
Abstract
Landscape genetics affords a potential analysis framework to evaluate the effect of contemporary land use on endangered species at a population level. However, historical patterns of population connectivity need to be accounted for prior to testing for the contemporary effect of threats. The goals of this study were to (1) optimally describe historical patterns in population connectivity for a diadromous fish species before (2) evaluating whether residual genetic variation was correlated with ecological changes arising from several types of land use. Using endangered Atlantic salmon populations as a case study, we evaluated whether historical patterns in population connectivity were more likely to result from dispersal limitation (isolation by distance) relative to habitat choice and reproductive success (isolation by environment). Second, we used Reciprocal Causal Modeling to identify the types of land use contributing to three threat indices, and subsequently Multiple Regression on Distance Matrices to evaluate the relative severity of each. These analyses suggest that straying Atlantic salmon avoid watersheds with reduced water quality (resulting from acidification and abandoned mines) and higher road density, yet are not responding to watershed fragmentation (from road-river crossings and dams) at a population level. This study is among the first to explicitly compare alternate behavioural hypotheses leading to dispersal patterns for diadromous fishes and to quantitatively assess freshwater threats for Atlantic salmon at a population level using landscape genetics.
Similar content being viewed by others
Introduction
Anthropogenic changes to the environment, particularly in terms of land use, are one of the major causes of species decline worldwide (Foley et al. 2005; Dudgeon et al. 2006; Venter et al. 2006). Effective recovery planning to address these declines relies on understanding the relative importance and severity of multiple threats in order to prioritize among potential recovery actions (Norris 2004). However, in many situations the quantitative links between population-level productivity and the specific land use activities identified as threats are not known (Lawler et al. 2002; Roni et al. 2002). For example, forestry, urbanization, agriculture, roads, industrial corridors or mining activities have all been shown to be related to processes governing sedimentation rates, hydrological flows, temperature regimes and other environmental characteristics of watersheds (Allan 2004; Poff et al. 2010). The relationships between such environmental processes (e.g. sedimentation) and changes in freshwater fish survival or other vital rates are often understood quantitatively (e.g. Bunn and Arthington 2002), yet typically cannot be linked back quantitatively to the underlying threat (e.g. extent of forestry activity; Lawler et al. 2002). This means that while it is possible to understand the relative severity of a single threat among watersheds, it is not possible to assess the relative severity of multiple threats affecting a single population within a watershed. Even when abundance time series are available, land use patterns and other types of spatial data on threats are typically aggregated over multiple years (due to the time and labour-intensive nature of data collection) thus preventing time series analyses with population abundance. Additionally, different threats can be measured in different units (e.g. proportions, counts, or densities), complicating direct comparison. For these reasons, alternate ways of evaluating multiple concurrent threats and their influence on populations are needed.
One possibility is to use landscape genetics to investigate whether or not specific threats influence population genetic structuring. Measured variation at neutral markers like microsatellites largely arises from within-population genetic drift causing divergence, tempered by the homogenizing effects of gene flow among populations (Allendorf et al. 2013). Threats that have fragmented populations or caused substantial reductions in abundance would be expected to reduce the rate of gene flow (fewer available migrants) while concurrently increasing the rate of genetic drift (increased isolation among small or declining populations) in a manner potentially dependent on the extent of the threat. One well-studied example is fragmentation of populations in response to roads, where roads have been found to increase genetic isolation of adjacent populations by acting as a barrier to movement (e.g. Arens et al. 2007; Clark et al. 2010; Epps et al. 2005). Such population-level responses to contemporary threats are expected to be relatively rapid. Simulation studies have found that contemporary landscape changes leading to reduced population connectivity are detectable from microsatellite variation in as little as 1–5 generations (longer if dispersal rates are extremely low), provided dispersal distances are sufficiently large (Landguth et al. 2010) or population size is low (Dileo et al. 2013). This suggests that the influence of recent threats on populations would be detectable even if the threats had been acting for a relatively short period of time.
Identifying contemporary threats relies on accurately characterizing historical patterns of gene flow (i.e. those existing prior to the advent of the threat), in order to prevent biased inference (Singh et al. 2013). Therefore, understanding how landscape elements influence the dispersal process becomes paramount to understanding expected patterns of population connectivity prior to the advent of threats. The majority of research questions in landscape genetics focus on the importance of landscape elements occurring between the locations or populations of interest (Pfluger and Balkenhol 2014). Resistance surfaces that describe the tendency or ability of individuals to move through this landscape matrix are compared to genetic variability when evaluating spatial genetic structure (Sawyer et al. 2011; Zeller et al. 2012; Spear et al. 2010). More geographically proximate habitats or populations are expected to be more genetically similar owing to a reduction in the number of migrants with increasing distance (Guillot et al. 2009). Ecologically, this isolation by distance (IBD) pattern is thought to result from cumulative fitness costs associated with movement or dispersal limitation, thereby making long-distance dispersal events rare (Bowler and Benton 2005; Bonte et al. 2012; Sexton et al. 2013). For non-migratory species, where individual dispersal distances are small relative to the area occupied by the population, it might be expected that IBD would be a good approximation of historical population connectivity (Guillot et al. 2009). However, migratory species like diadromous fishes are unlikely to be regulated by cumulative fitness costs associated with movement among spawning habitat, given that lifetime dispersal distances are orders of magnitude greater than the area occupied by breeding or reproducing individuals. An alternative model for historical patterns in gene flow could be an Isolation by Environment or IBE model, which predicts that gene flow is greatest among similar environments owing to either non-random mating or local adaptation (Sexton et al. 2013; Wang et al. 2013). In this instance, genetic variability among populations would be expected to be related to the degree of similarity in recipient environments (promoting local adaptation) or possibly to the degree of difference (counter-gradient gene flow against local adaptation) (Sexton et al. 2013). To maximize reproductive fitness, dispersing individuals would be expected to respond to the environmental characteristics of recipient habitats through habitat selection (Kubisch et al. 2013) rather than to the characteristics of the matrix between habitats.
The overall goals of this study were to use a landscape genetics approach to: (1) evaluate the IBD and IBE hypotheses relative to population structuring in a diadromous fish species and (2) evaluate whether residual genetic variation could be explained relative to the magnitude or importance of multiple contemporary threats. To achieve this, we used a case study of genetic variation among 11 endangered Atlantic salmon (Salmo salar) populations, where the results could be used directly to facilitate recovery planning. These analyses are structured into four main parts. The first part describes the methods used to obtain and summarize the genetic and landscape data sources, which were transformed into genetic and environmental distance matrices for subsequent analyses. The second component compares the relative support for the IBD and IBE hypotheses of gene flow. The third part explores whether residual genetic variation could be explained by anthropogenic threats, which relied on developing optimal indicators of three general categories of anthropogenic effects (those related to sedimentation, fragmentation and water quality). The fourth component evaluated the relative magnitude and significance of any correlation between residual genetic variation and each threat category. From a theoretical standpoint, the results potentially provide insight into the behavioural processes that may be leading to gene flow among watersheds for Atlantic salmon. From a practical standpoint, they also provide one of the first quantitative comparisons of the relative magnitude of threats among these populations, and can be used to facilitate recovery planning.
Methods
Study area
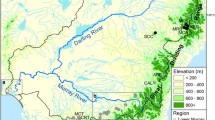
The Southern Upland (SU) region of Nova Scotia includes all rivers that flow into the Atlantic Ocean from Cape Split to the Canso causeway (Fig. 1) and contains 72 watersheds thought to support (or to have previously supported) distinct Atlantic salmon populations (Bowlby et al. 2014). The region is relatively homogeneous in terms of watershed characteristics as well as Atlantic salmon life history characteristics (Amiro et al. 2006; Chaput et al. 2006), and thus is considered to be distinct from population assemblages in other areas (COSEWIC 2010). Population dynamics research (Gibson and Bowlby 2013) as well as ongoing abundance declines and evidence of extirpations (Gibson et al. 2011) all indicate that extinction risk is high. Collectively, the Southern Upland population assemblage has been designated as endangered by the Committee on the Status of Endangered Wildlife in Canada (COSEWIC 2010). This study uses data from 11 SU watersheds (Fig. 1). These tend to be the larger systems and are well distributed throughout the region (i.e. they are not clustered geographically). As such, they are thought to largely represent the range of variability that exists among populations.

