidants and serve an important biological role in human health.
In a word, metal homeostasis is a dynamic process that each metal ion in the body is inextricably relevant and impactful to each other. The roles of metal ions and their transport channels in the CNS are summarized in Table 1.
Metal ion-related pathogenesis
Neuroinflammation
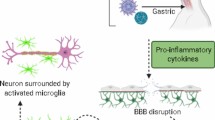
Inflammation in the CNS, also termed neuroinflammation, is a vital segment of neurological pathology intended to repair brain injuries and restore brain homeostasis (Candelario-Jalil et al. 2022). However, excessive neuroinflammation inflicts destructive damage to the CNS (Hou et al. 2021). Mounting evidence highlights that COVID-19 causes a range of neurological symptoms by inducing neuroinflammation (Fig. 2), and the pathogenesis of stroke is also closely associated with neuroinflammation (Anthony et al. 2022; Beckman et al. 2022). In general, neuroinflammation is induced by the release of damage associated molecular patterns (DAMPs) from injured or dead cells. Later, free DAMPs in the form of adenosine, heat shock proteins (HSPs), high mobility group box 1 (HMGB1), and interleukin-1α (IL-1α) are enrolled by associated immune cells that can induce various downstream signaling pathways (Liu et al. 2021; Villadiego et al. 2018; Zhang et al. 2022b). A number of immune cells, such as microglia, astrocytes, macrophages, and T lymphocytes, are activated during the release of DAMPs (Garofalo et al. 2022). In parallel, inflammation-related cytokines, interferons or chemokines including chemokine-chemokine ligand 2 (CCL2) and monocyte chemoattractant protein-1 (MCP-1) are spurred to recruit immune cells, leading to infiltration of leukocytes (Gutiérrez et al. 2022; Zhang et al. 2018).

Schematic diagram of COVID-19-induced neuroinflammation. SARS-CoV-2 activates different immune cells in the brain via infecting other organs and inducing inflammatory storms on the one hand, and directly invading the brain through the BBB on the other hand. These activated immune cells then secrete cytokines to induce neuroinflammation. TNF tumor necrosis factor, IL interleukin, MMPs matrix metalloproteinases, CCL C–C motif chemokine, CXCL C-X-C motif chemokine, TGF protransforming growth factor, MCP-1 monocyte chemoattractant protein-1, IFN interferon, GM-CSF granulocyte–macrophage colony-stimulating factor, VEGF vascular endothelial growth factor, G-CSF granulocyte colony-stimulating factor, FasL factor related apoptosis ligand, TRAIL tumor necrosis factor related apoptosis inducing ligand, BAFF B-cell activating factor of the TNF family, MIP-1β macrophage inflammatory protein 1 beta, RANTES regulated upon activation normal T cell expressed and secreted factor
Metal ions are thought to be relevant to neuroinflammation. Some pathways such as voltage and receptor gated Ca2+ influx promote a large increase in free cytosolic calcium, which causes mitochondrial calcium overload and in turn compromises the ATP production, further promoting ROS release (Bertero et al. 2021). In addition, elevated levels of intracellular Ca2+ activate various proteases, lipases, kinases, phosphatases, and endonucleases (Metwally et al. 2021; Tone et al. 2022). These toxic elements thus induce a series of inflammatory cascades, leading to mobilization of microglia and astrocytes and increased production of cytokines and chemokines (Mehta et al. 2023). As a calcium antagonist, Mg2+ can facilitate the alleviation of neuroinflammation by suppressing calcium influx via NMDARs (Zhu et al. 2018).
Iron accumulation is a major signature of activated microglia and neuroinflammation, which has been observed in several neurological disorders (Lu et al. 2022). It has been proposed that excess iron activates microglia by promoting NF-κB-mediated transcription of pro-inflammatory cytokines (Feng et al. 2021b). A recent study has further confirmed that excess irons provoke both morphological activation and transcriptomic changes of microglia using induced pluripotent stem cells (iPSCs) derived microglia, and this activation reduced both pro- and anti-inflammatory pathways (Kenkhuis et al. 2022). Additional studies have identified the interaction between inflammation and iron accumulation in neuronal cells. For example, Urrutia et al. have reported that the rise in inflammatory factors including TNF-α and IL-6 causes the upregulation of DMT1 and downregulation of FPN, resulting in iron accumulation (Urrutia et al. 2013).
Zinc is indispensable for immune stability, and it is able to work as a transcriptional repressor, suppressing NF-κB and restricting the function of TNF-α (Foster and Samman 2012; Voelkl et al. 2018). Therefore, intracellular zinc deficiency may boost NF-κB expression and trigger deleterious neuroinflammation. Consistently, subsequent evidence has indicated that changes in immune markers, including a decrease in MCP-1 and an increase in naive CD4+ T cell markers, worsen during zinc deficiency (Lu et al. 2012). In addition, zinc contributes to the suppression of IFN-γ, IL-17 and TNF-α in immune activated T cells (Guttek et al. 2018). These findings suggest that zinc deficiency is associated with immune dysfunction and neuroinflammation. However, Kauppinen et al. have reported that Zn2+ promotes the activation of microglia at the late stage of cerebral ischemic injury, indicating the double-edged sword effect of zinc (Kauppinen et al. 2008). Therefore, the effect of zinc on neuroinflammation depends on its basal level and the different disease courses.
Both COVID-19 and stroke can cause varying degrees of BBB damage, while heavy metals such as lead and cadmium are able to shuttle through the BBB and eventually accumulate in the brain, inducing neuroinflammation. It has been shown that cadmium induces the dismission of IL-6 and IL-8 from astrocytes through activating MAPK and NF-κB pathways, leading to neuroinflammation and neuronal death (Phuagkhaopong et al. 2017). In a rat model, Liu et al. have reported that lead exposure contributes to increased microglial activation and inferior long-term potentiation (LTP) (Liu et al. 2015). This might be one of the explanations that COVID-19 is able to trigger more severe neurological symptoms in patients with metal ion disorders.
Oxidative stress
Oxidative stress is provoked by overproduction of ROS, which leads to oxidative damage on lipids, biological membranes, proteins, and DNA (Feng et al. 2021a). ROS are highly reactive molecules comprising superoxide anion radicals (O2·−), hydroxyl radical (OH.), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl) (Knaus 2021). They are products mainly from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, mitochondria electron transport chain, and the Fenton or Haber–Weiss reaction (Wu et al. 2022). Metal ions play an essential role in the production, distribution and metabolism of ROS. There is emerging evidence that ROS are generated in mitochondria via an electron transport chain consisting of complex I-IV and mobile carriers (coenzyme Q [CoQ] and cytochrome c [Cytc]) (Nolfi-Donegan et al. 2020). Under physiological conditions, the electrons derived from nicotinamide adenine diphosphate hydride (NADH) and flavin adenine dinucletide (FAD) in the complex I and II, are transferred to complex III, and ultimately converge on complex IV, where oxygen is reduced to water (Turrens 2003). Iron is involved in the electron transport chain as a component of complex I, transferring electrons. Then, redox-active metal ions encompassing Cu+/2+ or Fe2+/3+ are able to convert H2O2 into the more harmful OH· following overproduction of O2·− in the electron transport chain, which is known as the Fenton reaction (Husain et al. 2008; Thiriveedi et al. 2020).
In response to ischemic and hypoxic stimuli, the metabolism of neuronal cells shifts from aerobic to anaerobic glycolysis with the production of hydrogen ions and lactic acid (Chen et al. 2018; Tan et al. 2021). Excessive production of these byproducts lowers intracellular pH, leading to a buildup of intracellular Na+ levels through the export of intracellular H+ and import of extracellular Na+ by the Na+/H+ exchangers (NHE) (Rotte et al. 2012). Subsequently, the plasma membrane Na+/Ca2+ exchanger is triggered to expel redundant Na+ and transport extracellular Ca2+ into the cytoplasm (Schnetkamp 1995). Ca2+ overload can provoke mitochondrial permeability transition pore (mPTP) opening, which further triggers ROS generation (Seidlmayer et al. 2015). Simultaneously, the opened mPTP is capable of damaging mitochondrial membrane potential, liberating Cytc and metal ions such as Ca2+, Cu+/2+ or Fe2+/3+, Mg2+ and Zn2+, inhibiting ATP production, causing a cascade reaction in neighboring mitochondria, and eventually inducing neuronal cell death.
Under physiological circumstances, antioxidants are in charge of scavenging ROS via enzymatic reactions that transform toxic free radicals into less-toxic or non-toxic species, involving superoxide dismutase (SOD), catalase and glutathione peroxidase (GPx) (Bai et al. 2021; Wen et al. 2021). In terms of mechanisms, SOD catabolizes O2·− into O2 and H2O2, peroxidase breaks down not only H2O2 but also other organic hydroperoxides, and GPx converts H2O2 into H2O and O2 with the assistance of glutathione (Chen et al. 2022; Rattanawong et al. 2021). It has been demonstrated that a few metal ions play crucial roles in composing these antioxidants, including Zn-Cu-SOD and Ca-peroxidase (Mohandass et al. 2021; Weydert and Cullen 2010). In addition, the enzyme synthesizing SOD is Mn-dependent (Zelko et al. 2002). When metal ion homeostasis is disrupted in COVID-19 or stroke, the antioxidants fail to remove extra ROS, ultimately causing oxidative stress.
Excitotoxicity
Glutamate and aspartate are the dominant excitatory neurotransmitters in the CNS and high concentrations of glutamate or aspartate lead to the damage of brain cells, referring to as excitotoxicity (Lai et al. 2014). Emerging evidence has emphasized the ionic underpinnings of excitotoxicity. K+ overload and acidosis have been reported as prodromal events in the ischemic cascade resulting in ionic disturbances (Lipton 1999). Elevated K+ facilitates the release of glutamate, thereby catalyzing Na+/Ca2+ channels linked to NMDARs and further increasing intracellular Na+ and Cl− concentrations as well as the passive influx of H2O that causes cytotoxic edema (Dumuis et al. 1993). Furthermore, extracellular glutamate can also activate AMPARs and mGluRs, a critical step in the inflammatory cascade (Lu et al. 2017; Ribeiro et al. 2017). Thereafter, NMDARs work through a single ion channel, which in turn improves intracellular Ca2+ levels. In the physiological state, Mg2+ obstructs the channel pore of NMDARs. Whereas with the release of glutamate from presynaptic sites and AMPARs activation, Mg2+ is completely depleted from NMDARs as the postsynaptic membrane is partially depolarized, resulting in an influx of Na+ and Ca2+ into the cells and mitochondrial calcium overload, further impairing ATP production (Arvanian et al. 2004; Wollmuth et al. 1998). Calcium overload in neurons generates a cascade of downstream death signaling pathways, involving calpain activation, ROS production, and mitochondrial insults.
