
Abstract
The vitamin E-bonded polysulfone membrane hemodialyzer (ViE™-21) was evaluated in a clinical study for regulatory submission. Seventeen patients on hemodialysis were treated with conventional high-flux hemodialyzers for 2 weeks (Pre-ViE phase) and switched to the ViE-21 for 36 sessions (ViE phase) followed by an additional 2 weeks on conventional hemodialyzers (Post-ViE phase). Reduction ratios of urea, creatinine, beta-2-microglobulin, albumin, and ultrafiltration coefficients (KUF) were measured once during the Pre-ViE phase and twice during the ViE phase. Moreover, biocompatibility markers [leucocyte count, platelet count, and activated complement factor (C3a) levels] were evaluated pre-dialysis, 15 min after initiation, and post-dialysis. During the study, type and number of adverse events (AEs), and device malfunctions were recorded. ViE-21 reduction ratios and KUF were not noticeably different than those of conventional hemodialyzers. Fluctuations of leucocyte counts and C3a concentrations were similar using ViE-21 and conventional hemodialyzers; however, the platelet count fluctuation was lower in ViE-21 sessions. The frequency of episodes of hypotension occurring during the ViE phase was lower than that occurring during the Pre- and Post-ViE phases. In conclusion, this study provided performance and safety data of the ViE-21 for regulatory application. The data suggest that vitamin E-bonded hemodialyzers are beneficial in lowering platelet activation and frequency of intradialytic hypotension. Larger randomized controlled trials are needed to confirm these findings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patients suffering from end-stage renal disease (ESRD) require renal replacement therapy to remove uremic toxins from their blood. Hemodialysis, an extra-corporeal blood purification therapy using a hemodialyzer, is a well-accepted treatment for ESRD patients. In the early stages, hollow fibers of cellulose were used as membrane materials for hemodialyzers. However, when blood components and cells come in to contact with the cellulose membrane, it causes significant activation of complement and reduction of leucocyte counts from the peripheral blood during the dialysis treatment. To overcome such bio-incompatibility, synthetic polymer membranes such as a polysulfone or polyether sulfone membrane were developed [1, 2].
Another challenge of hemodialysis is to reduce oxidative stress during the extracorporeal treatments. Anemia, cardiovascular disease, chronic inflammation, and intradialytic hypotension are among the common complications in patients receiving hemodialysis, which show a causal relationship with oxidative stress [3]. To reduce oxidative stress, vitamin C and E are well-accepted supplements having antioxidant activities.
Consequently, polysulfone-based vitamin E-bonded membranes were developed [4,5,6,7,8] to achieve a synergistic effect of the biocompatibility of synthetic membranes and the antioxidant activity of vitamin E. This clinical study was designed to evaluate the vitamin E-bonded polysulfone hemodialyzer, ViE™-21, manufactured by Asahi Kasei Medical Co., Ltd., at a clinical site in Canada to obtain performance and safety data for regulatory submission.
Methods
Design of the study
The study titled “Clinical Study of Asahi ViE Dialyzer in Canada (AVID)” was conducted as a prospective, open-label, non-randomized, single-arm study to satisfy regulatory requirements for single-use hemodialyzer in accordance with the Food and Drug Administration (FDA) guidance [9] of the United States of America, and in compliance with Good Clinical Practice of International Organization for Standardization (ISO) 14155:2011 guidelines [10]. The study was approved by the research ethics board of the University of British Columbia–Providence Health Care Research Institute, Vancouver, British Columbia, Canada, and registered at ClinicalTrials.gov with the registration number NCT 02292212. For each patient, data were collected over six dialysis sessions on a conventional hemodialyzer, which was the same hemodialyzer used for routine dialysis therapy (Pre-ViE phase), and then patients were switched to ViE-21 (surface area: 2.1 m2) for 36 dialysis sessions (ViE phase) for further data collection. After completion of the ViE phase, patients were switched to the original hemodialyzer used during the Pre-ViE phase for a final 6 dialysis sessions (Post-ViE phase) to observe safety issue which might be originated from ViE-21 treatments.