Geographical boundaries of the watersheds (coloured) containing the Atlantic salmon populations and rivers sampled for this study as well as other watersheds (grey) within the Southern Upland region of Nova Scotia. The two Salmon Rivers are in Digby County and Guysborough County, respectively
Part 1: genetic and landscape data sources
For microsatellite amplification, a total of 709 genetic samples from juveniles (young-of-the-year, age 1 or age 2) were collected opportunistically during electrofishing surveys, with the timing of each collection and the number of samples varying slightly among rivers (Table 1). During data collection, individual samples were identified by the river of origin instead of by individual, which necessitated an analysis based on populations. Older juveniles were sampled preferentially to minimize the likelihood of multiple sampling of individual families. DNA was extracted and purified from fin clips stored in 95 % ethanol using DNeasy tissue kits (QIAGEN), following the manufacturer’s specifications. Microsatellite amplification was carried out at 17 individual loci (O’Reilly et al. 2012), with references for the primer sequences given in the Supplementary Information (file: S1 Microsat validation.xlsx). Polymerase Chain Reaction (PCR) amplifications were carried out for each locus separately, using the following thermal cycling conditions: one step of 94 °C for 3 min; nine steps of 94 °C for 45 s, 58 °C for 30 s, and 74 °C for 1 min; 27 steps of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min; and lastly a 30-min extension step at 72 °C. Alleles from individual loci were size-fractionated without purification; products from multiple loci had salt, unincorporated dNTP’s and non-labeled primers removed using either MinElute plates (QIAGEN; as specified by the manufacturer) or ethanol precipitation methods, prior to size fractionation by denaturing gradient electrophoresis. Alleles were detected using either an MJ Research Base station or an Applied Biosystems 3130XL. In each batch of 84 samples, we included a positive control of 2 standard samples with known allele sizes across all loci. In conjunction with allelic ladder standards, these controls were used to ensure consistency among batches, days and platforms in the size determination of fragments. Additionally, 10 samples from each batch of 84 (12 %) were analyzed twice to identify potential sample placement errors, strip inversions, plate inversions and other laboratory mistakes.
From the microsatellite dataset, the potential for null alleles (non-amplified alleles that result in apparent homozygotes), allele dropout (preferential amplification of short alleles) and scoring errors due to stuttering (failure to discriminate adjacent alleles) was assessed using Micro-checker version 2.2.3 (van Oosterhout et al. 2004). We used 8000 Monte-Carlo randomizations of the observed data to calculate the p values for the χ 2 test of observed versus predicted homozygote frequencies and heterozygote size differences. Based on this analysis, there was no evidence for scoring error or allele dropout, yet null alleles were predicted in 5 populations at Locus 9 (Supplementary file: S1 Microsat validation.xlsx). We also tested for Hardy-Weinburg (HW) equilibrium over loci in all sampled populations using the R package ‘adegenet’ (Jombart 2008). At each locus within each population, 8000 Monte-Carlo simulations were used to calculate p values for the χ 2 test of observed versus predicted gene frequencies. After Bonferroni correction, significant departures and any patterns of departures over loci or within populations were assessed (Supplementary file: S1 Microsat validation.xlsx). Potential linkage disequilibrium (non-random association of alleles at multiple loci) was assessed using LinkDos (Garnier-Gere and Dillmann 1992) from GenePop 4.2 (Raymond and Rousset 1995; Rousset 2008) (Supplementary file: S1 Microsat validation.xlsx). After the above (further detail given in the Results section), five within-population measures of genetic variability (averaged over loci) were calculated to better understand the characteristics of individual populations: allelic richness (i.e. the number of alleles), the effective number of alleles (allelic richness standardized to account for sample size), expected heterozygosity (the expected probability that an individual will be heterozygous at a given locus), observed heterozygosity, and Wright’s inbreeding coefficient (Fis) within the R package ‘gstudio’ (Dyer 2012).
For watershed characterization, freely available data sources describing the physical, geological and land use characteristics of the watersheds sampled in this study were combined and analyzed using ArcGIS (ESRI® ArcGIS 10.0 software service pack 3). A detailed description of the GIS methods is provided in Bowlby et al. (2014). In brief, we first created a topographically connected hydrological network for each watershed (i.e. one that represented the direction of water flow) and delineated the drainage area of each stream network as the watershed boundary. Intersecting a Digital Elevation Model with the hydrological network enabled topographical evaluation of each watershed (i.e. stream segments could be characterized by gradient). Several additional data layers were intersected with watershed boundaries or stream segments to calculate: (1) the proportion of multiple geological features within a watershed (e.g. surficial geology types), (2) the proportion of watershed area affected by multiple land use types (e.g. forestry) or (3) the prevalence (i.e. a count) of a specific type of threat (e.g. road/river crossings) in each watershed. In the case of counts, values used in these analyses were expressed as a density, scaled either by the area of the watershed or by the length of the stream network. The sources of geographic data that were used are provided in Table S1, while the calculated landscape variables describing the watersheds are provided in Table S2 and the land use activities (i.e. threats) in Table S3 of the Supplementary Information.
It was necessary to determine the environmental metrics that would be most likely to influence habitat quality for Atlantic salmon in order to parameterize the IBE model. Given the hierarchical nature of watersheds, where landscape characteristics affect processes at successively smaller scales, (Allan 2004), it is thought that the evolution of life history differences as well as population dynamics of salmon are primarily governed by large-scale characteristics of watersheds (Ugedal and Finstad 2011). Therefore, the data types used in the IBE model included: surficial and bedrock geology, natural forest disturbance regimes, area metrics (e.g. watershed area, length of stream network), and topography (Table S2). To avoid weighting subsequent analyses due to different numbers of variables contributing to each information type (e.g. 4 variables describe natural disturbance regime while 14 describe surficial geology), each of the 5 data types were scaled to ensure comparability and then independently transformed into two non-metric multidimensional scaling (NMDS) axes (Bocard et al. 2011) using the metaMDS function of the R package ‘vegan’ (Oksanen et al. 2015). The Euclidean distance matrix representing landscape variation among watersheds was calculated from these 10 NMDS axes (i.e. the sum of 2 axes representing each of 5 data types).
Part 2: historical patterns of gene flow
If dispersal limitation was the primary determinant of gene flow among populations (as predicted by the IBD hypothesis), it would be expected that the geographic distance among watersheds would be positively correlated with the degree of genetic difference among populations (Guillot et al. 2009). Alternately, if habitat selection and local adaptation to recipient environments was the primary determinant of gene flow (as predicted by the IBE hypothesis), a stronger correlation may exist between environmental similarity and genetic distance (Sexton et al. 2013). It would also be expected that the better model would have significant explanatory power after any correlation with the other hypothesis was accounted for. To evaluate these hypotheses, we first developed genetic, geographical and environmental distance matrices and compared those using Mantel tests and two-step Reciprocal Causal Modeling. The Mantel tests evaluate whether a single model can explain a significant amount of genetic variation. The Reciprocal modeling evaluates whether IBD or IBE can be considered significantly better than the other model. Looking forward, there are many exciting statistical techniques that are being proposed as more appropriate and rigorous alternatives to Mantel and partial Mantel tests for evaluating IBD relative to IBE as well as other questions in landscape genetics. These include Generalized Dissimilarity Modelling of distance matrices (Ferrier et al. 2007; Fitzpatrick and Keller 2015), spatial regression methods based on conditional or simultaneous autoregressive models of individual-based data (Wagner and Fortin 2016), and Bayesian methods like BEDASSLE that model the covariance in allele frequencies from SNPs as functions of geographic or environmental data (Bradburd et al. 2013), among others. Unfortunately, none of these could have been applied here as they require a much more comprehensive data set than was available.