Interestingly, extracellular and intracellular zinc and copper signaling may play a neuroprotective role against glutamate-induced excitotoxicity. As we mentioned before, copper and zinc were coupled to glutamate for transmission during synaptic activity (Gasperini et al. 2015; Krall et al. 2020). In the synaptic cleft, the two metals modulate excitatory neurotransmission by suppressing the activity of NMDARs and AMPARs. Other metals such as Fe, Mn, Al, and Pb also seem to engage in excitotoxicity through regulating intracellular levels of Ca2+ or related receptor activity. Nevertheless, more work needs to be done to identify the roles of metals in excitotoxicity.
Neuronal cell death
As a consequence of oxidative stress, excitotoxicity and/or neuroinflammation, neuronal cells will ultimately die and the death is classified into three main different types: apoptosis, necrosis and autophagic cell death. For a long time, necrosis was considered passive and unregulated. However, in recent years, specific forms of necrosis have been identified as highly regulated and are referred to as regulated necrosis, including necroptosis, pyroptosis, ferroptosis, parthanatos, and cyclophilin D (CypD)-dependent necrosis.
There is no doubt that metal ions play an important role in regulating neuronal cell death. Novel evidence demonstrates that exposure to excessive amounts of metals, such as Ca, Fe, Zn, Cu, Pb, and Cd, leads to neuronal cell death (Gleitze et al. 2021; Lu et al. 2022; Tsvetkov et al. 2022). It is well accepted that the occurrence of ferroptosis depends on iron. Actually, zinc is also critical for ferroptosis in breast and renal cancer cells (Chen et al. 2021). It has been shown that ferroptosis is inhibited when zinc chelators are administered. Conversely, additional zinc treatment boosted ferroptosis despite the presence of an iron chelator. Mechanistically, by investigating the genes associated with zinc and ferroptosis, the authors identified SLC39A7, which encoded ZIP7 that regulated zinc transport from ER to cytosol, as a novel genetic determinant of ferroptosis (Chen et al. 2021). In addition to iron-dependent ferroptosis, a recent authoritative study uncovered a copper-dependent programmed cell death that, unlike known death mechanisms, relied on mitochondrial respiration. In this study, the authors presented robust evidence confirming copper-dependent death through direct binding of copper to lipoylated components of the tricarboxylic acid (TCA) cycle, resulting in accumulation of lipid acylated proteins and proteotoxic stress and ultimately cell death (Tsvetkov et al. 2022). It is still unknown whether there is a unique mechanism for neuronal cell death induced by zinc or copper. Nevertheless, there is substantial evidence that metal dysregulation disrupts the CNS in various ways, including releasing excessive ROS, triggering excitotoxicity, inducing neuroinflammation, and ultimately leading to neuronal cell death (Fig. 3).

Schematic diagram of the pathophysiology involved in COVID-19-induced neurological symptoms. A Neuroinflammation: excessive immune cells activation and chemokines and cytokines release promote inflammatory cell infiltration and BBB damage. B Oxidative stress: mitochondrial Ca2+ overload leads to excessive ROS production and mPTP opening causes excessive ROS and Cyt C release. C Excitotoxicity: excessive glutamate release activates NMDARs, which subsequently induces calcium overload and activates downstream death signaling pathways. D Neuronal cell death: involving apoptosis, necroptosis, pyroptosis, ferroptosis, parthanatos, CypD-mediated necrosis, and autophagy. NF-κB nuclear factor kappa-B, MAPK mitogen-activated protein kinase, NLRP3 NOD-like receptor thermal protein domain associated protein 3, MMPs matrix metalloproteinases, BBB blood–brain barrier, ETC electron transport chain, mPTP mitochondrial permeability transition pore, ROS reactive oxygen and species, NMDARs N-methyl-d-aspartate receptors, CypD cyclophilin D
Metal dysregulation in COVID-19-induced neurological symptoms
COVID-19-induced neurological symptoms
There is evidence that SARS-CoV-2 invades host cells via utilizing prominent spike proteins that attach to cell membrane receptors, including angiotensin-converting enzyme 2 (ACE-2) and CD147 (basigin), in combination with the processing of primer/transmembrane serine proliferase 2 (TMPRSS2) involving the viral spike protein (Scialo et al. 2020; Wang et al. 2020; Wu et al. 2021). SARS-CoV-2 is also able to access 293/hACE2 cells in a receptor non-dependent manner via endocytosis, in which PIKfyve, TPC2, and cathepsin L are essential for this entry (Ou et al. 2020).
A large number of reports have provided a full description of the pulmonary symptoms of COVID-19 (Guan and Zhong 2020; Huang et al. 2020; Zhu et al. 2020). However, besides the typical respiratory and gastrointestinal symptoms, COVID-19 infection might be also accompanied by neurological manifestations that may persist for a long time (Ellul et al. 2020). In the period of COVID-19, patients with no previous neuropsychiatric history suffered attention difficulties, insomnia, fatigue, hysteria and delusions, even changes in behavior such as suicide (Becker et al. 2020; Ceban et al. 2022; Farooq et al. 2021; Fricchione et al. 2022; Pappa et al. 2020). Patients admitted to the intensive care unit (ICU) have been revealed with agitation (69%) and corticospinal tract signs (67%), suggesting a definite association between COVID-19 and encephalopathy (Helms et al. 2020).
A number of recent studies have highlighted that adult patients with COVID-19 further developed a variety of neurological symptoms, such as stroke, dementia, hallucinations, seizures, and encephalopathy (Poloni et al. 2021; Stein et al. 2021; Uginet et al. 2021; Xu et al. 2022). In addition, Frontera et al. conducted a prospective study on the prevalence of new neurological disorders among COVID-19 patients in the New York City metropolitan area, which showed that 13.5% of COVID-19 patients developed neurological disorders (Frontera et al. 2021). These reports indicate a specific propensity of COVID-19 for the central and peripheral nervous system.
Radiographic evidence showed that a reduction in gray matter thickness and tissue contrast were observed in up to 80% of hospitalized patients (Chou et al. 2021; Douaud et al. 2022). From autopsies of COVID-19 decedents, many investigators have reported postmortem neuropathological findings: hypoxia, microglia activation, astrocyte lesions, mild lymphocytic infiltration, as well as microhemorrhages and hemorrhages (Egbert et al. 2020; Mesci et al. 2022; Samudyata et al. 2022; Thakur et al. 2021). Moreover, Khan et al. have recently discovered that ciliary cells of the respiratory mucosa and parasitic cells (non-neurons) of the olfactory mucosa are the major target cells of COVID-19, which is supposed to be the main pathway affecting olfactory sensory neurons, resulting in the sequelae of COVID-19 olfactory dysfunction (Khan et al. 2021). However, whether COVID-19 can directly infect the CNS remained controversial.
Some studies did not detect viral RNAs or proteins in the CNS tissue of COVID-19 decedents although their brains exhibited postmortem neuropathology, whereas some studies detected COVID-19 virus in autopsies of patients who died from COVID-19 (Mesci et al. 2022; Pellegrini et al. 2020; Soung et al. 2022; Thakur et al. 2021). The reason for this discrepancy might be due to the differences in basic diseases and severity of infection. In support of this view, another report has demonstrated that the S1 spike protein of COVID-19 virus is able to cross the blood brain barrier (BBB) in mice and enter the CNS, initiating a neuroinflammatory process (Pellegrini et al. 2020; Zhang et al. 2021b). These data suggested that COVID-19 virus was capable of infecting the brain and triggering long-term neurological manifestations. Patients with neurodegenerative disorders suffer from increased BBB permeability, which potentially facilitates the neural invasion of SARS-CoV-2. Based on these reports, it is reasonable to speculate that SARS-CoV-2-induced neuroinflammation may increase susceptibility to neurodegeneration in patients who do not yet develop these diseases. Despite the emerging evidence, a comprehensive overview of the cellular and molecular mechanisms of SARS-CoV-2 brain infection and the consequent impact on CNS is still not available.
Metal dysregulation in COVID-19-induced neurological symptoms
It is well accepted that virus is able to disrupt Ca2+ homeostasis in host cells and in turn to modulate signal transduction (Danta 2020a). Although there is no sufficient laboratory data to support the direct relationship between Ca2+ and SARS-CoV-2, several recent studies on other sequentially homologous coronaviruses, including severe acute respiratory syndrome CoV (SARS-CoV) and Middle East respiratory syndrome CoV (MERS-CoV), have revealed the enhanced fusion with host cells via Ca2+, thereby increasing the infection rate (Danta 2020b; Straus et al. 2020). Furthermore, viruses can also seize calcium channels and pumps, such as voltage-gated calcium channels (VGCCs), receptor-operated calcium channels, store-operated calcium channels, TRP channels, and Ca2+-ATPase in host cells to release more intracellular Ca2+ for their life cycle (Qu et al. 2022). In agreement with these findings, Chen et al. have shown that interactions between viruses and VGCCs contribute to virus-host cell fusion entry (Chen et al. 2019). Therefore, it is very likely that SARS-CoV-2 can also distort Ca2+ homeostasis when it attacks the body, which facilitates disease progression. Some highly lipophilic calcium channel blockers (CCBs), such as nimodipine and memantine, have been reported to be effective on AD dementia due to their ability to easily pass the BBB (López-Arrieta and Birks 2000; McShane et al. 2019). In particular, some CCBs have also been found to inhibit various viral infections such as Japanese encephalitis virus (JEV), dengue virus (DENV), West Nile virus (WNV), Zika virus (ZIKV), and even Ebola virus (EBOV) (Chen et al. 2019). Ribeiro et al. have also proposed that P2X7 receptor may be over-activated via SARS-CoV-2-induced extracellular ATP overload, leading to an increase in Ca2+ influx and glutamate release (Ribeiro et al. 2021). As a result, severe oxidative stress and cytokine storm occurs in the brain, eventually causing neuronal death. These reports shed some light on novel strategies for prevention of SARS-CoV-2 infection or the neurological diseases it causes via targeting calcium signaling pathways.
Zinc also exhibits a Yin-Yang role in COVID-19. Numerous reports have revealed that zinc deficiency facilitates the risk of viral infections and the development of severe forms of disease (Balboni et al. 2022; Dhawan et al. 2022). Observational clinical studies and animal experiments have demonstrated that appropriate zinc administration could counteract the disease progression (Ben Abdallah et al. 2022). For example, in a randomized clinical study, 470 newly diagnosed COVID-19 patients were enrolled and the effect of high-dose zinc (50 mg per day divided into two doses) was examined on the two primary endpoints, admission to the ICU and death. The results showed that the risk ratio for ICU admission in the zinc supplementation group was 0.43 and short-term mortality was 0.68, with additional benefits for the length of stay and the duration of symptoms in outpatients (Vinceti et al. 2022). In addition, a recent survey revealed that taste impairment was associated with the reduced salivary zinc levels in COVID-19 patients (Abdelmaksoud et al. 2021). These reports indicate that zinc should be considered as a preventive and adjuvant treatment for COVID-19 to reduce susceptibility and disease severity. Besides the transient protection of COVID-19 by zinc supplementation, its long-term effects on the CNS remain to be determined.