Patients
Up to 20 patients were to be enrolled at the start of the study to ensure that at least 12 patients completed 36 treatments each on the ViE-21 to satisfy regulatory requirements [9]. The inclusion criteria were: age between 18 and 80 years; stable condition on maintenance hemodialysis for at least 12 weeks; expectation to remain on hemodialysis for at least 24 weeks; on hemodialysis for more than 3 h per treatment and on a 3 times-per week schedule; having vascular access either as a native arteriovenous fistula or graft; well-maintained and capable of obtaining blood flow rate equal to or more than 350 mL/min; use of high-flux dialyzers (KUF ≥ 40 mL/h/mmHg) with surface area ≥ 1.5 m2 and ≤ 2.2 m2; capable of understanding the informed consent form; and provision of written consent and willingness to participate in the study. Blood flow rate was specified to meet criteria of the FDA guidance [9]. Exclusion criteria included: medical conditions requiring regular blood transfusion; history of more than 1-week hospitalization related to infection; inflammation or surgery within the past 12 weeks; previous participation in another clinical investigation within the previous 12 weeks; difficulty in maintaining vascular access function within the previous 12 weeks; known to be hepatitis B, C, or human immunodeficiency virus-positive; female patients who were pregnant, breast feeding, or planning a pregnancy within the study period; received blood purification therapy other than conventional hemodialysis within the past 12 weeks; intolerant to heparin; and any serious medical, social or psychological condition that in the opinion of the investigator would disqualify a patient from participation.
Reduction ratio evaluation
Blood samples were taken to assess reduction ratios of urea, creatinine, beta-2-microglobulin (B2M), and albumin once in the Pre-ViE phase during the 2nd (Wednesday or Thursday) or 3rd (Friday or Saturday) session of the 1st week for the conventional hemodialyzer, and twice in the ViE phase during weeks 7 and 13 at the 2nd or 3rd session for the ViE-21. The basis of selecting 2nd or 3rd session of the weeks was to unify interdialytica period before sampling. Pre-dialysis blood samples were collected within 15 min prior to commencing dialysis and post-dialysis blood samples were collected within 15 min after the completion of dialysis. The blood samples were immediately centrifuged to separate plasma, and then the plasma was used to assay the concentrations of urea, creatinine, B2M, and albumin. Reduction ratios of urea and creatinine were calculated using the following formula:
where, Cpre and Cpost were the urea or creatinine concentrations in peripheral blood at pre- and post-dialysis phases, respectively. The reduction ratios of B2M and albumin were calculated using the following formula with hematocrit (HCT) correction.
where, Cpre and Cpost were the concentrations of B2M and albumin in peripheral plasma, and HCTpre and HCTpost were HCT values (in %) measured at pre- and post-dialysis sessions, respectively.
For urea reduction evaluation, the values of Kt/V were determined using the following formula:
where, t was dialysis treatment time (in hours), BW was body weight (dry weight in kg), and dBW was the difference in body weight (in kg) between pre- and post-dialysis.
KUF evaluation
The KUF values were measured once in the Pre-ViE phase at the 1st or 2nd week for the conventional hemodialyzer, twice during the ViE phase at weeks 3–8, and at weeks 9–14 for the ViE-21. Each evaluation was performed at either the 2nd or 3rd treatment session but not on the same day of blood sampling for reduction ratio evaluation. The measurements were performed in accordance with the FDA guidance [9] as the recording of transmembrane pressure values 10 min after adjustment of ultrafiltration rates to 600, 1000, 1400, and 1800 mL/h in 10 min intervals, respectively. KUF values were then computed by measuring the slope of the ultrafiltration rate versus the transmembrane pressure for each patient using least squares estimates.