To evaluate the strength of the correlation between genetic distance and either the IBD or IBE models relative to a null distribution of no association, we used Mantel tests calculated from 10,000 permutations of the data, where significance was assessed relative to the 95 % quantile of the distribution of permutations (i.e. the test statistic would be significant only if it was greater than 95 % of the permuted values). The genetic distance matrix was calculated from Nei’s pairwise Fst (Nei 1978) using the R package ‘gstudio’ (Dyer 2012) (Supplementary file: S4 Distance matrices.xlsx). This distance is given by:
where \(\widehat{{G_{x} }}\), \(\widehat{{G_{y} }}\) and \(\widehat{{G_{xy} }}\) represent the bias-corrected averages of allele frequencies over the r loci sampled (Nei 1978). The correction prevents overestimating distances based on observed gene frequencies when sample sizes are small. For this model, differentiation among populations is assumed to arise through constant mutation rates and genetic drift; however, sensitivity to this assumption for our analyses was assessed by substituting Bray-Curtis dissimilarity (equivalent of 1 minus the proportion of shared alleles) as in Castillo et al. (2014). The Euclidean matrix representing pairwise geographical distances (IBD model) was calculated from the coastal straight-line distance between each pair of rivers, which represents a minimum distance over water; beginning and ending on the latitude and longitude between each pair of river mouths (Supplementary file: S4 Distance matrices.xlsx). The Euclidean matrix representing pairwise environmental distances (IBE model) was calculated from the NMDS axes described above (Supplementary file: S4 Distance matrices.xlsx). This Isolation by Environment (IBE) model explicitly did not include any positional information representing the geographical location of watersheds. The IBD and IBE distance matrices were highly correlated (Spearman correlation coefficient = 0.92), which was expected because environmental gradients tend to be spatially autocorrelated (Legendre 1993).
Second, we used the two-step Reciprocal Causal Modeling framework proposed by Wasserman et al. (2010), in which competing models (IBD or IBE) are compared directly to each-other using partial Mantel tests based on the same distance matrices as above. Reciprocal Causal Modeling has been advocated as a method for formal significance testing among highly correlated distance matrices (Wasserman et al. 2010; Cushman et al. 2006). Previous evaluation of the technique using simulated data has suggested that it is efficient in identifying the correct model from a range of alternatives in individual-based landscape genetic analyses (Cushman et al. 2013). However, it suffers from elevated Type I error rates, as do most analyses on highly correlated data (Balkenhol et al. 2009; Legendre 1993). The magnitude of Type I error can be related to the degree of second-order autocorrelation (i.e. the covariance function between observations) in the matrices being compared (Guillot and Rousset 2013); however, as advocated by Cushman et al. (2013), we have used the relative magnitudes of the Mantel r values for assessing the comparative support among models (e.g. Castillo et al. 2014), rather than the predicted significance. Reciprocal Causal Modeling works by comparing a partial Mantel test of a given hypothesis after parcelling out the influence of a competing hypothesis and vice versa (e.g. comparing the r statistic of Gene ~ Model 1|Model 2 with Gene ~ Model 2|Model 1). The null hypothesis for these partial Mantel tests is that there is no additional variation captured in the second distance matrix relative to the first. The expectation would be that a better model would have a large and significant r value relative to the alternate hypotheses in the first instance, while the r value would be small and non-significant in the second instance (although see comments related to Type I error above). In other words, competing models would not show a significant correlation with genetic structure after the influence of the chosen model was accounted for and vice versa (Wasserman et al. 2010; Cushman et al. 2013). All Mantel tests and partial Mantel tests were carried out using the R package ‘vegan’ (Oksanen et al. 2015).
Part 3: identification of variables contributing to threats indices
In general, the land use activities identified as threats to Atlantic salmon have been linked to changes in the environmental processes governing watersheds, rather than to changes in population dynamics (Allan 2004). In addition, different types of threats are expected to have the same overall effect on environmental processes, as detailed in the Introduction. Therefore, to quantitatively assess the influence of threats on genetic connectivity among populations, it becomes necessary to first determine which environmental process a threat is most likely to contribute to, and then to assess the relative magnitude of effect from multiple environmental processes. Here we consider a suite of eight land use variables as potential threats and evaluate their independent contributions to three more general environmental processes: sedimentation (S), fragmentation (F) and water quality (WQ) (hereafter called the three threats indices; Table 2). This was done both as a method for variable reduction and as a way to group activities that would be expected to influence populations in similar ways. Albeit indirectly, retention of a specific land use variable in a threat index provides support for both the hypotheses on how specific land use types influence populations as well as whether or not these activities can be considered to be substantial threats for these specific populations. A literature review was used to identify the manner in which specific land use types are thought to influence Atlantic salmon populations or their habitat characteristics. For example, agricultural activities, industrial sites (like gravel quarries) and roads are considered to be chronic sources of sediments (Gilvear et al. 2002), where increased sedimentation has been linked to reduced habitat quality for salmonids as well as to acute mortality of eggs or juveniles (Soulsby et al. 2001; Julien and Bergeron 2006), particularly during storm events (Lisle 1989). Furthermore, the bedrock geology of the Southern Upland region has little buffering capacity (Watt et al. 1983; Korman et al. 1994), which makes Southern Upland salmon particularly vulnerable acid deposition from either precipitation or land-based sources (Farmer et al. 1980; Watt 1987). Atmospheric deposition in the form of acid rain has substantially reduced pH in many rivers throughout the Southern Upland (Watt 1987). Lastly, salmonids depend on unobstructed movement in a watershed to access spawning and rearing areas, avoid predators, and respond to changing environmental conditions such as temperature, flow, or inter- and intra-species competition (Poplar-Jeffers et al. 2009). Threats that fragment watersheds, such as road/river crossings containing culverts or impassable dams, would be expected to reduce habitat productivity as well as accessibility for Atlantic salmon. Although other types of land use would be expected to influence freshwater environments (e.g. urbanization), the incidence of such threats in the Southern Upland region was very low (e.g. <1 % of watershed area affected) and quite similar among watersheds (data not shown), and so were not considered here. In terms of duration, each of the land use variables considered here would have become significant prior to the 1970s (Bowlby et al. 2014), which corresponds to more than five generations prior to genetics sampling for these populations.
The preferred model of each threat index was identified using the two-step Reciprocal Causal Modeling framework proposed by Wasserman et al. (2010), introduced above. A separate Euclidean distance matrix was calculated for each potential combination of the land use variables hypothesized to be contributing to each threat index (Table 2). Because land use variables were similarly scaled prior to calculating Euclidean distance, included land use types contribute equally in the model. As in Shirk et al. (2010), the model that had the highest correlation with genetic distance was initially identified using Mantel tests based on Spearman correlations (e.g. Gene ~ Model) and partial Mantel tests (Smouse et al. 1986) controlling for the influence of underlying genetic structure (e.g. Gene ~ Model|IBE). Here we used the IBE model only in order to avoid overestimating the effect of land use on gene flow, given that spatially correlated landscape features can mask or confound the independent contribution of contemporary land use effects on genetic distance (Dileo et al. 2013). However, we recognize that essentially equivalent results could have been obtained in this study by controlling for IBD. Next, the relative support for the chosen model was assessed using reciprocal comparisons with each competing threat model (e.g. Gene ~ Model 1|Model 2 vs. Gene ~ Model 2|Model 1). Again, the expectation would be that a better model would have a large and significant r value relative to all of the other hypotheses in the third test, while the r value would be small and non-significant in the fourth test. Using Reciprocal Causal Modeling to evaluate the contribution of land use variables to each of three threats indices necessitated two assumptions: (1) that the effects of land use were additive and (2) that specific types of land use contributed to one main threat index. By this we mean that road density (for example) would be mainly related to sedimentation, rather than having nearly equivalent contributions to the fragmentation and sedimentation indices. This assumption was evaluated a posteri by calculating the Spearman correlation coefficient among the threat indices as well as by substituting alternate land use variables into each threat index (e.g. considering road density rather than road crossings in the fragmentation index).