There is evidence that iron metabolism disturbances also occur in COVID-19 patients. COVID-19-induced inflammation promotes an accumulation of intracellular iron, resulting in low levels of circulating iron available for metabolism (Habib et al. 2021; Mahroum et al. 2022). A clinical study from Muhammad et al. showed a significant decrease in serum iron, total iron binding capacity (TIBC) and transferrin levels as well as a notable increase in ferritin and transferrin saturation in COVID-19 patients (Muhammad et al. 2022). In another study, iron and transferrin were reported to be associated with mortality in COVID-19 patients (Suriawinata and Mehta 2022). Even though not confirmed, it can be hypothesized that iron-related biomarkers play important roles in causing oxidative stress in various tissues of COVID-19 patients, further triggering the activation of cytokine storm, and ultimately inducing ferroptosis.
The role of selenium in fighting against COVID-19 and relevant neurological symptoms should not be underestimated. On the one hand, selenoproteins and selenium-containing organisms can prevent SARS-CoV-2 infection via suppressing the expression of ACE-2 receptors; on the other hand, they can exert anti-COVID-19 effects by inhibiting inflammation, oxidative stress, cytokine storm and 3CLPro, the main protease of SARS-CoV-2 (Alshammari et al. 2022). Clinical evidence showed that serum selenium concentrations were significantly higher in COVID-19 survivors than that in non-survivors (Moghaddam et al. 2020). Moreover, selenium concentrations in patient toenail samples were correlated with the COVID-19 severity index (CSI) (Larvie et al. 2023). These results suggest that selenium plays a potential role as a neuroprotective agent in treating COVID-19. In summary, long-term inflammation, BBB breakdown, and microglia activation may contribute to neurotransmitter alterations, and neuronal impairment, thus interpreting the neuropsychiatric symptoms of COVID-19. Dysregulation of metal homeostasis is involved in a range of neurological complications and sequelae in COVID-19 patients and targeting metal-related signaling pathways for treating COVID-19 infection should be performed with appropriate compatibility. Thus, appropriate drug combinations may be a promising approach for improving neurological injury caused by COVID-19. The roles of metal ions in COVID-19-induced neurological symptoms are summarized in Table 2.
Treating COVID-19-induced neurological symptoms via targeting metal dysregulation
Due to the pivotal roles of metal ions in COVID-19-induced neurological symptoms, targeting the metal-related signaling pathways might be a potential strategy to mitigate the neurological symptoms. The initial thought was to directly remediate the neural metal content by giving appropriate metals or metal chelator. A recent study has shown that zinc administration may ameliorate disease progression in COVID-19 by reducing the inflammatory factor levels of patients (Dhawan et al. 2022). In another report, patients with low selenium intake exhibited more severe COVID-19-related symptoms and selenium intake below 65 μg/day might increase the severity of COVID-19 (Zhang et al. 2020). Growing evidence has also demonstrated the role of iron chelators in COVID-19. Poonkuzhi Naseef et al. revealed that deferasirox, an iron chelator, was able to save COVID-19 critically ill patients, mitigated disease progression, and improved survival (Poonkuzhi Naseef et al. 2022). In addition, deferiprone (L1) might adjunctively treat COVID-19 through binding transferrin, involving in immune response and mobilization of iron in highly inflamed cells (Poonkuzhi Naseef et al. 2022). Unfortunately, single administration of iron chelators could cause a variety of serious neuropathological consequences, such as myelinopathy (McCarthy et al. 2022). These data indicated that it was extremely difficult to master the proper timing and dosage of metals in vivo by simple supplementation or chelation, and that inadequate or excessive amounts of these drugs gave rise to insufficient efficacy or more serious side effects. Therefore, it is necessary to monitor the dynamics of metal ions in vivo so as to adjust therapeutic strategies timely.
With the clarification of metal metabolic mechanisms, therapeutic protocols via targeting metal ions have become increasingly diverse. One possible strategy is to target the metal ion channels. Various antagonists or agonists targeting different metal ion channels have been developed (Table 3). Unfortunately, all of them are still only used as research tools so far. Therefore, selectively targeting the downstream pathways might be another feasible option. For instance, targeting the pro-death signaling of NMDARs downstream has shown great promise in neuroprotection (Yan et al. 2020). Different downstream pathways of NMDARs have been identified, including the GluN2B-PSD95-nNOS pathway, the GluN2B-DAPK1 pathway, the NMDAR-PTEN complex, and the NMDAR-Src-Panx1 complex (Sun et al. 2015; Tu et al. 2010; Weilinger et al. 2016; Zhou et al. 2010). Through developing small molecule inhibitors and interfering peptides, many experiments have shown that targeting these signaling pathways improved the neurological function after stroke and reduced the ischemia/hypoxia-induced neuronal death. Since many scholars assumes COVID-19 as a pathological model of human inflammation combined with hypoxia, analogous to the pathological features of ischemic stroke (Fig. 4), it is reasonable to speculate that targeting the downstream pathways of NMDARs also works for treating COVID-19-induced neurological symptoms.

Similarities between COVID-19-induced neurological symptoms and ischemic stroke. The pathological similarities between the neurological symptoms caused by COVID-19 and ischemic stroke mainly include: microglia and astrocyte activation and BBB breakdown; Ca2+ overload activates GluN2B and its downstream death signaling pathway; mitochondrial dysfunction leads to impaired ATP synthesis, ROS overproduction and neuronal apoptosis; iron overload results in lipid peroxidation and neuronal ferroptosis. BBB blood–brain barrier, GluN2B NMDA receptor 2B, DAPK1 death-associated protein kinase 1, PTEN phosphatase and tensin homolog, PSD95 postsynaptic density protein-95, nNOS neuronal nitric oxide synthase, Src proto-oncogene tyrosine-protein kinase Src, Panx1 Pannexin-1, mPTP mitochondrial permeability transition pore, Cyt C cytochrome C, ROS reactive oxygen species
Growing evidence indicates that iron deposition-induced ferroptosis causes neurological disorders (Ou et al. 2022). Thereby, targeting ferroptosis-relevant pathways represents a novel therapeutic approach for treating COVID-19-induced neurological symptoms. Currently available ferroptosis inhibitors mainly focus on preventing iron overload and inhibiting lipid peroxidation (Liang et al. 2022b). A recent study by Tuo et al. has shown that intranasal administration of ferritinase inhibitors, ferritinase-1 and lipoproteinase-1, immediately after cerebral ischemia in mice reduced neuronal damage, and the protective effect continued until 6 h after reperfusion (Tuo et al. 2022). By intravenous injection, Feng et al. have revealed that tirazol mesylate, an inhibitor of iron-dependent lipid peroxidation, alleviated hypoxic-ischemic brain injury in neonatal piglets (Feng et al. 2000). Furthermore, desferrioxamine (DFO), a powerful iron chelator, has also been reported to improve neurological function in ischemic rats and stroke patients (Papazisis et al. 2008; Selim 2010). It is worth mentioning that different organic and inorganic selenium compounds including methylselenocysteine, selenocystine, selenomethionine, selenocystamine, ebselen, sodium selenite, and sodium selenate are all effective in preventing erastin- and RSL3-induced iron toxicity and exerting antioxidant effects. Among them, methylselenocysteine or selenocystamine could reduce brain injury in stroke mice (Tuo et al. 2021). Due to similarity between stroke-induced brain injury and COVID-19-induced neurological symptoms as we mentioned before, it is reasonable to posit that targeting the iron-relevant pathways and applying the relevant antioxidants might also benefit the COVID-19-induced neurological symptoms.
Conclusion
In summary, maintenance of metal homeostasis is essential for neurological function. Metal metabolic dysregulation, aberrant activation or suppression of metal ion channels, and disruption of the ion-relevant signaling pathways may contribute to COVID-19-induced neurological symptoms (Balboni et al. 2022; Habib et al. 2021; Poonkuzhi Naseef et al. 2022; Suriawinata and Mehta 2022). So far, significant advances have been achieved in understanding different metal-relevant signaling pathways, offering us new directions to combat COVID-19-induced neurological symptoms (Gleitze et al. 2021; Sun et al. 2022; Thirupathi and Chang 2019). Thus, restoration of metal homeostasis is an effective strategy to prevent neurological damage. Despite the fact that preclinical studies have demonstrated the potential neuroprotective effects of a few metallo-modulators, they have obvious limitations, such as side effects and narrow therapeutic windows. This can be explained by the fact that multiple metal dysregulation can be triggered simultaneously in complex disease processes. The crosstalk between different metal dysregulation and the downstream signaling pathways merits further investigation, which implies that any single metal modulator may require combination with other drugs. However, the application of current discoveries based on animal or cellular experiments to the clinical setting (such as COVID-19-induced neurological symptoms) requires considerable effort.
Data availability
Enquiries about data availability should be directed to the authors.