Biocompatibility evaluation
Biocompatibility was evaluated by measuring blood leucocyte and platelet counts as well as C3a concentrations in plasma. Blood samples were taken pre-dialysis, 15 min after initiation, and post-dialysis. Blood samples were collected once in the Pre-ViE phase at the 2nd or 3rd session of the 1st week for the conventional hemodialyzer, twice in the ViE phase during weeks 7 and 13, and at the 2nd or 3rd treatment sessions for the ViE-21 during the same day of blood sample collection for reduction ratio evaluation. Blood samples collected for C3a determinations were centrifuged to separate plasma, and then the separated plasma was stored frozen until all samples from all patients in the study were collected. The samples for C3a testing were all analyzed at one time to minimize test variability.
AE and device malfunction
All AEs were recorded for all treatments and interdialytic days in a precise manner. For example, minor events such as a patient’s feeling of discomfort or muscle cramps requiring minimal intervention, such as changing ultrafiltration flow rate, were recorded as an AE in the case report forms. The dialysis treatment condition parameters such as flow rates of blood and dialysate, vital signs at pre- and post-dialysis, dry weight, volume of water removed, arterial and venous pressure of the extracorporeal circuit, use of anticoagulant therapy, and concomitant medications were recorded. All device malfunctions occurring during any of the treatment sessions were also recorded.
Statistical analysis
In the study protocol, statistical analysis was not foreseen, however, for the post hoc analysis the following statistical analyses were conducted. The two sets of \(\text{Kt}/\text{V}\), KUF, as well as reduction ratios of urea, creatinine, B2M, and albumin data at the ViE phase were compared to the corresponding data at the Pre-ViE phase using paired t test, respectively. For the biocompatibility markers, after normalization by setting the pre-dialysis levels as baseline (100%) and after HCT correction, the two sets of ViE phase data of leucocyte, platelet, and C3a levels were compared to those of the Pre-ViE phase data using the paired t test. No adjustment for multiple comparisons was performed due to the post hoc nature of the analysis. The P values were indicated in the results section without judging statistical significance. The statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., USA).
Results
Patient enrollment and analysis population
A total of 74 patients were screened for enrollment. Fifty-one patients were determined not to be eligible and an additional 6 patients declined to participate. Therefore, the study population consisted of 17 patients. Of the 17 patients enrolled, all completed the baseline assessments and Pre-ViE phase. Three patients began the ViE phase but exited the study prior to completing this phase. One patient was withdrawn after the 14th session because the patient was incorrectly given hemodiafiltration, while this study was restricted to hemodialysis only. A second patient was withdrawn because the patient accidently received 4 consecutive dialysis treatments using the conventional hemodialyzer instead of the ViE-21 during the ViE phase. The third patient withdrew consent at the 29th session. Therefore, 14 patients participated in all 3 phases of the study as per the established protocol. None of the patients were lost to follow-up and no deaths occurred during the study. In the following evaluations, the data for all 17 patients were defined as the safety analysis (SAA) population and the data for the 14 patients who participated in all 3 study phases were defined as the intent-to-treat (ITT) population. The first treatment of the study began on January 5, 2015 and the last treatment was completed on November 6, 2015.