Part 4: influence of anthropogenic land use
To evaluate if any of the threats indices explained significant residual genetic variation (over and above that explained by IBE), we used Multiple Regression on Distance Matrices (MRDM; Legendre et al. 1994; Lichstein 2007). For the retained predictors, a positive correlation would indicate increased genetic isolation relative to that expected historically and would provide evidence that the variables contributing to the threat index have had measureable effects on population dynamics. Here, the relative support for various models was assessed using the Akaike Information Criterion for small samples (AICc; Burnham and Anderson 2002). Although the underlying data are not independent, this non-independence is the same in each model and thus does not affect model ranking, making information theoretic approaches (Johnson and Omland 2004) valid for model selection (Engler et al. 2014).
Results
Population genetic structure
Meaningful comparisons of genetic diversity at neutral markers require that data are not biased by genotyping errors (e.g. null alleles), that all populations have been sampled representatively, and that the tested loci are not under selection (i.e. that they are in Hardy–Weinburg equilibrium (HWE); Allendorf et al. 2013). Round Hill River and Salmon River (Guysborough County) were unlikely to have been sampled representatively, given that juvenile salmon were only captured at one of the two sites electrofished on either river. Clustering among genetically similar individuals would lead to a non-representative sample at the population level (Schoville et al. 2012) and would be expected given the population structuring exhibited by Atlantic salmon and the tendency to home to specific places within a watershed (Keefer and Caudill 2014). The low genetic diversity in Round Hill River relative to the other 10 population sampled (Table 1) supports this conclusion, as does the prediction that one quarter of tested loci were out of HWE for Salmon River (Guysborough County) (Supplementary file: S1 Microsat validation.xlsx). Therefore, these rivers were removed from further analyses. The remaining rivers exhibited no systematic departures from HWE either within loci or populations. The tested loci and alleles appear to sort independently (i.e. do not exhibit linkage disequilibrium), given that predicted correlations among alleles were extremely low (<0.1) and significant results were not systematically distributed among samples (Supplementary file: S1 Microsat validation.xlsx). Null alleles were predicted at locus 9 in 5 populations (Country Harbour, LaHave, Gold, Moser, and St. Mary’s) and at locus 7 in Salmon River (Digby County) (Supplementary file: S1 Microsat validation.xlsx). Although accounting for null alleles would slightly change the predicted allele frequencies, the relative differences tend to be preserved (i.e. the predicted frequencies all decline, rather than some increasing and some decreasing (Supplementary file: S1 Microsat validation.xlsx)). Therefore, null alleles would not be expected to bias analyses based on pairwise distances, an expectation that we confirmed by both including and excluding locus 9 in the analyses. The results we present are based on the unadjusted allele frequencies at all loci. Measures of within-population genetic variation were similar (Table 1) and indicated relatively high levels of genetic diversity. Allelic richness was similar, and both expected and observed heterozygosity were higher than have been previously reported for anadromous fishes (DeWoody and Avise 2000).
Genetic variation among populations
The Mantel test predicts a higher correlation (and one much less likely to have occurred by chance) between Nei’s pairwise Fst and the landscape distance matrix (r = 0.392, p value = 0.007) as compared to coastal geographic distance (r = 0.373, p value = 0.020) (Table 3). Comparing the IBD and IBE hypotheses using Reciprocal Causal Modeling suggests that the IBE model may be better supported by the data, given that the partial Mantel r correlation is substantially higher between genetic isolation and the landscape distance matrix after controlling for any correlation with geographic distance (Gene ~ IBE|IBD; r = 0.176), as compared to the reverse (Gene ~ IBD|IBE; r = < −0.001) although neither p-value is significant at a level of 0.05 (Table 3). However, relatively little of the permuted null distribution is larger than 0.176 in the Gene ~ IBE|IBD comparison, while half of the permuted null distribution is larger than the r value in the Gene ~ IBD|IBE comparison. Re-analysis using Bray-Curtis dissimilarity of the proportion of shared alleles did not change the above pattern for the Mantel r (i.e. substantially higher for Gene ~IBE|IBD relative to Gene ~ IBD|IBE), although the actual values of the Mantel r correlations were lower and again, the p values were not significant (data not shown).
Identification of variables contributing to threats indices
Mantel and partial Mantel tests between genetic distance and a fragmentation index, F, (where each possible combination of road crossings (Rc), dams (D) and pH (pH) were assessed independently) showed the largest correlation with the RcD model (road crossings and dams; Table 4). Comparisons of this model to genetic distance while controlling for each of the other candidate models (Model 1|Model 2) had Mantel r values ranging from 0.229 to 0.283; although each of the p-values were greater than 0.05 (Table 4). Conversely, none of the other models retained any significant correlation with genetic distance once the effect of the RcD model was accounted for, and the absolute values of the Mantel r correlations were substantially lower (<0.135; Table 4). Although these results do not suggest that the RcD model can be considered significantly better than each of the other candidate models, the relatively large Mantel r correlations when the competing models are accounted for (and vice versa) suggests that this model is the best representation of the fragmentation index.
The Mantel and partial Mantel tests relative to the water quality index (WQ) were unexpected, in that the correlations tended to be strongly negative, suggesting greater population connectivity among more dissimilar watersheds related to mine density, forestry or mean annual pH. The largest negative Mantel r correlation was with the MP model (mines and pH). Relative to the competing models, the MP model retained a large negative correlation with genetic distance (r values <−0.33; Table 4), while the competing models had smaller correlations with genetic distance when the effect of the MP model was accounted for. Based on a comparison of the p values between Model 1|Model 2 (expected to be significant) and Model 2| Model 1 (expected to be non-significant), the MP model was significantly better than all of the competing models. It was chosen as the best representation of the water quality index.
Unlike the water quality and fragmentation indices, there was no single model that clearly had the largest correlation with genetic distance for the sedimentation index. With the exception of the A model (agriculture only), the partial Mantel tests (Model 1|IBE) demonstrated correlations above 0.48 for each of the other candidate models (Table 4). The IRdA model (industry, road density and agriculture) had among the highest r values for both the Mantel and partial Mantel tests, and so was chosen as the best representation of the sedimentation index (S) for Reciprocal Causal Modeling. However, sensitivity to this choice was also assessed by evaluating the IRd, ARd, IA and Rd models using Reciprocal Causal Modeling (data not shown). The IRdA model had no additional explanatory power relative to the IRd model (c.f. r = 0.146 and 0.148; Table 4), the ARd model (c.f. r = 0.191 and 0.126; Table 4) and the Rd model (c.f. r = 0.174 and 0.177; Table 4); there are essentially equivalent correlations with residual genetic variation in the reciprocal tests. In addition, there is still a correlation predicted for the IRdA model after accounting for the influence of the IA model (r = 0.195) while there is not for the IA model after accounting for IRdA (r = 0.008; Table 4). Similar patterns are found when the IRd, ARd, IA and Rd models are evaluated. All models excluding Rd have essentially no additional explanatory power, while models including one or both of I and A (in addition to Rd) have essentially equivalent explanatory power in the reciprocal tests. Taken together, these results suggest to us that road density is the primary land use variable contributing to the sedimentation index and that industry and agriculture have comparatively little contribution. Therefore, following the principle of parsimony, the Rd model was chosen as the best representation of the sedimentation index, although we cannot discount the possibility that industry and agriculture have a lesser effect.
