
Abstract
We analyze the influence of different groups on the intermolecular energy of aromatic homodimers and on the interaction between a single aromatic molecule and a graphene surface. The analysis is performed for benzene, phenol, catechol, and dopamine. For calculating the energies, we employ density functional theory within the local density approximation (LDA-DFT). Our results show that the lowest intermolecular energies between the aromatic molecules are related to the T-shaped configurations. This lower energy results from the quadrupole interaction. In the case of the interaction between the graphene sheet and the aromatic molecules, the lowest energy configuration is the face to face. The adsorption energy of a molecule on a graphene surface involves π − π interactions that explain the face to face arrangement. These results provide insight into the manner by which substituents can be utilized in crystal engineering, supramolecular chemistry, bioinspired materials, formation of various molecular clusters, parameterization of force fields suitable for classical simulations, and design of novel sensing, drug delivery, and filters based on graphene.








Similar content being viewed by others

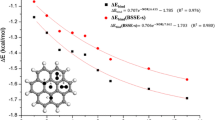
References
Iijima S, Ichihashi T (1993) Single-shell carbon nanotubes of 1-nm diameter. Nature 363:603
Moraes EE, Manhabosco TM, De Oliveira AB, Batista RJ (2012) Tunable band gap of boron nitride interfaces under uniaxial pressure. J Phys: Condens Matter 24:475502
Moraes EE, Coutinho-Filho MD, Batista RJ (2017) Transport properties of hydrogenated cubic boron nitride nanofilms with gold electrodes from density functional theory. ACS Omega 2(4):1696–1701
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Electric field effect in atomically thin carbon films. Science 306:666–669
Neto AC, Guinea F, Peres NM, Novoselov KS, Geim AK (2009) The electronic properties of graphene. Rev Mod Phys 81:109
Rao C, Sood A, Subrahmanyam K, Govindaraj A (2009) Graphene: the new two-dimensional nanomaterial. Angew Chem Int Ed 48:7752–7777
Gan T, Hu S (2011) Electrochemical sensors based on graphene materials. Microchim Acta 175:1
Tonel M, Lara I, Zanella I, Fagan S (2017) The influence of the concentration and adsorption sites of different chemical groups on graphene through first principles simulations. Phys Chem Chem Phys 19:27374–27383
Tonel MZ, Martins MO, Zanella I, Pontes RB, Fagan SB (2017) A first-principles study of the interaction of doxorubicin with graphene. Comput Theor Chem 1115:270–275
Chen RJ, Bangsaruntip S, Drouvalakis KA, Kam NWS, Shim M, Li Y, Kim W, Utz PJ, Dai H (2003) Noncovalent functionalization of carbon nanotubes for highly specific electronic biosensors. Proc Natl Acad Sci 100:4984–4989
Kang HS (2005) Theoretical study of binding of metal-doped graphene sheet and carbon nanotubes with dioxin. J Am Chem Soc 127:9839–9843
Gowtham S, Scheicher RH, Ahuja R, Pandey R, Karna SP (2007) Physisorption of nucleobases on graphene: density-functional calculations. Phys Rev B 76:033401
Varghese N, Mogera U, Govindaraj A, Das A, Maiti PK, Sood AK, Rao C (2009) Binding of dna nucleobases and nucleosides with graphene. Chem Phys Chem 10:206–210
Cazorla C (2010) Ab initio study of the binding of collagen amino acids to graphene and a-doped (a= h, ca) graphene. Thin Solid Films 518:6951–6961
Umadevi D, Sastry GN (2011) Quantum mechanical study of physisorption of nucleobases on carbon materials: graphene versus carbon nanotubes. J Phys Chem Lett 2:1572–1576
Cazorla C, Rojas-Cervellera V, Rovira C (2012) Calcium-based functionalization of carbon nanostructures for peptide immobilization in aqueous media. J Mat Chem 22:19684– 19693
Vovusha H, Sanyal S, Sanyal B (2013) Interaction of nucleobases and aromatic amino acids with graphene oxide and graphene flakes. J Phys Chem Lett 4:3710–3718
Chen L, Li X, Tanner EE, Compton RG (2017) Catechol adsorption on graphene nanoplatelets: isotherm, flat to vertical phase transition and desorption kinetics. Chem Sci 8:4771–4778
Li D, Müller MB, Gilje S, Kaner RB, Wallace GG (2008) Processable aqueous dispersions of graphene nanosheets. Nat Nanotechnol 3:101
Dong X, Fu D, Fang W, Shi Y, Chen P, Li L-J (2009) Doping single-layer graphene with aromatic molecules. Small 5:1422–1426
Wu T, Cai X, Tan S, Li H, Liu J, Yang W (2011) Adsorption characteristics of acrylonitrile, p-toluenesulfonic acid, 1-naphthalenesulfonic acid and methyl blue on graphene in aqueous solutions. Chem Eng J 173:144–149
Hwang YH, Chun HS, Ok KM, Lee K-K, Kwak K (2015) Density functional investigation of graphene doped with amine-based organic molecules. J Nanomater 2015:5