References
Abdelmaksoud AA, Ghweil AA, Hassan MH, Rashad A, Khodeary A, Aref ZF, Sayed MAA, Elsamman MK, Bazeed SES (2021) Olfactory disturbances as presenting manifestation among Egyptian patients with COVID-19: possible role of zinc. Biol Trace Elem Res 199:4101–4108. https://doi.org/10.1007/s12011-020-02546-5
Alim I, Teves L, Li R, Mori Y, Tymianski M (2013) Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci 33:17264–17277. https://doi.org/10.1523/JNEUROSCI.1729-13.2013
Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ et al (2019) Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177:1262-1279.e1225. https://doi.org/10.1016/j.cell.2019.03.032
Almutairi MMA, Xu G, Shi H (2019) Iron pathophysiology in stroke. Adv Exp Med Biol 1173:105–123. https://doi.org/10.1007/978-981-13-9589-5_6
Alshammari MK, Fatima W, Alraya RA, Khuzaim Alzahrani A, Kamal M, Alshammari RS, Alshammari SA, Alharbi LM, Alsubaie NS, Alosaimi RB et al (2022) Selenium and COVID-19: a spotlight on the clinical trials, inventive compositions, and patent literature. J Infect Public Health 15:1225–1233. https://doi.org/10.1016/j.jiph.2022.09.011
Anthony S, Cabantan D, Monsour M, Borlongan CV (2022) Neuroinflammation, stem cells, and stroke. Stroke 53:1460–1472. https://doi.org/10.1161/strokeaha.121.036948
Arvanian VL, Bowers WJ, Petruska JC, Motin V, Manuzon H, Narrow WC, Federoff HJ, Mendell LM (2004) Viral delivery of NR2D subunits reduces Mg2+ block of NMDA receptor and restores NT-3-induced potentiation of AMPA-kainate responses in maturing rat motoneurons. J Neurophysiol 92:2394–2404. https://doi.org/10.1152/jn.00278.2004
Bai LL, Zhang LQ, Ma J, Li J, Tian M, Cao RJ, He XX, He ZX, Yu HL, Zhu XJ (2021) DIP2A is involved in SOD-mediated antioxidative reactions in murine brain. Free Radic Biol Med 168:6–15. https://doi.org/10.1016/j.freeradbiomed.2021.03.027
Balboni E, Zagnoli F, Filippini T, Fairweather-Tait SJ, Vinceti M (2022) Zinc and selenium supplementation in COVID-19 prevention and treatment: a systematic review of the experimental studies. J Trace Elem Med Biol 71:126956. https://doi.org/10.1016/j.jtemb.2022.126956
Bar-Am O, Amit T, Kupershmidt L, Aluf Y, Mechlovich D, Kabha H, Danovitch L, Zurawski VR, Youdim MB, Weinreb O (2015) Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol Aging 36:1529–1542. https://doi.org/10.1016/j.neurobiolaging.2014.10.026
Becker SP, Breaux R, Cusick CN, Dvorsky MR, Marsh NP, Sciberras E, Langberg JM (2020) Remote learning during COVID-19: examining school practices, service continuation, and difficulties for adolescents with and without attention-deficit/hyperactivity disorder. J Adolesc Health 67:769–777. https://doi.org/10.1016/j.jadohealth.2020.09.002
Beckman D, Bonillas A, Diniz GB, Ott S, Roh JW, Elizaldi SR, Schmidt BA, Sammak RL, Van Rompay KKA, Iyer SS et al (2022) SARS-CoV-2 infects neurons and induces neuroinflammation in a non-human primate model of COVID-19. Cell Rep 41:111573. https://doi.org/10.1016/j.celrep.2022.111573
Ben Abdallah S, Mhalla Y, Trabelsi I, Sekma A, Youssef R, Bel Haj Ali K, Ben Soltane H, Yacoubi H, Msolli MA, Stambouli N et al (2022) Twice-daily oral zinc in the treatment of patients with coronavirus disease 2019: a randomized double-blind controlled trial. Clin Infect Dis. https://doi.org/10.1093/cid/ciac807
Bertero E, Nickel A, Kohlhaas M, Hohl M, Sequeira V, Brune C, Schwemmlein J, Abeßer M, Schuh K, Kutschka I et al (2021) Loss of mitochondrial Ca(2+) uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in barth syndrome cardiomyopathy. Circulation 144:1694–1713. https://doi.org/10.1161/circulationaha.121.053755
Black MM (1998) Zinc deficiency and child development. Am J Clin Nutr 68:464s–469s. https://doi.org/10.1093/ajcn/68.2.464s
Borisov VB, Forte E (2021) Terminal oxidase cytochrome bd protects bacteria against hydrogen sulfide toxicity. Biochemistry (mosc) 86:22–32. https://doi.org/10.1134/s000629792101003x
Bothwell SW, Janigro D, Patabendige A (2019) Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 16:9. https://doi.org/10.1186/s12987-019-0129-6
Cai H, Cheng X, Wang XP (2022) ATP7B gene therapy of autologous reprogrammed hepatocytes alleviates copper accumulation in a mouse model of Wilson’s disease. Hepatology 76:1046–1057. https://doi.org/10.1002/hep.32484
Candelario-Jalil E, Dijkhuizen RM, Magnus T (2022) Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke 53:1473–1486. https://doi.org/10.1161/strokeaha.122.036946
Carrillo E, Bhatia NK, Akimzhanov AM, Jayaraman V (2020) Activity dependent inhibition of AMPA receptors by Zn(2). J Neurosci 40:8629–8636. https://doi.org/10.1523/jneurosci.1481-20.2020
Ceban F, Ling S, Lui LMW, Lee Y, Gill H, Teopiz KM, Rodrigues NB, Subramaniapillai M, Di Vincenzo JD, Cao B et al (2022) Fatigue and cognitive impairment in Post-COVID-19 syndrome: a systematic review and meta-analysis. Brain Behav Immun 101:93–135. https://doi.org/10.1016/j.bbi.2021.12.020
Chaudhuri R, Arora H, Seth P (2021) Mitochondrial calcium signaling in the brain and its modulation by neurotropic viruses. Mitochondrion 59:8–16. https://doi.org/10.1016/j.mito.2021.03.010
Chen J, Zhang DM, Feng X, Wang J, Qin YY, Zhang T, Huang Q, Sheng R, Chen Z, Li M et al (2018) TIGAR inhibits ischemia/reperfusion-induced inflammatory response of astrocytes. Neuropharmacology 131:377–388. https://doi.org/10.1016/j.neuropharm.2018.01.012
Chen X, Cao R, Zhong W (2019) Host calcium channels and pumps in viral infections. Cells. https://doi.org/10.3390/cells9010094
Chen PH, Wu J, Xu Y, Ding CC, Mestre AA, Lin CC, Yang WH, Chi JT (2021) Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis 12:198. https://doi.org/10.1038/s41419-021-03482-5
Chen H, Lee J, Lee JM, Han M, Emonet A, Lee J, Jia X, Lee Y (2022) MSD2, an apoplastic Mn-SOD, contributes to root skotomorphogenic growth by modulating ROS distribution in Arabidopsis. Plant Sci 317:111192. https://doi.org/10.1016/j.plantsci.2022.111192
Chou SH, Beghi E, Helbok R, Moro E, Sampson J, Altamirano V, Mainali S, Bassetti C, Suarez JI, McNett M (2021) Global incidence of neurological manifestations among patients hospitalized with COVID-19-a report for the GCS-NeuroCOVID consortium and the ENERGY consortium. JAMA Netw Open 4:e2112131. https://doi.org/10.1001/jamanetworkopen.2021.12131
Chow LWC, Leung YM (2020) The versatile Kv channels in the nervous system: actions beyond action potentials. Cell Mol Life Sci 77:2473–2482. https://doi.org/10.1007/s00018-019-03415-8
Chuang SH, Reddy DS (2019) Zinc reduces antiseizure activity of neurosteroids by selective blockade of extrasynaptic GABA-A receptor-mediated tonic inhibition in the hippocampus. Neuropharmacology 148:244–256. https://doi.org/10.1016/j.neuropharm.2018.11.031
Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU (2011) CaMKII in cerebral ischemia. Acta Pharmacol Sin 32:861–872. https://doi.org/10.1038/aps.2011.68
D’Ambrosi N, Rossi L (2015) Copper at synapse: release, binding and modulation of neurotransmission. Neurochem Int 90:36–45. https://doi.org/10.1016/j.neuint.2015.07.006
Danta CC (2020a) Calcium channel blockers: a possible potential therapeutic strategy for the treatment of Alzheimer’s dementia patients with SARS-CoV-2 infection. ACS Chem Neurosci 11:2145–2148. https://doi.org/10.1021/acschemneuro.0c00391
Danta CC (2020b) CNS penetration ability: a critical factor for drugs in the treatment of SARS-CoV-2 brain infection. ACS Chem Neurosci 11:2137–2144. https://doi.org/10.1021/acschemneuro.0c00335
Dattilo M, Penington NJ, Williams K (2008) Inhibition of TRPC5 channels by intracellular ATP. Mol Pharmacol 73:42–49. https://doi.org/10.1124/mol.107.040899
Denechaud M, Geurs S, Comptdaer T, Bégard S, Garcia-Núñez A, Pechereau LA, Bouillet T, Vermeiren Y, De Deyn PP, Perbet R et al (2022) Tau promotes oxidative stress-associated cycling neurons in S phase as a pro-survival mechanism: possible implication for Alzheimer’s disease. Prog Neurobiol. https://doi.org/10.1016/j.pneurobio.2022.102386
Dhawan M, Emran TB, Choudhary OP (2022) Immunomodulatory effects of zinc and its impact on COVID-19 severity. Ann Med Surg (lond) 77:103638. https://doi.org/10.1016/j.amsu.2022.103638
Dixon RE, Navedo MF, Binder MD, Santana LF (2022) Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol Rev 102:1159–1210. https://doi.org/10.1152/physrev.00022.2021
Doleschal B, Primessnig U, Wölkart G, Wolf S, Schernthaner M, Lichtenegger M, Glasnov TN, Kappe CO, Mayer B, Antoons G et al (2015) TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc Res 106:163–173. https://doi.org/10.1093/cvr/cvv022
Donnelly DF (2013) Voltage-gated Na(+) channels in chemoreceptor afferent neurons–potential roles and changes with development. Respir Physiol Neurobiol 185:67–74. https://doi.org/10.1016/j.resp.2012.08.009
Douaud G, Lee S, Alfaro-Almagro F, Arthofer C, Wang C, McCarthy P, Lange F, Andersson JLR, Griffanti L, Duff E et al (2022) SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 604:697–707. https://doi.org/10.1038/s41586-022-04569-5
Dumuis A, Sebben M, Fagni L, Prézeau L, Manzoni O, Cragoe EJ Jr, Bockaert J (1993) Stimulation by glutamate receptors of arachidonic acid release depends on the Na+/Ca2+ exchanger in neuronal cells. Mol Pharmacol 43:976–981
Egbert AR, Cankurtaran S, Karpiak S (2020) Brain abnormalities in COVID-19 acute/subacute phase: a rapid systematic review. Brain Behav Immun 89:543–554. https://doi.org/10.1016/j.bbi.2020.07.014