Influence of anthropogenic land use
The three threats indices were relatively uncorrelated, with Spearman correlation coefficients of <0.4, and thus unlikely to violate assumptions regarding collinearity for inclusion in multiple regressions (Zuur et al. 2010). In isolation, the IBE model explained very little of the variation in genetic distances (R-square = 0.164, p-value = 0.005; Table 5), even though the variable was significant in the regression. Results were very similar between the full model including all of the indices (IBE + S + F + WQ) and the model that excluded the fragmentation index (IBE + S + WQ), although the latter was selected as the preferred model on the basis of AICc; c.f. R-square = 0.547 and 0.536, p values = 0.001 and <0.001, AICc = −108.71 and −110.01, respectively (Table 5). As expected, the fragmentation index was not a significant predictor in the full model (slope = 0.11; p value = 0.529). All other candidate models had R-square values of less than 0.4 (Table 5). It is important to remember that R-square values from MRDM regression (as well as r statistics from Mantel tests) cannot be interpreted as the proportion of variance in the response explained by the predictors, but only as a measure of the fit of a linear model to the paired sets of distances (Legendre and Fortin 2010). Given that each of the Euclidean distance matrices included in the MRDM models were similarly scaled, the slope estimates from each regression model give a direct comparison of the relative magnitude of each threat index on genetic distance among watersheds. Absolute values of the slope estimates from the sedimentation (slope = 0.44, p value = 0.012) and water quality (slope = −0.39, p value = 0.013) indices were similar to those from the landscape distance matrix (slope = 0.41, p value = 0.007).
Discussion
Our analyses do not significantly support the hypothesis that environmental variation is the primary determinant of gene flow in Atlantic salmon at a regional scale, as opposed to a stepping-stone pattern of movement. However, the behavioural mechanism proposed here to lead to an IBE model is consistent with the interpretation of genetic variation arising from contemporary threats, while the IBD model is not. In relation to population-level responses to threats, findings are consistent with multiple experimentally-predicted behavioural or physiological changes and suggest that straying behaviour and subsequent reproductive success is affected by anthropogenic land use in watersheds. Based on these results, we argue that landscape genetics appears to provide a powerful basis from which to develop future research priorities and remediation strategies to address population declines in Atlantic salmon and potentially other endangered species.
There is a strong theoretical basis by which to argue that IBD may be insufficient to describe straying behaviour, effective dispersal and historical patterns of population connectivity in Atlantic salmon, and potentially other migratory species. This contrasts most research on population structuring in diadromous fishes, where IBD is taken to be the expectation or null model of gene flow (e.g. Palstra et al. 2007; King et al. 2001; Bradbury et al. 2014). However, the theoretical concept of IBD was developed to represent a specific type of dispersal process, one limited by the cumulative fitness costs associated with movement (Slatkin 1993; Guillot et al. 2009). Such a pattern is unlikely to describe movement among watersheds in Atlantic salmon and other diadromous fishes, given that oceanic migrations are vast relative to any separation among river mouths (Hansen and Quinn 1998; Ritter 1989). Alternatively, the evolution of homing behaviour as well as substantial life history variation among populations provides indirect evidence of local adaptation (Garcia de Leaniz et al. 2007; Fraser et al. 2011), which implies that individuals have higher reproductive success in environments similar to their natal environment (Pfluger and Balkenhol 2014) and could be motivated to seek out similar habitats (Bonte et al. 2012). Thus, among returning adults, habitat selection would be a more likely behavioural motivation for watershed choice while straying, where subsequent reproduction (i.e. effective dispersal) would also be expected to be higher in more similar recipient watersheds. Proximate cues to assess habitat quality could be olfactory (e.g. dissolved mineral content, oxygen saturation) or environmental (e.g. flow rate, temperature), factors that are linked to landscape characteristics and environmental variation among watersheds (reviewed in Allan 2004). If straying adults are using such proximate cues, gene flow among watersheds should correlate with the environmental characteristics of watersheds. Although a marginally stronger correlation existed with IBE as IBD in this study, our IBE model did not explain significantly more genetic variation. Variability in this relatively limited data set coupled with the low power of Mantel tests (Legendre et al. 2015) would hinder the description of any underlying relationship, as could strong spatial autocorrelation between environmental variation and geographic distance (here estimated at 0.92), making IBE closely approximate IBD. This latter pattern would be expected to break down under weak spatial autocorrelation, and could explain why IBD is both supported (e.g. Dionne et al. 2009; King et al. 2001) and not (e.g. Bradbury et al. 2014) in the literature for Atlantic salmon. Given the theoretical basis for the underlying assumptions, we propose that future research on gene flow among populations (i.e. among watersheds) in diadromous species should develop an IBE model in addition to IBD. The potential exits for environmental variation to affect the dispersal process directly, rather than acting exclusively on within population processes like genetic drift or adaptation.
The negative Mantel r correlation predicted between genetic distance and the WQ can only be explained if returning adults are actively assessing the characteristics of the watersheds they encounter (implicit in the IBE model) as opposed to incurring cumulative fitness costs associated with movement behaviour among watersheds (implicit in the IBD model). Most landscape genetics questions dealing with a species’ response to threats test for the effect of barriers or some type of land use that increases resistance to movement (through degrading habitat quality) and thus reduces the ability or tendency to move through areas more heavily affected by the threat (e.g. Sawyer et al. 2011; Zeller et al. 2012). From these examples, IBD is predicted to become stronger than what would be expected historically and any Mantel correlation between genetic distance and the magnitude of the threat would be inherently positive (i.e. high levels of the threat would cause a greater reduction in movement among populations). However, if individuals have the ability to avoid areas more heavily affected by a threat (e.g. particular watersheds) while moving freely through a uniform habitat mosaic (e.g. the marine environment), negative correlations become possible, like the one seen here between the WQ and genetic distance.
The negative correlation suggests increased gene flow among environmentally dissimilar watersheds (based on mines +pH), which would be consistent with individuals preferentially entering or experiencing higher spawning success in rivers that are less acidified or contain fewer contaminants relative to their natal watershed. One hypothesis is that observed patterns of genetic divergence could result from a combination of: (1) a reduced capacity to imprint using olfactory cues (leading to an increase in the proportion of a population that strays), and (2) relatively lower survival in watersheds with poor water quality (leading to higher survival of individuals straying to watersheds with good water quality). Besides the potential for acute mortality caused by low pH (e.g. Lacroix and Townsend 1987), acidification is known to interfere with chemosensory functions related to the detection and response to chemical signals (Leduc et al. 2010). One chemosensory function would be the olfactory imprinting that occurs during emigration to the marine environment and enables individuals to return to natal rivers (McCormick et al. 1998). Similarly, a range of chemical compounds (including insecticides used in forestry practices in Nova Scotia; Fairchild et al. 1999) as well as heavy metals (like those typically present in acid rock drainage from old mine sites; Akcil and Koldas 2006) can have comparable effects on chemosensory function (reviewed in Lurling and Scheffer 2007). Preliminary tag-based research on sub-lethal exposure to an insecticide corroborates this hypothesis, in that homing success was lower for exposed Pacific salmon relative to controls (Scholz et al. 2000). Similarly, higher straying rates among localized stream reaches were observed when conditions at the natal site were less favorable (Dittman et al. 2010; Cram et al. 2013). A recent review of homing behaviour in anadromous salmonids indicates that environmental conditions influence attraction to non-natal sites and that any chemicals interfering with olfactory imprinting or sensory development would be expected to increase the incidence of straying as well as to influence adult habitat choice (Keefer and Caudill 2014).