Kong L, Román-Pérez G, Soler JM, Langreth DC (2009) Energetics and dynamics of h 2 adsorbed in a nanoporous material at low temperature. Phys Rev Lett 103:096103
Cacelli I, Cinacchi G, Prampolini G, Tani A (2004) Computer simulation of solid and liquid benzene with an atomistic interaction potential derived from ab initio calculations. J Am Chem Soc 126:14278–14286
Amovilli C, Cacelli I, Cinacchi G, De Gaetani L, Prampolini G, Tani A (2007) Structure and dynamics of mesogens using intermolecular potentials derived from ab initio calculations. Theor Chem Acc 117:885–901
Cacelli I, Cimoli A, Livotto PR, Prampolini G (2012) An automated approach for the parameterization of accurate intermolecular force-fields: pyridine as a case study. J Comput Chem 33:1055–1067
Hobza P, Selzle HL, Schlag EW (1994) Structure and properties of benzene-containing molecular clusters: nonempirical ab initio calculations and experiments. Chem Rev 94:1767–1785
Bieske EJ, Dopfer O (2000) High-resolution spectroscopy of cluster ions. Chem Rev 100:3963–3998
Tarakeshwar P, Kim KS, Brutschy B (2001) σ to π conformational transition: interactions of the water trimer with π systems. J Chem Phys 114:1295–1305
Guin M, Patwari GN, Karthikeyan S, Kim KS (2009) A π-stacked phenylacetylene and 1, 3, 5-triazine heterodimer: a combined spectroscopic and ab initio investigation. Phys Chem Chem Phys 11:11207–11212
Hoeben FJ, Jonkheijm P, Meijer E, Schenning AP (2005) About supramolecular assemblies of π-conjugated systems. Chem Rev 105:1491–1546
Cerný J, Kabelác M, Hobza P (2008) Double-helical ladder structural transition in the b-dna is induced by a loss of dispersion energy. J Am Chem Soc 130:16055–16059
Meyer EA, Castellano RK, Diederich F (2003) Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed 42:1210–1250
Lee JY, Hong BH, Kim WY, Min SK, Kim Y, Jouravlev MV, Bose R, Kim KS, Hwang I-C, Kaufman LJ et al (2009) Near-field focusing and magnification through self-assembled nanoscale spherical lenses. Nature 460:498
Vaupel S, Brutschy B, Tarakeshwar P, Kim KS (2006) Characterization of weak nh- π intermolecular interactions of ammonia with various substituted π-systems. J Am Chem Soc 128:5416–5426
Tarakeshwar P, Choi HS, Kim KS (2001) Olefinic vs aromatic π- h interaction: a theoretical investigation of the nature of interaction of first-row hydrides with ethene and benzene. J Am Chem Soc 123:3323–3331
Hohenstein EG, Sherrill CD (2009) Effects of heteroatoms on aromatic π- π interactions: benzene- pyridine and pyridine dimer. J Phys Chem A 113:878–886
Piacenza M, Grimme S (2005) Van der waals complexes of polar aromatic molecules: unexpected structures for dimers of azulene. J Am Chem Soc 127:14841–14848
Ringer AL, Sherrill CD (2009) Substituent effects in sandwich configurations of multiply substituted benzene dimers are not solely governed by electrostatic control. J Am Chem Soc 131:4574–4575
Geronimo I, Singh NJ, Kim KS (2011) Can electron-rich π systems bind anions. J Chem Theory Comput 7:825–829
Kołaski M, Kumar A, Singh NJ, Kim KS (2011) Differences in structure, energy, and spectrum between neutral, protonated, and deprotonated phenol dimers: comparison of various density functionals with ab initio theory. Phys Chem Chem Phys 13:991–1001
Hohenberg P (1964) Inhomogeneous electron gas. Phys Rev B 136:864
Kohn W (1965) Self-consistent equations including exchange and correlation effects. Phys Rev A 140:1133
Soler JM, Artacho E, Gale JD, García A, Junquera J, Ordejón P, Sánchez-Portal D (2002) The siesta method for ab initio order-n materials simulation. J Phys: Condens Matter 14:2745
Perdew JP, Zunger A (1981) Self-interaction correction to density-functional approximations for many-electron systems. Phys Rev B 23:5048
Boys SF, Bernardi Fd (1970) The calculation of small molecular interactions by the differences of separate total energies. some procedures with reduced errors. Molec Phys 19:553–566
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865
Artacho E, Anglada E, Diéguez O, Gale JD, García A, Junquera J, Martin RM, Ordejón P, Pruneda JM, Sánchez-Portal D et al (2008) The siesta method; developments and applicability. J Phys: Condens Matter 20:064208
Grimme S (2006) Semiempirical gga-type density functional constructed with a long-range dispersion correction. J Comput Chem 27:1787–1799
Prampolini G, Livotto PR, Cacelli I (2015) Accuracy of quantum mechanically derived force-fields parameterized from dispersion-corrected dft data: the benzene dimer as a prototype for aromatic interactions. J Chem Theory Comput 11:5182–5196