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S, Sejvar J, Solomon T (2020) Neurological associations of COVID-19. Lancet Neurol 19:767–783. https://doi.org/10.1016/s1474-4422(20)30221-0
Farooq S, Tunmore J, Wajid Ali M, Ayub M (2021) Suicide, self-harm and suicidal ideation during COVID-19: A systematic review. Psychiatry Res 306:114228. https://doi.org/10.1016/j.psychres.2021.114228
Feng Y, LeBlanc MH, LeBlanc EB, Parker CC, Fratkin JD, Qian XB, Patel DM, Huang M, Smith EE, Vig PJ (2000) Desmethyl tirilazad improves neurologic function after hypoxic ischemic brain injury in piglets. Crit Care Med 28:1431–1438. https://doi.org/10.1097/00003246-200005000-00029
Feng L, Dou C, Xia Y, Li B, Zhao M, Yu P, Zheng Y, El-Toni AM, Atta NF, Galal A et al (2021a) Neutrophil-like cell-membrane-coated nanozyme therapy for ischemic brain damage and long-term neurological functional recovery. ACS Nano 15:2263–2280. https://doi.org/10.1021/acsnano.0c07973
Feng Z, Min L, Chen H, Deng W, Tan M, Liu H, Hou J (2021b) Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol 43:101984. https://doi.org/10.1016/j.redox.2021.101984
Foster M, Samman S (2012) Zinc and regulation of inflammatory cytokines: implications for cardiometabolic disease. Nutrients 4:676–694. https://doi.org/10.3390/nu4070676
Fricchione GL, Paul AB, Chemali Z, Kritzer MD (2022) Case 34–2022: a 57-year-old woman with Covid-19 and delusions. N Engl J Med 387:1795–1803. https://doi.org/10.1056/nejmcpc2115857
Frontera JA, Sabadia S, Lalchan R, Fang T, Flusty B, Millar-Vernetti P, Snyder T, Berger S, Yang D, Granger A et al (2021) A prospective study of neurologic disorders in hospitalized patients with COVID-19 in New York City. Neurology 96:e575–e586. https://doi.org/10.1212/wnl.0000000000010979
Garofalo S, Cocozza G, Bernardini G, Savage J, Raspa M, Aronica E, Tremblay ME, Ransohoff RM, Santoni A, Limatola C (2022) Blocking immune cell infiltration of the central nervous system to tame Neuroinflammation in Amyotrophic lateral sclerosis. Brain Behav Immun 105:1–14. https://doi.org/10.1016/j.bbi.2022.06.004
Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G (2015) Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 22:772–784. https://doi.org/10.1089/ars.2014.6032
Genchi G, Lauria G, Catalano A, Sinicropi MS, Carocci A (2023) Biological activity of selenium and its impact on human health. Int J Mol Sci 24:2633. https://doi.org/10.3390/ijms24032633
Gleitze S, Paula-Lima A, Núñez MT, Hidalgo C (2021) The calcium-iron connection in ferroptosis-mediated neuronal death. Free Radic Biol Med 175:28–41. https://doi.org/10.1016/j.freeradbiomed.2021.08.231
Golovina VA, Bambrick LL, Yarowsky PJ, Krueger BK, Blaustein MP (1996) Modulation of two functionally distinct Ca2+ stores in astrocytes: role of the plasmalemmal Na/Ca exchanger. Glia 16:296–305. https://doi.org/10.1002/(sici)1098-1136(199604)16:4%3C296::aid-glia2%3E3.0.co;2-z
Gotru SK, Chen W, Kraft P, Becker IC, Wolf K, Stritt S, Zierler S, Hermanns HM, Rao D, Perraud AL et al (2018) TRPM7 kinase controls calcium responses in arterial thrombosis and stroke in mice. Arterioscler Thromb Vasc Biol 38:344–352. https://doi.org/10.1161/atvbaha.117.310391
Guan WJ, Zhong NS (2020) Clinical characteristics of Covid-19 in China. Reply N Engl J Med 382:1861–1862. https://doi.org/10.1056/nejmc2005203
Gutiérrez IL, Novellino F, Caso JR, García-Bueno B, Leza JC, Madrigal JLM (2022) CCL2 inhibition of pro-resolving mediators potentiates neuroinflammation in astrocytes. Int J Mol Sci. https://doi.org/10.3390/ijms23063307
Guttek K, Wagenbrett L, Reinhold A, Grüngreiff K, Reinhold D (2018) Zinc aspartate suppresses proliferation and Th1/Th2/Th17 cytokine production of pre-activated human T cells in vitro. J Trace Elem Med Biol 49:86–90. https://doi.org/10.1016/j.jtemb.2018.05.003
Habib HM, Ibrahim S, Zaim A, Ibrahim WH (2021) The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed Pharmacother 136:111228. https://doi.org/10.1016/j.biopha.2021.111228
Halcrow PW, Lynch ML, Geiger JD, Ohm JE (2021) Role of endolysosome function in iron metabolism and brain carcinogenesis. Semin Cancer Biol 76:74–85. https://doi.org/10.1016/j.semcancer.2021.06.013
Hatori Y, Lutsenko S (2016) The role of copper chaperone atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants (basel). https://doi.org/10.3390/antiox5030025
Hell JW (2016) How Ca2+-permeable AMPA receptors, the kinase PKA, and the phosphatase PP2B are intertwined in synaptic LTP and LTD. Sci Signal 9:e2. https://doi.org/10.1126/scisignal.aaf7067
Hellman NE, Gitlin JD (2002) Ceruloplasmin metabolism and function. Annu Rev Nutr 22:439–458. https://doi.org/10.1146/annurev.nutr.22.012502.114457
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C, Fafi-Kremer S, Ohana M et al (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 382:2268–2270. https://doi.org/10.1056/nejmc2008597
Hernández RJ (1992) Na+/K(+)-ATPase regulation by neurotransmitters. Neurochem Int 20:1–10. https://doi.org/10.1016/0197-0186(92)90119-c
Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, Mattson MP, Croteau DL, Bohr VA (2021) NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.2011226118
Huang L, Xue Y, Feng D, Yang R, Nie T, Zhu G, Tao K, Gao G, Yang Q (2017) Blockade of RyRs in the ER attenuates 6-OHDA-induced calcium overload, cellular hypo-excitability and apoptosis in dopaminergic neurons. Front Cell Neurosci 11:52. https://doi.org/10.3389/fncel.2017.00052
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. https://doi.org/10.1016/s0140-6736(20)30183-5
Husain M, Bourret TJ, McCollister BD, Jones-Carson J, Laughlin J, Vázquez-Torres A (2008) Nitric oxide evokes an adaptive response to oxidative stress by arresting respiration. J Biol Chem 283:7682–7689. https://doi.org/10.1074/jbc.m708845200
Jaywant A, Vanderlind WM, Alexopoulos GS, Fridman CB, Perlis RH, Gunning FM (2021) Frequency and profile of objective cognitive deficits in hospitalized patients recovering from COVID-19. Neuropsychopharmacology 46:2235–2240. https://doi.org/10.1038/s41386-021-00978-8
Jin J, Ma M, Shi S, Wang J, Xiao P, Yu HF, Zhang C, Guo Q, Yu Z, Lou Z et al (2022) Copper enhances genotoxic drug resistance via ATOX1 activated DNA damage repair. Cancer Lett 536:215651. https://doi.org/10.1016/j.canlet.2022.215651
Kalappa BI, Anderson CT, Goldberg JM, Lippard SJ, Tzounopoulos T (2015) AMPA receptor inhibition by synaptically released zinc. Proc Natl Acad Sci USA 112:15749–15754. https://doi.org/10.1073/pnas.1512296112
Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, St Croix CM, Mikulska-Ruminska K, Liu B, Shrivastava IH et al (2020) Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 16:278–290. https://doi.org/10.1038/s41589-019-0462-8
Kauer JA, Gibson HE (2009) Hot flash: TRPV channels in the brain. Trends Neurosci 32:215–224. https://doi.org/10.1016/j.tins.2008.12.006
Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA (2008) Zinc triggers microglial activation. J Neurosci 28:5827–5835. https://doi.org/10.1523/jneurosci.1236-08.2008
Kawabata T (2022) Iron-induced oxidative stress in human diseases. Cells. https://doi.org/10.3390/cells11142152
Kenkhuis B, van Eekeren M, Parfitt DA, Ariyurek Y, Banerjee P, Priller J, van der Weerd L, van Roon-Mom WMC (2022) Iron accumulation induces oxidative stress, while depressing inflammatory polarization in human iPSC-derived microglia. Stem Cell Reports 17:1351–1365. https://doi.org/10.1016/j.stemcr.2022.04.006
Khaliq ZM, Raman IM (2006) Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci 26:1935–1944. https://doi.org/10.1523/jneurosci.4664-05.2006
Khan M, Yoo SJ, Clijsters M, Backaert W, Vanstapel A, Speleman K, Lietaer C, Choi S, Hether TD, Marcelis L et al (2021) Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 184:5932-5949.e5915. https://doi.org/10.1016/j.cell.2021.10.027
Knaus UG (2021) Oxidants in physiological processes. Handb Exp Pharmacol 264:27–47. https://doi.org/10.1007/164_2020_380
Kovács G, Környei Z, Tóth K, Baranyi M, Brunner J, Neubrandt M, Dénes Á, Sperlágh B (2018) Modulation of P2X7 purinergic receptor activity by extracellular Zn(2+) in cultured mouse hippocampal astroglia. Cell Calcium 75:1–13. https://doi.org/10.1016/j.ceca.2018.07.010
Krall RF, Moutal A, Phillips MB, Asraf H, Johnson JW, Khanna R, Hershfinkel M, Aizenman E, Tzounopoulos T (2020) Synaptic zinc inhibition of NMDA receptors depends on the association of GluN2A with the zinc transporter ZnT1. Sci Adv. https://doi.org/10.1126/sciadv.abb1515
Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188. https://doi.org/10.1016/j.pneurobio.2013.11.006
Larvie DY, Perrin MT, Donati GL, Armah SM (2023) COVID-19 severity is associated with selenium intake among young adults with low selenium and zinc intake in North Carolina(1). Curr Dev Nutr. https://doi.org/10.1016/j.cdnut.2023.100044
Lavoie N, Jeyaraju DV, Peralta MR 3rd, Seress L, Pellegrini L, Tóth K (2011) Vesicular zinc regulates the Ca2+ sensitivity of a subpopulation of presynaptic vesicles at hippocampal mossy fiber terminals. J Neurosci 31:18251–18265. https://doi.org/10.1523/jneurosci.4164-11.2011
Lee JH, Han JH, Kim H, Park SM, Joe EH, Jou I (2019) Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun 7:68. https://doi.org/10.1186/s40478-019-0716-4
Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C et al (2017) Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. https://doi.org/10.1172/jci.insight.90777
Li M, Liu E, Zhou Q, Li S, Wang X, Liu Y, Wang L, Sun D, Ye J, Gao Y et al (2018) TRPC1 null exacerbates memory deficit and apoptosis induced by amyloid-β. J Alzheimers Dis 63:761–772. https://doi.org/10.3233/jad-180077
Li Y, Lu YX, Chi HL, Xiao T, Chen YM, Fu LY, Zibrila AI, Qi J, Li HB, Su Q et al (2021) Chronic blockade of NMDAR subunit 2A in the hypothalamic paraventricular nucleus alleviates hypertension through suppression of MEK/ERK/CREB pathway. Am J Hypertens 34:840–850. https://doi.org/10.1093/ajh/hpab047
Liang CF, Wu SH, Wang YL, Xu Z, Liu Y, Ren HT, Jia SY, Han X (2022a) The fast redox cycle of Cu(II)-Cu(I)-Cu(II) in the reduction of Cr(VI) by the Cu(II)-thiosulfate system. Chemosphere 293:133584. https://doi.org/10.1016/j.chemosphere.2022.133584
Liang D, Minikes AM, Jiang X (2022b) Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell 82:2215–2227. https://doi.org/10.1016/j.molcel.2022.03.022
Lin Y, Li LL, Nie W, Liu X, Adler A, Xiao C, Lu F, Wang L, Han H, Wang X et al (2019) Brain activity regulates loose coupling between mitochondrial and cytosolic Ca(2+) transients. Nat Commun 10:5277. https://doi.org/10.1038/s41467-019-13142-0
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79:1431–1568. https://doi.org/10.1152/physrev.1999.79.4.1431
Liu CC, Karimi Galougahi K, Weisbrod RM, Hansen T, Ravaie R, Nunez A, Liu YB, Fry N, Garcia A, Hamilton EJ et al (2013) Oxidative inhibition of the vascular Na+-K+ pump via NADPH oxidase-dependent β1-subunit glutathionylation: implications for angiotensin II-induced vascular dysfunction. Free Radic Biol Med 65:563–572. https://doi.org/10.1016/j.freeradbiomed.2013.06.040
Liu JT, Chen BY, Zhang JQ, Kuang F, Chen LW (2015) Lead exposure induced microgliosis and astrogliosis in hippocampus of young mice potentially by triggering TLR4-MyD88-NFκB signaling cascades. Toxicol Lett 239:97–107. https://doi.org/10.1016/j.toxlet.2015.09.015
Liu DL, Hong Z, Li JY, Yang YX, Chen C, Du JR (2021) Phthalide derivative CD21 attenuates tissue plasminogen activator-induced hemorrhagic transformation in ischemic stroke by enhancing macrophage scavenger receptor 1-mediated DAMP (peroxiredoxin 1) clearance. J Neuroinflammation 18:143. https://doi.org/10.1186/s12974-021-02170-7
López-Arrieta JM, Birks J (2000) Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.cd000147
Lu H, Xin Y, Tang Y, Shao G (2012) Zinc suppressed the airway inflammation in asthmatic rats: effects of zinc on generation of eotaxin, MCP-1, IL-8, IL-4, and IFN-γ. Biol Trace Elem Res 150:314–321. https://doi.org/10.1007/s12011-012-9493-7
Lu HW, Balmer TS, Romero GE, Trussell LO (2017) Slow AMPAR synaptic transmission is determined by stargazin and glutamate transporters. Neuron 96:73-80.e74. https://doi.org/10.1016/j.neuron.2017.08.043
Lu J, Zhou W, Dou F, Wang C, Yu Z (2021) TRPV1 sustains microglial metabolic reprogramming in Alzheimer’s disease. EMBO Rep 22:e52013. https://doi.org/10.15252/embr.202052013
Lu C, Tan C, Ouyang H, Chen Z, Yan Z, Zhang M (2022) Ferroptosis in intracerebral haemorrhage: a panoramic perspective of the metabolism, mechanism and theranostics. Aging Dis 13:1348–1364. https://doi.org/10.14336/ad.2022.01302
Mahroum N, Alghory A, Kiyak Z, Alwani A, Seida R, Alrais M, Shoenfeld Y (2022) Ferritin - from iron, through inflammation and autoimmunity, to COVID-19. J Autoimmun 126:102778. https://doi.org/10.1016/j.jaut.2021.102778
Maret W (2015) Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics 7:202–211. https://doi.org/10.1039/c4mt00230j
Maret W (2019) The redox biology of redox-inert zinc ions. Free Radic Biol Med 134:311–326. https://doi.org/10.1016/j.freeradbiomed.2019.01.006
Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C (2019) Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med 133:221–233. https://doi.org/10.1016/j.freeradbiomed.2018.09.033
McCarthy EK, Murray DM, Kiely ME (2022) Iron deficiency during the first 1000 days of life: are we doing enough to protect the developing brain? Proc Nutr Soc 81:108–118. https://doi.org/10.1017/s0029665121002858
McDaid J, Mustaly-Kalimi S, Stutzmann GE (2020) Ca(2+) dyshomeostasis disrupts neuronal and synaptic function in Alzheimer’s disease. Cells. https://doi.org/10.3390/cells9122655
McIntosh A, Mela V, Harty C, Minogue AM, Costello DA, Kerskens C, Lynch MA (2019) Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol 29:606–621. https://doi.org/10.1111/bpa.12704
McShane R, Westby MJ, Roberts E, Minakaran N, Schneider L, Farrimond LE, Maayan N, Ware J, Debarros J (2019) Memantine for dementia. Cochrane Database Syst Rev 3:Cd003154. https://doi.org/10.1002/14651858.cd003154.pub6
Mehta SL, Chokkalla AK, Bathula S, Arruri V, Chelluboina B, Vemuganti R (2023) CDR1as regulates α-synuclein-mediated ischemic brain damage by controlling miR-7 availability. Mol Ther Nucleic Acids 31:57–67. https://doi.org/10.1016/j.omtn.2022.11.022
Méndez R, Balanzá-Martínez V, Luperdi SC, Estrada I, Latorre A, González-Jiménez P, Bouzas L, Yépez K, Ferrando A, Reyes S et al (2022) Long-term neuropsychiatric outcomes in COVID-19 survivors: a 1-year longitudinal study. J Intern Med 291:247–251. https://doi.org/10.1111/joim.13389
Mesci P, de Souza JS, Martin-Sancho L, Macia A, Saleh A, Yin X, Snethlage C, Adams JW, Avansini SH, Herai RH et al (2022) SARS-CoV-2 infects human brain organoids causing cell death and loss of synapses that can be rescued by treatment with Sofosbuvir. PLoS Biol 20:e3001845. https://doi.org/10.1371/journal.pbio.3001845
Metwally E, Zhao G, Zhang YQ (2021) The calcium-dependent protease calpain in neuronal remodeling and neurodegeneration. Trends Neurosci 44:741–752. https://doi.org/10.1016/j.tins.2021.07.003
Moghaddam A, Heller RA, Sun Q, Seelig J, Cherkezov A, Seibert L, Hackler J, Seemann P, Diegmann J, Pilz M et al (2020) Selenium deficiency is associated with mortality risk from COVID-19. Nutrients 12:2098. https://doi.org/10.3390/nu12072098
Mohandass S, Ragavan M, Gnanasekaran D, Lakshmanan U, Dharmar P, Saha SK (2021) Overexpression of Cu/Zn superoxide dismutase (Cu/Zn SOD) in Synechococcus elongatus PCC 7942 for enhanced azo dye removal through hydrogen peroxide accumulation. Biology (basel). https://doi.org/10.3390/biology10121313
Muhammad JS, ElGhazali G, Shafarin J, Mohammad MG, Abu-Qiyas A, Hamad M (2022) SARS-CoV-2-induced hypomethylation of the ferritin heavy chain (FTH1) gene underlies serum hyperferritinemia in severe COVID-19 patients. Biochem Biophys Res Commun 631:138–145. https://doi.org/10.1016/j.bbrc.2022.09.083
Nadolni W, Immler R, Hoelting K, Fraticelli M, Ripphahn M, Rothmiller S, Matsushita M, Boekhoff I, Gudermann T, Sperandio M et al (2020) TRPM7 kinase is essential for neutrophil recruitment and function via regulation of Akt/mTOR signaling. Front Immunol 11:606893. https://doi.org/10.3389/fimmu.2020.606893
Nam LB, Keum YS (2020) Regulation of NRF2 by Na(+)/K(+)-ATPase: implication of tyrosine phosphorylation of Src. Free Radic Res 54:883–893. https://doi.org/10.1080/10715762.2020.1735633
Neveu D, Zucker RS (1996) Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron 16:619–629. https://doi.org/10.1016/s0896-6273(00)80081-1
Nolfi-Donegan D, Braganza A, Shiva S (2020) Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol 37:101674. https://doi.org/10.1016/j.redox.2020.101674
Ogawa Y, Kinoshita M, Shimada S, Kawamura T (2018) Zinc and skin disorders. Nutrients. https://doi.org/10.3390/nu10020199
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J et al (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11:1620. https://doi.org/10.1038/s41467-020-15562-9
Ou M, Jiang Y, Ji Y, Zhou Q, Du Z, Zhu H, Zhou Z (2022) Role and mechanism of ferroptosis in neurological diseases. Mol Metab 61:101502. https://doi.org/10.1016/j.molmet.2022.101502
Papazisis G, Pourzitaki C, Sardeli C, Lallas A, Amaniti E, Kouvelas D (2008) Deferoxamine decreases the excitatory amino acid levels and improves the histological outcome in the hippocampus of neonatal rats after hypoxia-ischemia. Pharmacol Res 57:73–78. https://doi.org/10.1016/j.phrs.2007.12.003
Pappa S, Ntella V, Giannakas T, Giannakoulis VG, Papoutsi E, Katsaounou P (2020) Prevalence of depression, anxiety, and insomnia among healthcare workers during the COVID-19 pandemic: a systematic review and meta-analysis. Brain Behav Immun 88:901–907. https://doi.org/10.1016/j.bbi.2020.05.026
Park HS, Yoo MH, Koh JY (2020) Role of zinc dyshomeostasis in inflammasome formation in cultured cortical cells following lipopolysaccharide or oxygen-glucose deprivation/reperfusion exposure. Neurobiol Dis 137:104771. https://doi.org/10.1016/j.nbd.2020.104771
Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, James LC, Lancaster MA (2020) SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell 27:951-961.e955. https://doi.org/10.1016/j.stem.2020.10.001
Pereira ME, Souza JV, Galiciolli MEA, Sare F, Vieira GS, Kruk IL, Oliveira CS (2022) Effects of selenium supplementation in patients with mild cognitive impairment or Alzheimer’s disease: a systematic review and meta-analysis. Nutrients 14:3250. https://doi.org/10.3390/nu14153205