Performance evaluation
The reduction ratios of urea, creatinine, B2M, and albumin as well as Kt/V and KUF values for the ITT population including P values of paired t tests comparing the single Pre-ViE phase measurement with two ViE phase measurements are summarized in Table 1. As indicated in Table 1, there were no detectable differences among the ITT values with the exception of three values: the reduction ratio of urea for the second ViE phase was lower, the reduction ratio of albumin for the first ViE phase was lower, and the KUF value for the first ViE phase had a higher tendency compared to the values for the Pre-ViE phase with P values of 0.048, 0.038, and 0.028, respectively. These three tendencies were not observed for the other ITT values in ViE phase with P values of 0.395, 0.110, and 0.103, respectively. The differences of type and surface area of the conventional hemodialyzers at the Pre-ViE phase were FX 600 (n = 1), FX 800 (n = 7), FX 1000 (n = 5) (Fresenius Medical Care, polysulfone membrane, surface areas were 1.5, 1.8, and 2.2 m2, respectively), and Nephral ST500 (n = 1) (Baxter, polyacrylonitrile membrane, surface area was 2.15 m2). The mean and standard deviation of dry weights of the patients were 63.5, 66.5 ± 9.5, 93.5 ± 15.5, and 68.0 kg for the groups of FX 600, FX 800, FX 1000, and ST500, respectively. The ST500 was used for patients who had demonstrated sensitivity to the polysulfone membrane. The differences of hemodialyzers used at the Pre-ViE phase did not show noticeable differences in the values of reduction ratios, as well as Kt/V and KUF as shown in Table 1.
Biocompatibility evaluation
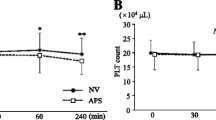
Fluctuations of leucocyte, platelet, and C3a levels during dialysis session are illustrated in Fig. 1. Both the Pre-ViE and ViE phase measurements showed a reduction of leucocyte levels at 15 min after the initiation of dialysis treatment averaging 10–20% and a return to the pre-dialysis level at post-dialysis. Regarding the platelet levels, the Pre-ViE phase measurement showed an approximately 5% reduction at both 15 min after initiation and at post-dialysis, while both ViE phase measurements showed smaller fluctuations. Both post-dialysis levels of the ViE phase measurements almost returned to pre-dialysis levels, while post-dialysis levels of the Pre-ViE phase measurements did not recover to pre-dialysis levels, and showed a greater than 5% reduction. For complement activation evaluated by C3a levels, all Pre-ViE and ViE phase measurements showed activation of about 100–150% at 15 min after initiation of treatments with the return to the pre-dialysis levels at post-dialysis. The values of these biomarkers showed no tendency of deterioration nor improvement throughout the study period as indicated in Table 2.

Fluctuation of biocompatibility markers during the dialysis sessions for the intent-to-treat (ITT) population. Figures a, b, and c correspond to fluctuations of leucocyte, platelet, and C3a, respectively. Open circles, filled circles, and filled squares indicate measurements at the Pre-ViE phase, ViE phase 1st measurement, and ViE phase 2nd measurement, respectively. The 15 min after initiation and post-dialysis values were first normalized by defining the pre-dialysis values as baseline (100%) and using the hematocrit correction equation. The changes (%) at 15 min and post-dialysis are shown in the figure by indicating the pre-dialysis point as 0%. The bars indicate standard errors and P values indicate the results of paired t test compared to the Pre-ViE phase
AE evaluation
The summary of the AEs for the SAA population is shown in Table 3. In all phases of the study, AEs were commonly observed with proportions varying between 78.6 and 100% of patients having at least one event. Most of the AEs were related to the dialysis procedures themselves and the remainder were related to other non-dialysis-related factors. Two of the AEs were related to the ViE-21 and occurred in a single patient. The patient began the study having 7 dialysis treatments with the conventional hemodialyzer, then switched to the ViE-21 for treatment session 8 and continued with the ViE-21 through treatment session 28. During treatment session 26, the patient experienced a mild event of pruritus. The pruritus progressed and during the next treatment session 27, the patient experienced a full-body rash. The patient was given 25 mg of diphenhydramine during the dialysis session. The next day, the patient was given another 50 mg of diphenhydramine. The patient also switched back to the conventional hemodialyzer for session 29 and then withdrew from the study by patient’s decision. The rash did not resolve after the diphenhydramine administration and the patient started taking prednisone 2 days after treatment session 29 for 10 days. The event was considered resolved after about 3 weeks from session 26. There were no other identified changes to the patient’s treatment to explain the body rash, and it is possible that this was a delayed hypersensitivity reaction to ViE-21. Among the AEs, one event was categorized in SAE. The patient complained of weakness, dizziness, and nausea during dialysis at session 47 in the Post-ViE phase. Additional descriptions of the symptoms included vertigo and generalized body weakness. The patient was treated with dimenhydrinate and discharged. This was coded as vertigo and was judged to be unrelated to the ViE-21 or the conventional hemodialyzer, but with an unknown relationship to the dialysis procedure.