During recovery planning for endangered species, threats that tend to be well-understood are often focused on for remediation at the expense of those that are not (Lawler et al. 2002; Norris 2004). The specific threats identified as contributing to the sedimentation, fragmentation and water quality indices are very interesting from a recovery planning perspective, and contradict some current perceptions. It is generally accepted that pH has an extremely large influence on Atlantic salmon in the Southern Upland, where increases in acid precipitation from the 1950s have been implicated in population extirpations and severe population decline (Watt et al. 1983; Lacroix 1989). However, the influence of past mining activities on current water quality has never been identified as a pressing issue for mitigation, although it has been identified as a potential threat (e.g. Bowlby et al. 2014). The present analysis suggests that mining activities affect population connectivity (movement and spawning success) in the same manner and with the same severity as acidification. Similarly, road crossings and fragmentation have been identified as extremely important determinants of freshwater habitat accessibility for salmon, and many user groups are interested in culvert and dam removal or remediation as a method for improving habitat accessibility and presumably increasing population size for Atlantic salmon (Langill and Zamora 2002; Gibson et al. 2005). Here, the larger concern appears to be road density itself, given that the fragmentation index had no significant effect on population connectivity while the sedimentation index did. The presence of roads has been linked to a suite of ecological changes in watersheds beyond sedimentation: including changes in thermal regimes, constriction of channel movement, and changes in channel morphology, contaminants, as well as the spread of invasives in watersheds (Trombulak and Frissell 2000). It has been argued for a wide variety of taxa that the effects of roads on natural populations are one of the most pressing current conservation issues (Forman and Alexander 1998; Clark et al. 2010). It is interesting that our analysis suggests that the presence of roads causes changes in population connectivity of a similar magnitude relative to changes in water quality (i.e. the slope estimate for the S index is comparable to that of the WQ index). However, we have not explicitly linked these results to a population dynamics model used to assess status, so it is unknown how the opposing effects of the two threats culminate into changes in abundance.
It is well-known that the influence of specific environmental factors on systems or populations can vary over different scales (e.g. Schneider 2001), which has implications for how well this population-level analysis can fully characterize the relative magnitude and influence of this suite of threats affecting adult Atlantic salmon populations, particularly the fragmentation index. In general, adult habitat choice can be considered to result from processes at two scales: first, the selection of a watershed to enter from the marine environment (large scale), and second, the selection of a spawning location within that watershed (small scale). Starting from the premise that adult salmon assess watershed characteristics when choosing a watershed for spawning (as suggested by IBE), only threats that influence the characteristics of the water being discharged from the river could be perceived by individuals at a large scale and used as proxies for habitat quality. Of the variables considered here, such threats would be contributing to the sedimentation and water quality indices; the two threat indices identified as significant predictors of population structuring in the regression analyses. At smaller scales, the manner in which adults would be expected to interact with their environment changes, in that movement behaviour is now influenced by the spatial arrangement of suitable spawning substrate as well as the relative connectivity among stream reaches (Waples et al. 2008; Sabo et al. 2010). Stated another way, gene flow within a population would be partially determined by an individual’s ability or motivation to move through the habitat mosaic, as well as by habitat suitability. It may be more appropriate to characterize threats like full or partial barriers (e.g. road-river crossings) as well as localized habitat degradation caused by land use (e.g. an agricultural field adjacent to a particular stream reach) using resistance surfaces developed for a dendritic landscape (i.e. one in which animals are constrained to move along the stream network; Fagan 2002; Hopken et al. 2013).
Detectability, sublethal effects and cumulative effects (among others) all complicate attempts to quantify relationships between abundance and environmental variability or anthropogenic effects on fish populations (Rose 2000), undermining effective conservation and hindering timely recovery planning (Lawler et al. 2002; Norris 2004). Using genetic variation (as opposed to abundance time series) to investigate population-level responses to threats addresses the latter two issues: multiple correlated hypotheses can be directly compared (assessing cumulative effects) and population-level responses can give indirect evidence of physiological change (sublethal effects), as seen here in relation to the water quality index. Secondary benefits are that these analyses can be done at the level most appropriate for recovery planning (i.e. for populations), can be completed relatively rapidly (i.e. they do not rely on the collection of time series data), and different threats measured in different units (e.g. proportions, counts, or densities) can be directly compared, with the caveat that relationships between genetic distance and environmental distance are assumed to be linear. Although landscape genetics would not be an appropriate method in situations where population decline was extremely rapid (given that there would not be adequate time for evolutionary effects to become detectable; Landguth et al. 2010; Dileo et al. 2013), it would be appropriate in the majority of situations where declines are observed for many years prior to their identification as a conservation concern, provided the same suite of threats are influencing populations over the entire time period. Although these analyses only provide a way to compare the influence and magnitude of threats relative to each-other among a group of watersheds (rather than developing predictive relationships between specific populations and a suite of threats), they appear to be a powerful basis from which to develop future research priorities and remediation strategies to address population declines.
References
Akcil A, Koldas S (2006) Acid mine drainage (AMD): causes, treatment and case studies. J Clean Prod 14:1139–1145
Allan JD (2004) Landscapes and riverscapes: the influence of land use on stream ecosystems. Annu Rev Ecol Evol Syst 35:257–284
Allendorf FW, Luikart G, Aitken SN (2013) Conservation and the genetics of populations, 2nd edn. Blackwell, London
Amiro PG, Gibson AJF, Bowlby HD (2006) Atlantic salmon (Salmo salar) overview for eastern Cape Breton, Eastern Shore, Southwest Nova Scotia and inner Bay of Fundy rivers (SFA 19 to 22) in 2005. DFO Can Sci Advis Sec Res Doc 2006/024
Arens P, van der Sluis T, van’t Westende WPC, Vosman B, Vos CC, Smulders MJM (2007) Genetic population differentiation and connectivity among fragmented Moor frog (Rana arvalis) population in the Netherlands. Landsc Ecol 22:1489–1500
Balkenhol N, Waits LP, Dezzani RJ (2009) Statistical approaches in landscape genetics: an evaluation of methods for linking landscape and genetic data. Ecography 32:818–830
Bocard D, Gillet F, Legendre P (2011) Numerical ecology with R. Springer, New York
Bonte D, van Dyck H, Bullock JM, Coulon A, Delgado M et al (2012) Costs of dispersal. Biol Rev 87:290–312
Bowlby HD, Horsman T, Mitchell SC, Gibson AJF (2014) Recovery potential assessment for southern upland atlantic salmon: habitat requirements and availability, threats to populations, and feasibility of habitat restoration. DFO Can Sci Advis Sec Res Doc 2013/006