Pezacki AT, Matier CD, Gu X, Kummelstedt E, Bond SE, Torrente L, Jordan-Sciutto KL, DeNicola GM, Su TA, Brady DC et al (2022) Oxidation state-specific fluorescent copper sensors reveal oncogene-driven redox changes that regulate labile copper(II) pools. Proc Natl Acad Sci USA 119:e2202736119. https://doi.org/10.1073/pnas.2202736119
Phuagkhaopong S, Ospondpant D, Kasemsuk T, Sibmooh N, Soodvilai S, Power C, Vivithanaporn P (2017) Cadmium-induced IL-6 and IL-8 expression and release from astrocytes are mediated by MAPK and NF-κB pathways. Neurotoxicology 60:82–91. https://doi.org/10.1016/j.neuro.2017.03.001
Pivovarov AS, Calahorro F, Walker RJ (2018) Na(+)/K(+)-pump and neurotransmitter membrane receptors. Invert Neurosci 19:1. https://doi.org/10.1007/s10158-018-0221-7
Poloni TE, Medici V, Moretti M, Visonà SD, Cirrincione A, Carlos AF, Davin A, Gagliardi S, Pansarasa O, Cereda C et al (2021) COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol 31:e12997. https://doi.org/10.1111/bpa.12997
Poonkuzhi Naseef P, Elayadeth-Meethal M, Mohammed Salim KT, Anjana A, Muhas C, Abdul Vajid K, Saheer Kuruniyan M (2022) Therapeutic potential of induced iron depletion using iron chelators in Covid-19. Saudi J Biol Sci 29:1947–1956. https://doi.org/10.1016/j.sjbs.2021.11.061
Pulido Perez P, Póndigo de Los Angeles JA, Perez Peralta A, Ramirez Mojica E, Torres Rasgado E, Hernandez-Hernandez ME, Romero JR (2022) Reduction in serum magnesium levels and renal function are associated with increased mortality in obese COVID-19 patients. Nutrients. https://doi.org/10.3390/nu14194054
Qu Y, Sun Y, Yang Z, Ding C (2022) Calcium ions signaling: targets for attack and utilization by viruses. Front Microbiol 13:889374. https://doi.org/10.3389/fmicb.2022.889374
Rao SS, Lago L, Volitakis I, Shukla JJ, McColl G, Finkelstein DI, Adlard PA (2021) Deferiprone treatment in aged transgenic tau mice improves Y-maze performance and alters tau pathology. Neurotherapeutics 18:1081–1094. https://doi.org/10.1007/s13311-020-00972-w
Rattanawong K, Koiso N, Toda E, Kinoshita A, Tanaka M, Tsuji H, Okamoto T (2021) Regulatory functions of ROS dynamics via glutathione metabolism and glutathione peroxidase activity in developing rice zygote. Plant J 108:1097–1115. https://doi.org/10.1111/tpj.15497
Ribeiro FM, Vieira LB, Pires RG, Olmo RP, Ferguson SS (2017) Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res 115:179–191. https://doi.org/10.1016/j.phrs.2016.11.013
Ribeiro DE, Oliveira-Giacomelli Á, Glaser T, Arnaud-Sampaio VF, Andrejew R, Dieckmann L, Baranova J, Lameu C, Ratajczak MZ, Ulrich H (2021) Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Mol Psychiatry 26:1044–1059. https://doi.org/10.1038/s41380-020-00965-3
Robinett NG, Culbertson EM, Peterson RL, Sanchez H, Andes DR, Nett JE, Culotta VC (2019) Exploiting the vulnerable active site of a copper-only superoxide dismutase to disrupt fungal pathogenesis. J Biol Chem 294:2700–2713. https://doi.org/10.1074/jbc.ra118.007095
Rotte A, Pasham V, Bhandaru M, Bobbala D, Zelenak C, Lang F (2012) Rapamycin sensitive ROS formation and Na(+)/H(+) exchanger activity in dendritic cells. Cell Physiol Biochem 29:543–550. https://doi.org/10.1159/000338508
Samudyata OAO, Malwade S, Rufino de Sousa N, Goparaju SK, Gracias J, Orhan F, Steponaviciute L, Schalling M, Sheridan SD et al (2022) SARS-CoV-2 promotes microglial synapse elimination in human brain organoids. Mol Psychiatry 27:3939–3950. https://doi.org/10.1038/s41380-022-01786-2
Scheiber IF, Mercer JF, Dringen R (2014) Metabolism and functions of copper in brain. Prog Neurobiol 116:33–57. https://doi.org/10.1016/j.pneurobio.2014.01.002
Schnetkamp PP (1995) Chelating properties of the Ca2+ transport site of the retinal rod Na-Ca+K exchanger: evidence for a common Ca2+ and Na+ binding site. Biochemistry 34:7282–7287. https://doi.org/10.1021/bi00021a045
Schulz K, Kroner A, David S (2012) Iron efflux from astrocytes plays a role in remyelination. J Neurosci 32:4841–4847. https://doi.org/10.1523/jneurosci.5328-11.2012
Schweizer U, Bohleber S, Zhao W, Fradejas-Villar N (2021) The neurobiology of selenium: looking back and to the future. Front Neurosci 15:652099. https://doi.org/10.3389/fnins.2021.652099
Scialo F, Daniele A, Amato F, Pastore L, Matera MG, Cazzola M, Castaldo G, Bianco A (2020) ACE2: the major cell entry receptor for SARS-CoV-2. Lung 198:867–877. https://doi.org/10.1007/s00408-020-00408-4
Scuri R, Lombardo P, Cataldo E, Ristori C, Brunelli M (2007) Inhibition of Na+/K+ ATPase potentiates synaptic transmission in tactile sensory neurons of the leech. Eur J Neurosci 25:159–167. https://doi.org/10.1111/j.1460-9568.2006.05257.x
Seidlmayer LK, Juettner VV, Kettlewell S, Pavlov EV, Blatter LA, Dedkova EN (2015) Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc Res 106:237–248. https://doi.org/10.1093/cvr/cvv097
Selim M (2010) Treatment with the iron chelator, deferoxamine mesylate, alters serum markers of oxidative stress in stroke patients. Transl Stroke Res 1:35–39. https://doi.org/10.1007/s12975-009-0001-0
Soung AL, Vanderheiden A, Nordvig AS, Sissoko CA, Canoll P, Mariani MB, Jiang X, Bricker T, Rosoklija GB, Arango V et al (2022) COVID-19 induces CNS cytokine expression and loss of hippocampal neurogenesis. Brain 145:4193–4201. https://doi.org/10.1093/brain/awac270
Specker E, Matthes S, Wesolowski R, Schütz A, Grohmann M, Alenina N, Pleimes D, Mallow K, Neuenschwander M, Gogolin A et al (2022) Structure-based design of xanthine-benzimidazole derivatives as novel and potent tryptophan hydroxylase inhibitors. J Med Chem 65:11126–11149. https://doi.org/10.1021/acs.jmedchem.2c00598
Stein LK, Mayman NA, Dhamoon MS, Fifi JT (2021) The emerging association between COVID-19 and acute stroke. Trends Neurosci 44:527–537. https://doi.org/10.1016/j.tins.2021.03.005
Straus MR, Tang T, Lai AL, Flegel A, Bidon M, Freed JH, Daniel S, Whittaker GR (2020) Ca(2+) ions promote fusion of middle east respiratory syndrome coronavirus with host cells and increase infectivity. J Virol. https://doi.org/10.1128/jvi.00426-20
Sun Y, Zhang L, Chen Y, Zhan L, Gao Z (2015) Therapeutic targets for cerebral ischemia based on the signaling pathways of the GluN2B C terminus. Stroke 46:2347–2353. https://doi.org/10.1161/strokeaha.115.009314
Sun R, Wang J, Feng J, Cao B (2022) Zinc in cognitive impairment and aging. Biomolecules. https://doi.org/10.3390/biom12071000
Suriawinata E, Mehta KJ (2022) Iron and iron-related proteins in COVID-19. Clin Exp Med. https://doi.org/10.1007/s10238-022-00851-y
Tan LL, Jiang XL, Xu LX, Li G, Feng CX, Ding X, Sun B, Qin ZH, Zhang ZB, Feng X et al (2021) TP53-induced glycolysis and apoptosis regulator alleviates hypoxia/ischemia-induced microglial pyroptosis and ischemic brain damage. Neural Regen Res 16:1037–1043. https://doi.org/10.4103/1673-5374.300453
Tanaka KI, Kawahara M (2017) Copper enhances zinc-induced neurotoxicity and the endoplasmic reticulum stress response in a neuronal model of vascular dementia. Front Neurosci 11:58. https://doi.org/10.3389/fnins.2017.00058
Taniguchi K, Yamamoto F, Amano A, Tamaoka A, Sanjo N, Yokota T, Kametani F, Araki W (2022) Amyloid-β oligomers interact with NMDA receptors containing GluN2B subunits and metabotropic glutamate receptor 1 in primary cortical neurons: relevance to the synapse pathology of Alzheimer’s disease. Neurosci Res 180:90–98. https://doi.org/10.1016/j.neures.2022.03.001
Tao W, Lee J, Chen X, Díaz-Alonso J, Zhou J, Pleasure S, Nicoll RA (2021) Synaptic memory requires CaMKII. Elife. https://doi.org/10.7554/elife.60360
Thakur KT, Miller EH, Glendinning MD, Al-Dalahmah O, Banu MA, Boehme AK, Boubour AL, Bruce SS, Chong AM, Claassen J et al (2021) COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 144:2696–2708. https://doi.org/10.1093/brain/awab148
Thapak P, Bishnoi M, Sharma SS (2022) Tranilast, a transient receptor potential vanilloid 2 channel (TRPV2) inhibitor attenuates amyloid β-induced cognitive impairment: possible mechanisms. Neuromolecular Med 24:183–194. https://doi.org/10.1007/s12017-021-08675-x
Thiriveedi VR, Mattam U, Pattabhi P, Bisoyi V, Talari NK, Krishnamoorthy T, Sepuri NBV (2020) Glutathionylated and Fe-S cluster containing hMIA40 (CHCHD4) regulates ROS and mitochondrial complex III and IV activities of the electron transport chain. Redox Biol 37:101725. https://doi.org/10.1016/j.redox.2020.101725
Thirupathi A, Chang YZ (2019) Brain Iron metabolism and CNS diseases. Adv Exp Med Biol 1173:1–19. https://doi.org/10.1007/978-981-13-9589-5_1
Tlili A, Corcoll N, Bonet B, Morin S, Montuelle B, Bérard A, Guasch H (2011) In situ spatio-temporal changes in pollution-induced community tolerance to zinc in autotrophic and heterotrophic biofilm communities. Ecotoxicology 20:1823–1839. https://doi.org/10.1007/s10646-011-0721-2
Tone D, Ode KL, Zhang Q, Fujishima H, Yamada RG, Nagashima Y, Matsumoto K, Wen Z, Yoshida SY, Mitani TT et al (2022) Distinct phosphorylation states of mammalian CaMKIIβ control the induction and maintenance of sleep. PLoS Biol 20:e3001813. https://doi.org/10.1371/journal.pbio.3001813
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler RD et al (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375:1254–1261. https://doi.org/10.1126/science.abf0529
Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W et al (2010) DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140:222–234. https://doi.org/10.1016/j.cell.2009.12.055