Occurrence proportions of AEs
The occurrence proportions of all or each symptom during the study for the SAA population are summarized in Table 4. The frequency of dialysis sessions with any AE was lower in the ViE phase (29.7%) compared to Pre- and Post-ViE phases (36.6%). Specifically, the frequency of sessions with hypotension, which was the most common AE in all phases, was lower in the ViE phase at 17.0% compared to that in the Pre- and Post-ViE phases at 24.7%.
Device malfunctions
There were three device malfunctions recorded in one patient in the ViE phase. These three device malfunctions were due to thrombus formation. These events occurred more than 3 h after the initiation of dialysis in a single patient with long dialysis sessions. The sessions were continued after replacement of the ViE-21 and completed without any further problems.
Anticoagulant-free sessions
During the study, there were three sessions performed without anti-coagulant administration. All these sessions occurred during the ViE phase using ViE-21 for three different patients. The decision not to use anticoagulation was made by the clinician to reduce the risk of bleeding due to recent falls and potential head trauma. All three sessions were completed with no problems, particularly, without thrombus formation or system clotting.
Discussion
Based on this study, the ViE-21 can be used as a high-flux hemodialyzer with essentially comparable performance to conventional high-flux hemodialyzers. Allergic type reactions were observed in the ViE phase in one patient; however, the symptoms were not severe. Such allergic-type reactions are commonly observed in these complex hemodialysis patients and can be observed with all hemodialyzer types. Although three episodes of dialyzer clotting were observed in one patient during the ViE-phase, retrospective chart review showed that this patient also had problems with dialyzer clotting when conventional hemodialyzers were used before the study. Furthermore, no serious AEs related to the ViE-21 were recorded. Therefore, ViE-21 are equally safe compared to conventional polysulfone high-flux hemodialyzers.
Regarding biocompatibility, ViE-21 showed no detectable differences compared to conventional hemodialyzers on leucocyte and C3a fluctuations during the dialysis treatments. However, ViE-21 showed more stability in platelet fluctuations compared to conventional hemodialyzers, and this may indicate a lower activation property of platelets. Previous studies have demonstrated that vitamin E-bonded polysulfone hemodialyzers were successful at reducing anticoagulation requirements in dialysis treatments [11, 12] and showed comparable anticoagulation with heparin-coated hemodialyzers [13]. An in vitro study reported that vitamin E-bonded membranes might reduce platelet activation driven by leucocytes [14]. During this study, three anticoagulation-free sessions were successfully performed using ViE-21. Therefore, vitamin E-bonding on polysulfone membranes may reduce platelet activation during dialysis sessions and may reduce the need for anticoagulant use during dialysis therapy.
Another interesting finding was that the occurrence of AEs during the ViE phase was lower than during Pre- and Post-ViE phases. In particular, the frequency of hypotensive events was reduced by about 30% in the ViE-phase compared to the Pre- and Post-ViE phases. In previous reports, symptoms of intradialytic hypotension were improved using vitamin E-bonded hemodialyzers [15, 16]; and thus, these hemodialyzers may potentially reduce the incidence of hypotension.
In this study, there were no findings showing a reduction of the dosage requirement for erythropoiesis stimulating agents (ESA) or improvement of hemoglobin level by post hoc analysis, even though previous studies demonstrated such improvement in ESA resistance [17,18,19,20,21,22,23,24]. This may be due to the observation period consisting of 3 months, which was not sufficient to optimally evaluate improvements in anemia. Also, previous studies of vitamin E-bonded polysulfone hemodialyzers have demonstrated a reduction in oxidative and inflammatory markers [18, 25,26,27]; however, this was not evaluated in the present study.
