
Abstract
Background
Autosomal recessive polycystic kidney disease (ARPKD) is genetically one of the least heterogeneous ciliopathies, resulting primarily from mutations of PKHD1. Nevertheless, 13–20% of patients diagnosed with ARPKD are found not to carry PKHD1 mutations by sequencing. Here, we assess whether PKHD1 copy number variations or second locus mutations explain these cases.
Methods
Thirty-six unrelated patients with the clinical diagnosis of ARPKD were screened for PKHD1 point mutations and copy number variations. Patients without biallelic mutations were re-evaluated and screened for second locus mutations targeted by the phenotype, followed, if negative, by clinical exome sequencing.
Results
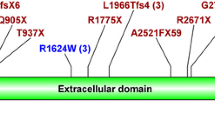
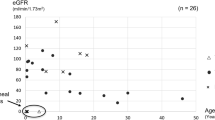
Twenty-eight patients (78%) carried PKHD1 point mutations, three of whom on only one allele. Two of the three patients harbored in trans either a duplication of exons 33–35 or a large deletion involving exons 1–55. All eight patients without PKHD1 mutations (22%) harbored mutations in other genes (PKD1 (n = 2), HNF1B (n = 3), NPHP1, TMEM67, PKD1/TSC2). Perinatal respiratory failure, a kidney length > +4SD and early-onset hypertension increase the likelihood of PKHD1-associated ARPKD. A patient compound heterozygous for a second and a last exon truncating PKHD1 mutation (p.Gly4013Alafs*25) presented with a moderate phenotype, indicating that fibrocystin is partially functional in the absence of its C-terminal 62 amino acids.
Conclusions
We found all ARPKD cases without PKHD1 point mutations to be phenocopies, and none to be explained by biallelic PKHD1 copy number variations. Screening for copy number variations is recommended in patients with a heterozygous point mutation.


Similar content being viewed by others
References
Kurschat CE, Muller RU, Franke M, Maintz D, Schermer B, Benzing T (2014) An approach to cystic kidney diseases: the clinician’s view. Nat Rev Nephrol 10:687–699
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30:259–269
Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, Kramer C, Ortiz-Bruchle N, Hilger N, Metzis V, Hiersche M, Tay SY, Tunningley R, Vij S, Courtney AD, Whittle B, Wuhl E, Vester U, Hartleben B, Neuber S, Frank V, Little MH, Epting D, Papathanasiou P, Perkins AC, Wright GD, Hunziker W, Gee HY, Otto EA, Zerres K, Hildebrandt F, Roy S, Wicking C, Bergmann C (2017) Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet 49:1025–1034
Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H, Anton-Gamero M, Argente J, Bussell AM, Brandli A, Cheshire C, Crowne E, Dumitriu S, Drynda R, Hamilton-Shield JP, Hayes W, Hofherr A, Iancu D, Issler N, Jefferies C, Jones P, Johnson M, Kesselheim A, Klootwijk E, Koettgen M, Lewis W, Martos JM, Mozere M, Norman J, Patel V, Parrish A, Perez-Cerda C, Pozo J, Rahman SA, Sebire N, Tekman M, Turnpenny PD, Hoff WV, Viering D, Weedon MN, Wilson P, Guay-Woodford L, Kleta R, Hussain K, Ellard S, Bockenhauer D (2017) Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol 28:2529–2539
Nagano J, Kitamura K, Hujer KM, Ward CJ, Bram RJ, Hopfer U, Tomita K, Huang C, Miller RT (2005) Fibrocystin interacts with CAML, a protein involved in Ca2+ signaling. Biochem Biophys Res Commun 338:880–889
Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP (2006) Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17:178–187
Wu M, Yu S (2016) New insights into the molecular mechanisms targeting tubular channels/transporters in PKD development. Kidney Dis (Basel) 2:128–135
Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E, Satlin LM (2005) Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Ren Physiol 289:F978–F988
Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD (2006) Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Ren Physiol 290:F1320–F1328
Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, Rudenhed B, Nasr SH, Torres VE, Torra R, Bogdanova N, Harris PC (2010) Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 21:1097–1102
Losekoot M, Ruivenkamp CA, Tholens AP, Grimbergen JE, Vijfhuizen L, Vermeer S, Dijkman HB, Cornelissen EA, Bongers EM, Peters DJ (2012) Neonatal onset autosomal dominant polycystic kidney disease (ADPKD) in a patient homozygous for a PKD2 missense mutation due to uniparental disomy. J Med Genet 49:37–40
Bellanne-Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, Dubois-Laforgue D, Dusselier L, Gautier JF, Jadoul M, Laloi-Michelin M, Jacquesson L, Larger E, Louis J, Nicolino M, Subra JF, Wilhem JM, Young J, Velho G, Timsit J (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54:3126–3132
Hiesberger T, Shao X, Gourley E, Reimann A, Pontoglio M, Igarashi P (2005) Role of the hepatocyte nuclear factor-1beta (HNF-1beta) C-terminal domain in Pkhd1 (ARPKD) gene transcription and renal cystogenesis. J Biol Chem 280:10578–10586
Williams SS, Cobo-Stark P, Hajarnis S, Aboudehen K, Shao X, Richardson JA, Patel V, Igarashi P (2014) Tissue-specific regulation of the mouse Pkhd1 (ARPKD) gene promoter. Am J Physiol Ren Physiol 307:F356–F368
Denamur E, Delezoide AL, Alberti C, Bourillon A, Gubler MC, Bouvier R, Pascaud O, Elion J, Grandchamp B, Michel-Calemard L, Missy P, Zaccaria I, Le Nagard H, Gerard B, Loirat C, Societe Francaise de F, Barbet J, Beaufrere AM, Berchel C, Bessieres B, Boudjemaa S, Buenerd A, Carles D, Clemenson A, Dechelotte P, Devisme L, Dijoud F, Esperandieu O, Fallet C, Gonzales M, Hillion Y, Jacob B, Joubert M, Kermanach P, Lallemand A, Laquerriere A, Laurent N, Liprandi A, Loeuillet L, Loget P, Martinovic J, Menez F, Narcy F, Roux JJ, Rouleau-Dubois C, Sinico M, Tantau J, Wann AR (2010) Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int 77:350–358
Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schoneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Buttner R, Zerres K, Apn (2005) Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int 67:829–848
Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler NG, Roque A, Douek DC, Graf J, Huizing M, Bryant JC, Mohan P, Gahl WA, Heller T (2013) Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology 144:112–121 e112
Shneider BL, Magid MS (2005) Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transplant 9:634–639
Turkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, Choyke PL, Gunay-Aygun M (2009) Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol 39:100–111
Zerres K, Mucher G, Becker J, Steinkamm C, Rudnik-Schoneborn S, Heikkila P, Rapola J, Salonen R, Germino GG, Onuchic L, Somlo S, Avner ED, Harman LA, Stockwin JM, Guay-Woodford LM (1998) Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 76:137–144
Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MT, Utsch B, Becker C, Nurnberg G, Nurnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F (2009) Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 46:663–670
Tory K, Rousset-Rouviere C, Gubler MC, Moriniere V, Pawtowski A, Becker C, Guyot C, Gie S, Frishberg Y, Nivet H, Deschenes G, Cochat P, Gagnadoux MF, Saunier S, Antignac C, Salomon R (2009) Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int 75:839–847
Arbeiter A, Buscher R, Bonzel KE, Wingen AM, Vester U, Wohlschlager J, Zerres K, Nurnberger J, Bergmann C, Hoyer PF (2008) Nephrectomy in an autosomal recessive polycystic kidney disease (ARPKD) patient with rapid kidney enlargement and increased expression of EGFR. Nephrol Dial Transplant 23:3026–3029
Rossetti S, Harris PC (2007) Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol 18:1374–1380
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN (2017) The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 136:665–677