Tuo QZ, Masaldan S, Southon A, Mawal C, Ayton S, Bush AI, Lei P, Belaidi AA (2021) Characterization of selenium compounds for anti-ferroptotic activity in neuronal cells and after cerebral ischemia-reperfusion injury. Neurotherapeutics 18:2682–2691. https://doi.org/10.1007/s13311-021-01111-9
Tuo QZ, Liu Y, Xiang Z, Yan HF, Zou T, Shu Y, Ding XL, Zou JJ, Xu S, Tang F et al (2022) Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct Target Ther 7:59. https://doi.org/10.1038/s41392-022-00917-z
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344. https://doi.org/10.1113/jphysiol.2003.049478
Uginet M, Breville G, Assal F, Lövblad KO, Vargas MI, Pugin J, Serratrice J, Herrmann FR, Lalive PH, Allali G (2021) COVID-19 encephalopathy: clinical and neurobiological features. J Med Virol 93:4374–4381. https://doi.org/10.1002/jmv.26973
Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, González-Billault C, Núñez MT (2013) Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem 126:541–549. https://doi.org/10.1111/jnc.12244
Villadiego J, Labrador-Garrido A, Franco JM, Leal-Lasarte M, De Genst EJ, Dobson CM, Pozo D, Toledo-Aral JJ, Roodveldt C (2018) Immunization with α-synuclein/Grp94 reshapes peripheral immunity and suppresses microgliosis in a chronic Parkinsonism model. Glia 66:191–205. https://doi.org/10.1002/glia.23237
Vinceti M, Filippini T, Fairweather-Tait S (2022) A potential role for zinc to enhance treatment for COVID-19? Clin Infect Dis. https://doi.org/10.1093/cid/ciac849
Voelkl J, Tuffaha R, Luong TTD, Zickler D, Masyout J, Feger M, Verheyen N, Blaschke F, Kuro OM, Tomaschitz A et al (2018) Zinc inhibits phosphate-induced vascular calcification through TNFAIP3-mediated suppression of NF-κB. J Am Soc Nephrol 29:1636–1648. https://doi.org/10.1681/asn.2017050492
Vogler NW, Betti VM, Goldberg JM, Tzounopoulos T (2020) Mechanisms underlying long-term synaptic zinc plasticity at mouse dorsal cochlear nucleus glutamatergic synapses. J Neurosci 40:4981–4996. https://doi.org/10.1523/jneurosci.0175-20.2020
Wang Z, Van den Berg RJ, Ypey DL (1994) Resting membrane potentials and excitability at different regions of rat dorsal root ganglion neurons in culture. Neuroscience 60:245–254. https://doi.org/10.1016/0306-4522(94)90218-6
Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, Wei D, Zhang Y, Sun XX, Gong L et al (2020) CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther 5:283. https://doi.org/10.1038/s41392-020-00426-x
Wang S, de Fabritus L, Kumar PA, Werner Y, Ma M, Li D, Siret C, Simic M, Li B, Kerdiles YM et al (2023) Brain endothelial CXCL12 attracts protective natural killer cells during ischemic stroke. J Neuroinflammation 20:8. https://doi.org/10.1186/s12974-023-02689-x
Weilinger NL, Lohman AW, Rakai BD, Ma EM, Bialecki J, Maslieieva V, Rilea T, Bandet MV, Ikuta NT, Scott L et al (2016) Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci 19:432–442. https://doi.org/10.1038/nn.4236
Weinreb O, Mandel S, Youdim MBH, Amit T (2013) Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic Biol Med 62:52–64. https://doi.org/10.1016/j.freeradbiomed.2013.01.017
Wen Y, Chen H, Zhang L, Wu M, Zhang F, Yang D, Shen J, Chen J (2021) Glycyrrhetinic acid induces oxidative/nitrative stress and drives ferroptosis through activating NADPH oxidases and iNOS, and depriving glutathione in triple-negative breast cancer cells. Free Radic Biol Med 173:41–51. https://doi.org/10.1016/j.freeradbiomed.2021.07.019
Weydert CJ, Cullen JJ (2010) Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc 5:51–66. https://doi.org/10.1038/nprot.2009.197
Wicher D, Berlau J, Walther C, Borst A (2006) Peptidergic counter-regulation of Ca(2+)- and Na(+)-dependent K(+) currents modulates the shape of action potentials in neurosecretory insect neurons. J Neurophysiol 95:311–322. https://doi.org/10.1152/jn.00904.2005
Williams TL, Serpell LC, Urbanc B (2016) Stabilization of native amyloid β-protein oligomers by copper and hydrogen peroxide induced cross-linking of unmodified proteins (CHICUP). Biochim Biophys Acta 1864:249–259. https://doi.org/10.1016/j.bbapap.2015.12.001
Wollmuth LP, Kuner T, Sakmann B (1998) Intracellular Mg2+ interacts with structural determinants of the narrow constriction contributed by the NR1-subunit in the NMDA receptor channel. J Physiol 506(Pt 1):33–52. https://doi.org/10.1111/j.1469-7793.1998.00033.x
Wu CT, Lidsky PV, Xiao Y, Lee IT, Cheng R, Nakayama T, Jiang S, Demeter J, Bevacqua RJ, Chang CA et al (2021) SARS-CoV-2 infects human pancreatic β cells and elicits β cell impairment. Cell Metab 33:1565-1576.e1565. https://doi.org/10.1016/j.cmet.2021.05.013
Wu B, Li P, Hong X, Xu C, Wang R, Liang Y (2022) The receptor-like cytosolic kinase RIPK activates NADP-malic enzyme 2 to generate NADPH for fueling ROS production. Mol Plant 15:887–903. https://doi.org/10.1016/j.molp.2022.03.003
Xu E, Xie Y, Al-Aly Z (2022) Long-term neurologic outcomes of COVID-19. Nat Med 28:2406–2415. https://doi.org/10.1038/s41591-022-02001-z
Yamanaka R, Shindo Y, Oka K (2019) Magnesium is a key player in neuronal maturation and neuropathology. Int J Mol Sci. https://doi.org/10.3390/ijms20143439
Yan J, Bengtson CP, Buchthal B, Hagenston AM, Bading H (2020) Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science. https://doi.org/10.1126/science.aay3302
Yao H, Peng F, Dhillon N, Callen S, Bokhari S, Stehno-Bittel L, Ahmad SO, Wang JQ, Buch S (2009) Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J Neurosci 29:1657–1669. https://doi.org/10.1523/jneurosci.2781-08.2009
Ye L, Zeng Q, Ling M, Ma R, Chen H, Lin F, Li Z, Pan L (2021) Inhibition of IP3R/Ca2+ dysregulation protects mice from ventilator-induced lung injury via endoplasmic reticulum and mitochondrial pathways. Front Immunol 12:729094. https://doi.org/10.3389/fimmu.2021.729094
Zelko IN, Mariani TJ, Folz RJ (2002) Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 33:337–349. https://doi.org/10.1016/s0891-5849(02)00905-x
Zhang T, Lu X, Li J, Chidiac P, Sims SM, Feng Q (2012) Inhibition of Na/K-ATPase promotes myocardial tumor necrosis factor-alpha protein expression and cardiac dysfunction via calcium/mTOR signaling in endotoxemia. Basic Res Cardiol 107:254. https://doi.org/10.1007/s00395-012-0254-8
Zhang K, Wang H, Xu M, Frank JA, Luo J (2018) Role of MCP-1 and CCR2 in ethanol-induced neuroinflammation and neurodegeneration in the developing brain. J Neuroinflammation 15:197. https://doi.org/10.1186/s12974-018-1241-2
Zhang J, Saad R, Taylor EW, Rayman MP (2020) Selenium and selenoproteins in viral infection with potential relevance to COVID-19. Redox Biol 37:101715. https://doi.org/10.1016/j.redox.2020.101715
Zhang H, Ostrowski R, Jiang D, Zhao Q, Liang Y, Che X, Zhao J, Xiang X, Qin W, He Z (2021a) Hepcidin promoted ferroptosis through iron metabolism which is associated with DMT1 signaling activation in early brain injury following subarachnoid hemorrhage. Oxid Med Cell Longev 2021:9800794. https://doi.org/10.1155/2021/9800794
Zhang L, Zhou L, Bao L, Liu J, Zhu H, Lv Q, Liu R, Chen W, Tong W, Wei Q et al (2021b) SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther 6:337. https://doi.org/10.1038/s41392-021-00719-9
Zhang YY, Yang XY, Liu HQ, Zhang Z, Hu CP, Peng J, Luo XJ (2022a) The weakened interaction between HECTD4 and GluN2B in ischemic stroke promotes calcium overload and brain injury through a mechanism involving the decrease of GluN2B and MALT1 ubiquitination. Mol Neurobiol. https://doi.org/10.1007/s12035-022-03169-8
Zhang Z, Jiang J, He Y, Cai J, Xie J, Wu M, Xing M, Zhang Z, Chang H, Yu P et al (2022b) Pregabalin mitigates microglial activation and neuronal injury by inhibiting HMGB1 signaling pathway in radiation-induced brain injury. J Neuroinflammation 19:231. https://doi.org/10.1186/s12974-022-02596-7
Zhou L, Li F, Xu HB, Luo CX, Wu HY, Zhu MM, Lu W, Ji X, Zhou QG, Zhu DY (2010) Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med 16:1439–1443. https://doi.org/10.1038/nm.2245
Zhu D, Su Y, Fu B, Xu H (2018) Magnesium reduces blood-brain barrier permeability and regulates amyloid-β transcytosis. Mol Neurobiol 55:7118–7131. https://doi.org/10.1007/s12035-018-0896-0
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R et al (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. https://doi.org/10.1056/nejmoa2001017
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 82073849 to Xiu-Ju Luo, China, No. 82204389 to Kai-Di Ren, No. 82173815 to Jun Peng, China), Natural Science Foundation of Hunan Province, China (No. 2021JJ30032 to Xiu-Ju Luo).
Funding
The study was supported by National Natural Science Foundation of China (grant nos. 82073849, 82204389, 82173815) and Natural Science Foundation of Hunan Province (grant no. 2021JJ30032).
Author information
Authors and Affiliations
Contributions
Yi-Yue Zhang, Jun Peng and Xiu-Ju Luo conceived this review. Yi-Yue Zhang and Kai-Di Ren wrote the manuscript. Jun Peng and Xiu-Ju Luo revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Consent for publication
This publication is approved by all the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, YY., Ren, KD., Luo, XJ. et al. COVID-19-induced neurological symptoms: focus on the role of metal ions. Inflammopharmacol 31, 611–631 (2023). https://doi.org/10.1007/s10787-023-01176-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-023-01176-2