Bowler DE, Benton TG (2005) Causes and consequences of animal dispersal strategies: relating individual behaviour to spatial dynamics. Biol Rev 80:205–225
Bradburd G, Ralph P, Coop GM (2013) Disentangling the effects of geographic and ecological isolation on genetic differentiation. Evolution. doi:10.1111/evo.12193
Bradbury IR, Hamilton LC, Robertson MJ, Bourgeois CE, Mansour A, Dempson JB (2014) Landscape structure and climatic variation determine Atlantic salmon genetic connectivity in the Northwest Atlantic. Can J Fish Aquat Sci 71:246–258
Bunn SE, Arthington AH (2002) Basic principles and ecological consequences of altered flow regimes for aquatic biodiversity. Environ Manage 30:492–507
Burnham KP, Anderson DR (2002) Model selection and multimodel inference: a practical information-theoretic approach. Springer, New York
Castillo JA, Epps CW, Davis AR, Cushman SA (2014) Landscape effects on gene flow for a climate-sensitive montane species, the American pika. Molec Ecol 23:843–856
Chaput G, Dempson JB, Caron F, Jones R, Gibson J (2006) A synthesis of life history characteristics and stock grouping of Atlantic salmon (Salmo salar L.) in eastern Canada. DFO Can Sci Advis Sec Res Doc 2006/015
Clark RW, Brown WS, Stechert R, Zamudio KR (2010) Roads, interrupted dispersal, and genetic diversity in timber rattlesnakes. Conserv Biol 24:1059–1069
COSEWIC (2010) COSEWIC assessment and status report on the Atlantic salmon Salmo salar Nunavik population, Labrador population, Northeast Newfoundland population, South Newfoundland population, Southwest Newfoundland population, Northwest Newfoundland population, Quebec Eastern North Shore population, Quebec Western North Shore population, Anticosti Island population, Inner St. Lawrence population, Lake Ontario population, Gaspe-Southern Gulf of St. Lawrence population, Eastern Cape Breton population, Nova Scotia Southern Upland population, Inner Bay of Fundy population Outer Bay of Fundy population in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa. www.sararegistry.gc.ca/status/status_e.cfm. Accessed 2 Feb 2014
Cram JM, Torgersen CE, Klett RS, Pess GR, May D, Pearsons TN, Dittman AH (2013) Tradeoffs between homing and habitat quality for spawning site selection by hatchery origin Chinook salmon. Environ Biol Fish 96:109–122
Cushman SA, McKelvey KS, Hayden J, Schwartz MK (2006) Gene-flow in complex landscapes: testing multiple models with causal modeling. Am Nat 168:486–499
Cushman SA, Wasserman TN, Landguth EL, Shirk AJ (2013) Re-evaluating causal modeling with Mantel tests in landscape genetics. Diversity 5:51–72
DeWoody JA, Avise JC (2000) Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J Fish Biol 56:461–473
Dileo MF, Rouse JD, Davila FA, Lougheed SC (2013) The influence of landscape on gene flow in the eastern massasauga rattlesnake (Sistrurus c. catenatus): insight from computer simulations. Molec Ecol 22:4483–4498
Dionne M, Caron F, Dodson J, Bernatchez L (2009) Comparative survey of within-river genetic structure in Atlantic salmon: relevance for management and conservation. Conserv Genet 10:869–879
Dittman AH, May D, Larsen DA, Moser ML, Johnston M, Fast D (2010) Homing and spawning site selection by supplemented hatchery- and natural-origin Yakima River spring Chinook salmon. Trans Am Fish Soc 139:1014–1028
Dudgeon D, Arthington AH, Gessner MO, Kawabata Z-I, Knowler DJ, Leveque C, Naiman RJ, Prieur-Richard A-H, Soto D, Stiassny MLJ, Sullivan CA (2006) Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev 81:163–182
Dyer RJ (2012) The gstudio package. Virginia Commonwealth University, Virginia
Engler JO, Balkenhol N, Filz KJ, Habel JC, Rödder D (2014) Comparative landscape genetics of three closely related sympatric hesperid butterflies with diverging ecological traits. PLoS One 9:e106526
Epps CW, Palsboll PJ, Wehausen JD, Roderick GK, Ramey RR, McCullough DR (2005) Highways block gene flow and cause a rapid decline in genetic diversity of desert bighorn sheep. Ecol Lett 8:1029–1038
Fagan WF (2002) Connectivity, fragmentation, and extinction risk in dendritic metapopulations. Ecology 83:3243–3249
Fairchild WL, Swansburg EO, Arsenault JT, Brown SB (1999) Does an association between pesticide use and subsequent declines in catch of Atlantic salmon (Salmo salar) represent a case of endocrine disruption? Environ Health Perspect 107:349–357
Farmer GJ, Goff TR, Ashfield D, Samant HS (1980) Some effects of the acidification of Atlantic salmon rivers in Nova Scotia. Can Tech Rep Fish Aquat Sci 972
Ferrier S, Manion G, Elith J, Richardson K (2007) Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers Distrib 13:252–264
Fitzpatrick MC, Keller SR (2015) Ecological genomics meets community-level modelling of biodiversity: mapping the genomic landscape of current and future environmental adaptation. Ecol Lett 18:1–16
Foley JA, DeFries R, Asner GP, Barford C, Bonan G et al (2005) Global consequences of land use. Science 309:570–574
Forman RTT, Alexander LE (1998) Roads and their major ecological effects. Annu Rev Ecol Syst 29:207–257
Fraser DJ, Weir LK, Bernatchez L, Hansen MM, Taylor EB (2011) Extent and scale of local adaptation in salmonid fishes: review and meta-analysis. Heredity 106:404–420
Garcia de Leaniz C, Fleming IA, Einum S, Verspoor E, Jordan WC et al (2007) A critical review of inherited adaptive variation in Atlantic salmon. Biol Rev 82:173–211
Garnier-Gere P, Dillmann C (1992) A computer program for testing pairwise linkage disequilibrium in subdivided populations. J Hered 83:239
Gibson AJF, Bowlby HD (2013) Recovery potential assessment for Southern Upland Atlantic salmon: population dynamics and viability. DFO Can Sci Advis Sec Res Doc 2012/142
Gibson RJ, Haedrich RL, Wernerheim CM (2005) Loss of fish habitat as a consequence of inappropriately constructed stream crossings. Fisheries 30:10–17
Gibson AJF, Bowlby HD, Hardie D, O’Reilly P (2011) Populations on the brink: atlantic salmon (Salmo salar) in the Southern Upland Region of Nova Scotia, Canada. North Am J Fish Manage 31:733–741
Gilvear DJ, Heal KV, Stephen A (2002) Hydrology and ecological quality of Scottish river ecosystems. Sci Total Environ 294:131–159
Guillot G, Rousset F (2013) Dismantling the Mantel tests. Methods Ecol Evol 4:336–344
Guillot G, LeBlois R, Coulon A, Frantz AC (2009) Statistical methods in spatial genetics. Molec Ecol 18:4734–4756
Hansen LP, Quinn TP (1998) The marine phase of the Atlantic salmon (Salmo salar) life cycle, with comparisons to Pacific salmon. Can J Fish Aquat Sci 55(Suppl. 1):104–118
Hopken MW, Douglas MR, Douglas ME (2013) Stream hierarchy defines riverscape genetics of a North American desert fish. Molec Ecol 22:956–971
Johnson JB, Omland KS (2004) Model selection in ecology and evolution. Trends Ecol Evol 19:101–108
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Julien HP, Bergeron NE (2006) Effect of fine sediment infiltration during the incubation period on Atlantic salmon (Salmo salar) embryo survival. Hydrobiologia 563:61–71
Keefer ML, Caudill CC (2014) Homing and straying by anadromous salmonids: a review of mechanisms and rates. Rev Fish Biol Fisher 24:333–368
King TL, Kalinowski ST, Schill WB, Spidle AP, Lubinski BA (2001) Population structure of Atlantic salmon (Salmo salar L.): a range-wide perspective from microsatellite DNA variation. Molec Ecol 10:807–821
Korman J, Marmorek DR, Lacroix GL, Amiro PG, Ritter JA, Watt WD, Cutting RE, Robinson DCE (1994) Development and evaluation of a biological model to assess regional-scale effects of acidification on Atlantic salmon (Salmo salar). Can J Fish Aquat Sci 51:662–680
Kubisch A, Holt RD, Poethke H-J, Fronhofer EA (2013) Where am I and why? Synthesizing range biology and the eco-evolutionary dynamics of dispersal. Oikos 123:5–22
Lacroix GL (1989) Production of juvenile Atlantic salmon (Salmo salar) in two acidic rivers of Nova Scotia. Can J Fish Aquat Sci 46:2003–2018
Lacroix GL, Townsend DR (1987) Responses of juvenile Atlantic salmon (Salmo salar) to episodic increases in acidity of Nova Scotia rivers. Can J Fish Aquat Sci 44:1475–1484