Conclusion
This study provides performance and safety data for the ViE-21. Vitamin E-bonded polysulfone membrane hemodialyzers may have a beneficial role in reducing platelet activation and lowering the frequency of AEs such as hypotension during hemodialysis treatment. Limitations of this study include the small sample size, and the variations in the surface area of the hemodialyzers used. To confirm these findings, randomized controlled trials with proper comparison design are required.
References
Bowry SK. Nano-controlled membrane spinning technology: regulation of pore size, distribution and morphology of a new polysulfone dialysis membrane. Contrib Nephrol. 2002;137:85–94.
Krane V, Krieter DH, Olschewski M, et al. Dialyzer membrane characteristics and outcome of patients with type 2 diabetes on maintenance hemodialysis. Am J Kidney Dis. 2007;49:267–75.
Del Vecchio L, Locatelli F, Carini M. What we know about oxidative stress in patients with chronic kidney disease on dialysis-clinical effects, potential treatment, and prevention. Semin Dial. 2011;24:56–64.
Galli F. Vitamin E-modified dialyzers. Contrib Nephrol. 2002;137:95–105.
Floridi A, Piroddi M, Pilolli F, Matsumoto Y, Aritomi M, Galli F. Analysis method and characterization of the antioxidant capacity of vitamin E-interactive polysulfone hemodialyzers. Acta Biomater. 2009;5:2974–82.
Aritomi M, Galli F. Review of the effectiveness of cellulose- and polysulfone-based vitamin E-bonded dialysis membranes. In: Technical problems in patients on hemodialysis (Intech open access book), 2011; 17, 295–302.
Piroddi M, Pilolli F, Aritomi M, Galli F. Vitamin E as a functional and biocompatibility modifier of synthetic hemodialyzer membranes: an overview of the literature on vitamin E-modified hemodialyzer membranes. Am J Nephrol. 2012;35:559–72.
Yamadera S, Nakamura Y, Inagaki M, et al. Vitamin E-coated dialyzer inhibits oxidative stress. Blood Purif. 2017;44:288–93.
FDA Guidance for Industry and CDRH Reviewers (1998) Guidance for the content of premarket notifications for conventional and high permeability hemodialyzers
ISO 14155 (2011) Clinical investigation of medical devices for human subjects—good clinical practice, Second edition
Aoun B, Janssen-Lozinska Y, Ulinski T. Effect of vitamin E coated dialyzers on anticoagulation requirement in hemodialyzed children. Saudi J Kidney Dis Transpl. 2010;21:466–70.
Torato T, Doi K, Negishi K, et al. Efficacy of vitamin E-bonded polysulfone dialyzer and polysulfone dialyzer on a series of non-anticoagulant hemodialysis. ASAIO J. 2013;59:284–5.
Islam MS, Hassan ZA, Chalmin F, et al. Vitamin E-coated and heparin-coated dialyzer membranes for heparin-free hemodialysis: A multicenter, randomized, crossover trial. Am J Kidney Dis. 2016;68:752–62.
Tsukao H, Kokubo K, Takahashi H, et al. Activation of platelets upon contact with a vitamin E-coated/non-coated surface. J Artif Organs. 2013;16:193–205.
Matsumura M, Sasaki H, Sekizuka K, et al. Improved management of intradialytic hypotension (IDH) using vitamin E-bonded polysulfone membrane dialyzer. Int J Artif Organs. 2010;33:147–53.
Koremoto M, Takahara N, Takahashi M, et al. Improvement of intradialytic hypotension in diabetic hemodialysis patients using vitamin E-bonded polysulfone membrane dialyzers. Artif Organs. 2012;36:901–10.