Miyazaki J, Ito M, Nishizawa H, Kato T, Minami Y, Inagaki H, Ohye T, Miyata M, Boda H, Kiriyama Y, Kuroda M, Sekiya T, Kurahashi H, Fujii T (2015) Intragenic duplication in the PKHD1 gene in autosomal recessive polycystic kidney disease. BMC Med Genet 16:98
Bergmann C, Kupper F, Schmitt CP, Vester U, Neuhaus TJ, Senderek J, Zerres K (2005) Multi-exon deletions of the PKHD1 gene cause autosomal recessive polycystic kidney disease (ARPKD). J Med Genet 42:e63
Sweeney WE Jr, Avner ED (2011) Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol 26:675–692
Krall P, Pineda C, Ruiz P, Ejarque L, Vendrell T, Camacho JA, Mendizabal S, Oliver A, Ballarin J, Torra R, Ars E (2014) Cost-effective PKHD1 genetic testing for autosomal recessive polycystic kidney disease. Pediatr Nephrol 29:223–234
Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, Rudnik-Schoneborn S, Furu L, Onuchic LF, De Baca M, Germino GG, Guay-Woodford L, Somlo S, Moser M, Buttner R, Zerres K (2003) Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol 14:76–89
Obeidova L, Seeman T, Elisakova V, Reiterova J, Puchmajerova A, Stekrova J (2015) Molecular genetic analysis of PKHD1 by next-generation sequencing in Czech families with autosomal recessive polycystic kidney disease. BMC Med Genet 16:116
Sharp AM, Messiaen LM, Page G, Antignac C, Gubler MC, Onuchic LF, Somlo S, Germino GG, Guay-Woodford LM (2005) Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet 42:336–349
Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG, Piwnica-Worms K, Bryant J, Bernardini I, Fischer R, Huizing M, Guay-Woodford L, Gahl WA (2010) PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab 99:160–173
Losekoot M, Haarloo C, Ruivenkamp C, White SJ, Breuning MH, Peters DJ (2005) Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum Genet 118:185–206
Javorszky E, Moriniere V, Kerti A, Balogh E, Piko H, Saunier S, Karcagi V, Antignac C, Tory K (2017) QMPSF is sensitive and specific in the detection of NPHP1 heterozygous deletions. Clin Chem Lab Med 55:809–816
Orosz O, Rajta I, Vajas A, Takacs L, Csutak A, Fodor M, Kolozsvari B, Resch M, Senyi K, Lesch B, Szabo V, Berta A, Balogh I, Losonczy G (2017) Myopia and late-onset progressive cone dystrophy associate to LVAVA/MVAVA exon 3 interchange haplotypes of opsin genes on chromosome X. Invest Ophthalmol Vis Sci 58:1834–1842
Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, Germino GG, Guay-Woodford LM, Somlo S (2003) Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol 14:2004–2014
Rossetti S, Torra R, Coto E, Consugar M, Kubly V, Malaga S, Navarro M, El-Youssef M, Torres VE, Harris PC (2003) A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int 64:391–403
Wang S, Wu M, Yao G, Zhang J, Zhou J (2014) The cytoplasmic tail of FPC antagonizes the full-length protein in the regulation of mTOR pathway. PLoS One 9:e95630
Follit JA, Li L, Vucica Y, Pazour GJ (2010) The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J Cell Biol 188:21–28
Outeda P, Menezes L, Hartung EA, Bridges S, Zhou F, Zhu X, Xu H, Huang Q, Yao Q, Qian F, Germino GG, Watnick T (2017) A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int 92:1130–1144
Funding
This work was supported by OTKA K109076 and Ministry of National Economy, Hungary GINOP-2.3.2-15-2016-00039 (to István Balogh, Zoltán Maróti and Tibor Kalmár), MTA-SE Lendulet Research Grant (LP2015-11/2015) of the Hungarian Academy of Sciences and NKFIA/OTKA K109718, KH125566 (to Kálmán Tory).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Parents and patients gave informed written consent.
Rights and permissions
About this article

Cite this article
Szabó, T., Orosz, P., Balogh, E. et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr Nephrol 33, 1713–1721 (2018). https://doi.org/10.1007/s00467-018-3992-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-018-3992-5