Tournus F, Charlier J-C (2005) Ab initio study of benzene adsorption on carbon nanotubes. Phys Rev B 71:165421
Sato T, Tsuneda T, Hirao K (2005) A density-functional study on π-aromatic interaction: benzene dimer and naphthalene dimer. J Chem Phys 123:104307
Chakarova-Käck SD, Schröder E, Lundqvist BI, Langreth DC (2006) Application of van der waals density functional to an extended system: Adsorption of benzene and naphthalene on graphite. Phys Rev Lett 96:146107
Sinnokrot MO, Sherrill CD (2003) Unexpected substituent effects in face-to-face π-stacking interactions. J Phys Chem A 107:8377–8379
Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K (2002) Origin of attraction and directionality of the π/π interaction: model chemistry calculations of benzene dimer interaction. J Am Chem Soc 124:104–112
Grover J, Walters E, Hui E (1987) Dissociation energies of the benzene dimer and dimer cation. J Phys Chem Chem 91:3233–3237
Sinnokrot MO, Sherrill CD (2004) Substituent effects in π- π interactions: sandwich and t-shaped configurations. J Am Chem Soc 126:7690–7697
Cozzi F, Siegel J (1995) Interaction between stacked aryl groups in 1, 8-diarylnaphthalenes: dominance of polar/π over charge-transfer effects. Pure Appl Chem 67:683–689
Cozzi F, Ponzini F, Annunziata R, Cinquini M, Siegel JS (1995) Polar interactions between stacked π systems in fluorinated 1, 8-diarylnaphthalenes: importance of quadrupole moments in molecular recognition. Angew Chem Int Ed 34:1019–1020
Barone V, Cacelli I, Ferretti A, Prampolini G (2017) Noncovalent interactions in the catechol dimer. Biomim 2:18
Purushotham U, Sastry GN (2012) Exploration of conformations and quantum chemical investigation of l-tyrosine dimers, anions, cations and zwitterions: a dft study. Theor Chem Acc 131:1093
Furlan A, Almarza NG, Barbosa M (2016) Lattice model for water-solute mixtures. J Chem Phys 145:144501
Furlan A, Lomba E, Barbosa M (2017) Temperature of maximum density and excess properties of short-chain alcohol aqueous solutions: a simplified model simulation study. J Chem Phys 146:144503
AlZahrani A (2010) First-principles study on the structural and electronic properties of graphene upon benzene and naphthalene adsorption. Appl Surf Sci 257:807–810
Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, Cavazzoni C, Ceresoli D, Chiarotti GL, Cococcioni M, Dabo I et al (2009) Quantum espresso: a modular and open-source software project for quantum simulations of materials. J Phys: Condens Matter 21:395502
Kong L, Enders A, Rahman TS, Dowben PA (2014) Molecular adsorption on graphene. J Phys: Condens Matter 26:443001
Hernández JMG, Anota EC, de la Cruz MTR, Melchor MG, Cocoletzi GH (2012) First principles studies of the graphene-phenol interactions. J Mol Mod 18:3857–3866
Giese TJ, York DM (2010) Density-functional expansion methods: evaluation of lda, gga, and meta-gga functionals and different integral approximations. J Chem Phys 133:244107
Boukhvalov D (2013) Dft modeling of the covalent functionalization of graphene: from ideal to realistic models. Rsc Adv 3:7150–7159
Avila Y, Cocoletzi GH, Romero MT (2014) First principles calculations of phenol adsorption on pristine and group iii (b, al, ga) doped graphene layers. J Mol Model 20:2112
Mian SA, Saha LC, Jang J, Wang L, Gao X, Nagase S (2010) Density functional theory study of catechol adhesion on silica surfaces. J Phys Chem C 114:20793–20800
Mian SA, Yang L-M, Saha LC, Ahmed E, Ajmal M, Ganz E (2014) A fundamental understanding of catechol and water adsorption on a hydrophilic silica surface: exploring the underwater adhesion mechanism of mussels on an atomic scale. Langmuir 30:6906–6914
Fernández ACR, Castellani NJ (2017) Noncovalent interactions between dopamine and regular and defective graphene. ChemPhysChem 18(15):2065–2080
Zhang H-p, Lin X-y, Lu X, Wang Z, Fang L, Tang Y (2017) Understanding the interfacial interactions between dopamine and different graphenes for biomedical materials. Mater Chem Front 1(6):1156–1164
Antony J, Grimme S (2008) Structures and interaction energies of stacked graphene–nucleobase complexes. Phys Chem Chem Phys 10:2722–2729
Li Y, Liao M, Zhou J (2018) Catechol and its derivatives adhesion on graphene: insights from molecular dynamics simulations. J Phys Chem C 122:22965–22974
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de Moraes, E. ., Tonel, M.Z., Fagan, S.B. et al. Density functional theory study of π-aromatic interaction of benzene, phenol, catechol, dopamine isolated dimers and adsorbed on graphene surface. J Mol Model 25, 302 (2019). https://doi.org/10.1007/s00894-019-4185-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4185-2