Andrulli S, Di Filippo S, Manzoni C, et al. Effect of synthetic vitamin E-bonded membrane on responsiveness to erythropoiesis-stimulating agents in hemodialysis patients: a pilot study. Nephron Clin Pract. 2010;115:c82–9.
Panichi V, Rosati A, Paoletti S, et al. A vitamin E-coated polysulfone membrane reduces serum levels of inflammatory markers and resistance to erythropoietin-stimulating agents in hemodialysis patients: results of a randomized cross-over multicenter trial. Blood Purif. 2011;32:7–14.
Mandolfo S, Corradi B, Bucci R, Farina M, Pilolli F, Galli F. Evaluation of the impact of a new synthetic vitamin E-bonded membrane on anemia and rHuEPO requirement in ESRD patients with central venous catheters: a pilot study. Int Urol Nephrol. 2012;44:1493–500.
Lines SW, Carter AM, Dunn EJ, Lindley EJ, Tattersall JE, Wright MJ. A randomized controlled trial evaluating the erythropoiesis stimulating agent sparing potential of a vitamin E-bonded polysulfone dialysis membrane. Nephrol Dial Transplant. 2014;29:649–56.
Sanaka T, Mochizuki T, Kinugasa E, et al. Randomized controlled open-label trial of vitamin E-bonded polysulfone dialyzer and erythropoiesis-stimulating agent response. Clin J Am Soc Nephrol. 2013;8:969–78.
Bargnoux AS, Cristol JP, Jaussent I, et al. Vitamin E-coated polysulfone membrane improved red blood cell antioxidant status in hemodialysis patients. J Nephrol. 2013;26:556–63.
Locatelli F, Andrulli S, Viganò SM, et al. Evaluation of the impact of a new synthetic vitamin E-bonded membrane on the hypo-responsiveness to the erythropoietin therapy in hemodialysis patients: a multicenter study. Blood Purif. 2017;43:338–45.
D'Arrigo G, Baggetta R, Tripepi G, Galli F, Bolignano D. Effects of vitamin E-coated versus conventional membranes in chronic hemodialysis patients: a systematic review and meta-analysis. Blood Purif. 2016;43:101–22.
Morimoto H, Nakao K, Fukuoka K, et al. Long-term use of vitamin E-coated polysulfone membrane reduces oxidative stress markers in hemodialysis patients. Nephrol Dial Transplant. 2005;20:2775–82.
Calò LA, Naso A, D'Angelo A, et al. Molecular biology-based assessment of vitamin E-coated dialyzer effects on oxidative stress, inflammation, and vascular remodeling. Artif Organs. 2011;35:E33–E39.
Kitamura Y, Kamimura K, Yoshioka N, et al. The effect of vitamin E-bonded polysulfone membrane dialyzer on a new oxidative lipid marker. J Artif Organs. 2013;16:206–10.
Acknowledgements
We are grateful to the hemodialysis staff of the dialysis unit and the clinical research coordinators at St. Paul’s Hospital, especially, Ms. Michele Trask, Mr. Dave Morrison, Mr. Luisito Sera Josep, Ms. Vela Katy, Mr. Perry Taylor, and Ms. Sheriff Zainab for their activities in conducting the study. We are also grateful for the staff of Boston Biomedical Associates for their activities as the contract research organization. Drs. Brian Pereira and Sally Hood are acknowledged for protocol preparation, and medical advice, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The study was financially supported by Asahi Kasei Medical Co., Ltd. M. Aritomi and M. Nagase are employees of Asahi Kasei Medical Co., Ltd. There are no other conflicts of interests for this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kiaii, M., Aritomi, M., Nagase, M. et al. Clinical evaluation of performance, biocompatibility, and safety of vitamin E-bonded polysulfone membrane hemodialyzer compared to non-vitamin E-bonded hemodialyzer. J Artif Organs 22, 307–315 (2019). https://doi.org/10.1007/s10047-019-01110-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10047-019-01110-w