Landguth EL, Cushman SA, Schwartz MK, McKelvey KS, Murphy M, Luikart G (2010) Quantifying the lag time to detect barriers in landscape genetics. Molec Ecol 19:4179–4191
Langill DA, Zamora PJ (2002) An audit of small culvert installations in Nova Scotia: habitat loss and habitat fragmentation. Can Tech Rep Fish Aquat Sci 2422
Lawler JJ, Campbell SP, Guerry AD, Kolozsvary MB, O’Connor RJ, Seward LC (2002) The scope and treatment of threats in endangered species recovery plans. Ecol Appl 12:663–667
Leduc AOHC, Roh E, MacNaughton CJ, Benz F, Rosenfeld J, Brown GE (2010) Ambient pH and the response to chemical alarm cues in juvenile Atlantic salmon: mechanisms of reduced behavioral responses. Trans Am Fish Soc 139:117–128
Legendre P (1993) Spatial autocorrelation: trouble or new paradigm? Ecology 74:1659–1673
Legendre P, Fortin M-J (2010) Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data. Molec Ecol Resour 10:831–844
Legendre P, Lapointe F-J, Casgrain P (1994) Modeling brain evolution from behavior: a permutational regression approach. Evolution 48:1487–1499
Legendre P, Fortin M-J, Borcard D (2015) Should the Mantel test be used in spatial analysis? Method Ecol Evol 6:1239–1247
Lichstein JW (2007) Multiple regression on distance matrices: a multivariate spatial analysis tool. Plant Ecol 188:117–131
Lisle TE (1989) Sediment transport and resulting deposition in spawning gravels, north coastal California. Water Resour Res 25:1303–1319
Lurling M, Scheffer M (2007) Info-disruption: pollution and the transfer of chemical information between organisms. Trends Ecol Evol 22:374–379
McCormick SD, Hansen LP, Quinn TP, Saunders RL (1998) Movement, migration and smolting of Atlantic salmon (Salmo salar). Can J Fish Aquat Sci 55(Suppl. 1):77–92
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Norris K (2004) Managing threatened species—the ecological toolbox, evolutionary theory and the declining-population paradigm. J Appl Ecol 41:413–426
O’Reilly P, Rafferty S, Gibson J (2012) Within- and among-population genetic variation in the Southern Upland Designatable Unit of Maritime Atlantic Salmon (Salmo salar L.). DFO Can Sci Advis Sec Res Doc 2012/077
Oksanen J, Guillaume Blanchet F, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H (2015) vegan: Community ecology package. R package version 2.2-1. http://CRAN.R-project.org/package=vegan
Palstra FP, O’Connell MF, Ruzzante DE (2007) Population structure and gene flow reversals in Atlantic salmon (Salmo salar) over contemporary and long-term temporal scales: effects of population size and life history. Molec Ecol 16:4504–4522
Pfluger FJ, Balkenhol N (2014) A plea for simultaneously considering matrix quality and local environmental conditions when analysing landscape impacts on effective dispersal. Molec Ecol 23:2146–2156
Poff NL, Richter BD, Arthington AH, Bunn SE, Naiman RJ et al (2010) The ecological limits of hydrologic alteration (ELOHA): a new framework for developing regional environmental flow standards. Freshwater Biol 55:147–170
Poplar-Jeffers IO, Petty JT, Anderson JT, Kite SJ, Stragler MP, Fortney RH (2009) Culvert replacement and stream habitat restoration: implications from brook trout management in an Appalachian watershed. USA. Restor Ecol 17:404–413
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Ritter JA (1989) Marine migration and natural mortality of North American Atlantic salmon (Salmo salar L.). Can Manuscr Rep Fish Aquat Sci 2041
Roni P, Beechie TJ, Bilby RE, Leonetti FE, Pollock MM, Pess GR (2002) A review of stream restoration techniques and a hierarchical strategy for prioritizing restoration in Pacific Northwest watersheds. North Am J Fish Manage 22:1–20
Rose KA (2000) Why are quantitative relationships between environmental quality and fish populations so elusive? Ecol Appl 10:367–385
Rousset F (2008) Genepop’007: a complete reimplementation of the Genepop software for Windows and Linux. Molec Ecol Resour 8:103–106
Sabo JL, Finlay JC, Kennedy T, Post DM (2010) The role of discharge variation in scaling of drainage area and food chain length in rivers. Science 330:965–967
Sawyer SC, Epps CW, Brashares JS (2011) Placing linkages among fragmented habitats: do least-cost models reflect how animals use landscapes? J Appl Ecol 48:668–678
Schneider DC (2001) The rise of the concept of scale in ecology. Bioscience 51:545–553
Scholz NL, Truelove NK, French BL, Berejikian BA, Quinn TP, Casillas E, Collier TK (2000) Diazinon disrupts antipredator and homing behaviors in Chinook salmon (Oncorhynchus tshawytscha). Can J Fish Aquat Sci 57:1911–1918
Schoville SD, Bonin A, Francois O, Lobeaux S, Melodelima C, Manel S (2012) Adaptive genetic variation on the landscape: methods and cases. Annu Rev Ecol Evol Syst 43:23–43
Sexton JP, Hangartner SB, Hoffman AA (2013) Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68:1–15
Shirk A, Wallin DO, Cushman SA, Rice RC, Warheit C (2010) Inferring landscape effects on gene flow: a new multi-scale model selection framework. Molec Ecol 19:3603–3619
Singh ND, Jensen JD, Clark AG, Aquadro CF (2013) Inferences of demography and selection in an African population of Drosophila melanogaster. Genetics 193:215–228
Slatkin M (1993) Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47:264–279
Smouse PE, Long JC, Sokal RR (1986) Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst Zool 35:627–632
Soulsby C, Youngson AF, Moir HJ, Malcolm IA (2001) Fine sediment influence on salmonid spawning habitat in a lowland agricultural stream: a preliminary assessment. Sci Total Environ 265:295–307
Spear SF, Balkenhol N, Fortin M-J, McRae BH (2010) Use of resistance surfaces for landscape genetic studies: considerations for parameterization and analysis. Molec Ecol 19:3576–3591
Trombulak SC, Frissell CA (2000) Review of ecological effects of roads on terrestrial and aquatic communities. Conserv Biol 14:18–30
Ugedal O, Finstad AG (2011) Landscape and land use effects on Atlantic salmon. Aas O, Einum S, Klemetsen A, Skurdal J [eds] Atlantic Salmon Ecology. Blackwell, London, pp 333–349
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molec Ecol Notes 4:535–538
Venter O, Brodeur NN, Nemiroff L, Belland B, Dolinsek IJ, Grant JWA (2006) Threats to endangered species in Canada. Bioscience 56:903–910
Wagner HH, Fortin M-J (2016) Basics of spatial data analysis: linking landscape and genetic data for landscape genetic studies. In: Balkenhol N, Cushman SA, Storfer AT, Waits LP (eds) Landscape genetics Concepts, methods, applications. Wiley, Hoboken
Wang IJ, Glor RE, Losos JB (2013) Quantifying the roles of ecology and geography in spatial genetic divergence. Ecol Lett 16:175–182
Waples RS, Aable RW, Scheuerell MD, Sanderson BL (2008) Evolutionary responses by native species to major anthropogenic changes to their ecosystems: pacific salmon in the Columbia River hydropower system. Molec Ecol 17:84–96
Wasserman TN, Cushman SA, Schwartz MK, Wallin DO (2010) Spatial scaling and multi-model inference in landscape genetics: martes americana in northern Idaho. Landsc Ecol 25:1601–1612
Watt WD (1987) A summary of the impact of acid rain on Atlantic salmon (Salmo salar) in Canada. Water Air Soil Poll 35:27–35
Watt WD, Scott CD, White WJ (1983) Evidence of acidification of some Nova Scotia rivers and its impact on Atlantic salmon, Salmo salar. Can J Fish Aquat Sci 40:462–473
Zeller KA, McGarigal K, Whiteley AR (2012) Estimating landscape resistance to movement: a review. Landsc Ecol 27:777–797
Zuur AF, Ieno EN, Elphick CS (2010) A protocol for data exploration to avoid common statistical problems. Method Ecol Evol 1:3–14
Acknowledgments
We would like to thank all staff and students involved in electrofishing surveys and the genetics lab, particularly Patrick O’Reilly for providing the microsatellite data as well as Tracy Horseman for GIS analyses. Comments from Jessica Sameoto, Rod Bradford and four anonymous reviewers substantially improved the quality of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bowlby, H.D., Fleming, I.A. & Gibson, A.J.F. Applying landscape genetics to evaluate threats affecting endangered Atlantic salmon populations. Conserv Genet 17, 823–838 (2016). https://doi.org/10.1007/s10592-016-0824-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0824-7